
Abstract
Objective
Respiratory syncytial virus (RSV) and rhinovirus (RV) are common viral infections that may result in post-viral airway/atopic disease. By understanding the antiviral immune response involved, and the mechanisms that translate/associate with post-viral airway disease, further research can be directed to potential treatments that affect these mechanisms.
Data sources
Utilized peer-reviewed manuscripts listed in PubMed that had relevance to RSV/RV and development of atopic/airway disease in both humans and mice.
Study selections
Studies that explained the mechanisms behind antiviral response were selected.
Results
RSV infections have been associated with post-viral airway disease primarily in those without preexisting atopy; however, the mechanistic link connecting the viral infection with atopy is less clear. Mouse models (in particular those using Sendai virus, a virus related to RSV) provide a potential mechanistic pathway that may explain the linkage between RSV and post-viral airway disease. RV infection also can drive post-viral airway disease, but unlike RSV, this seems to occur only in those with preexisting atopy. Studies explore this link by demonstrating an impaired interferon response in atopic individuals, which may make them more susceptible to development of post-viral airway disease with RV infection.
Conclusion
Both RSV and RV are associated with a risk for developing post-viral airway disease and atopy. However, the mechanisms that connect these viruses with post-viral disease appear to be disparate, suggesting that treatments to prevent post-viral airway disease may need to be specific to the viral etiology.
Introduction
Asthma is a chronic disorder that is consistently increasing among the global pediatric population; this has caused an impactful burden on the healthcare system (Citation1). Asthma not only causes recurrent hospital admissions for pediatric patients, but also presents a lifelong burden for these patients (Citation1). There are a few hypotheses for the increasing prevalence of asthma including environmental factors as well as host factors such as atopic disease (Citation1). Another possible explanation for the onset of asthma in the pediatric population is the prevalence of viral respiratory tract infections (Citation2). Almost all children by the age of 2 years have had a respiratory syncytial virus (RSV) infection and almost all have had a rhinovirus (RV) infection within one year of life (Citation3). These viral respiratory tract infections induce wheezing episodes, an early marker of asthma development, which may continue well past the course of the infection. This review will explore the differences between RSV (respiratory syncytial virus) and RV (rhinovirus) in the development of post-viral airway disease (asthma), and potential mechanisms responsible for the difference between these two viruses.
Methods
Primary database used was PubMed and searches included the following MeSH terms: [asthma AND RSV], [asthma AND RV], [allergic sensitization AND RV], [allergic sensitization AND RSV], [neutrophils AND RSV], [neutrophils AND RV], [atopy AND RSV], and [atopy AND RV]. Results were filtered to include only Clinical Trial, Randomized Controlled Trial, or Review. Using this search process, peer-reviewed journals and literature reviews were selected with around 800 search results with a total of 31 papers chosen for the review. The paper selection was informed by our prior literature reviews (Citation4,Citation5). If particular papers did not fit the premise of this review but were included in the MeSH term search, we examined other papers by the same authors to select papers that highlighted the mechanisms of RSV, RV, and development of post-viral wheeze. Potential flaws in this methodology include incorporating older sources and using MeSH terms that may be too broad or not specific to the particular review premise.
Results/discussion
Epidemiology
RSV and RV are the two main viral infections that induce viral wheezing in children and infants (Citation6). Both RSV and RV differ in their manifestations in the pediatric population as well as their effects on the development of asthma and other atopic disease including allergic sensitization.
Respiratory syncytial virus
Bronchiolitis is inflammation of the small airways of the lung, and there have been several studies suggesting that RSV is one of the main pathogens responsible for the onset of bronchiolitis in pediatric patients (Citation7,Citation8). Studies looking at RSV bronchiolitis have shown that having RSV related bronchiolitis in the first year of life is a risk factor for development of wheezing (Citation7). Further, studies describe that wheezing in the first 3 years of life is associated with an increased risk of asthma at ages 6, 8, and 11 years but not significantly at 13 years (Citation9). On the contrary, Sigurs’ group suggests that RSV bronchiolitis is a risk factor for asthma and recurrent wheezing in early adolescence and well into early adulthood (Citation10,Citation11). Further exploration suggests that the difference in findings from Sigurs’ study could be due to the severe viral disease in a patient population <1 year of age as well as “differences in populations, climatic factors, and also allergen load” (Citation10).
RSV is clearly associated with the development of asthma, but it is also associated with allergic sensitization and further development of atopic disease. Sigurs’ group proposes that patients with RSV had more positive skin tests as well as serum IgE tests (Citation10). Ultimately, Sigurs’ studies show that RSV bronchiolitis is a risk factor for post-viral allergic disease (Citation10).
Rhinovirus
Rhinovirus was found to be the most common pathogen for a patient’s viral-induced first wheeze and is one of the most common risk factors for recurrent wheezing (Citation6). An important connection with RV is the association with allergic sensitization. Several studies indicate that patients with atopic characteristics who are infected with RV are predisposed to development of post-viral wheeze (Citation6–8). One study shows that patients with high eosinophil count, eczema, atopic eczema, and parents with allergic rhinitis who are infected with RV may be more likely to develop post-viral wheeze (Citation6). Another study shows that patients ages 1 through 6 have an increased likelihood of wheezing with RV if these patients had allergic sensitization before viral disease (Citation8). Both studies delineate the idea that RV is associated with atopy, and preexisting atopy can result in a wheeze with RV infection.
Similar to RSV, RV has an association with age. According to Rubner, RV induces wheezing within the first 3 years of life is linked to an increased risk of asthma development at ages 6, 9, 11, and 13 years (Citation9). Rubner’s study exhibits a link between RV wheezing and early adolescence unlike Rubner’s finding with RSV (Citation9). Sigurs’ study, mentioned above, shows that RSV bronchiolitis has a link with increased asthma development well into early adulthood, but did not study the role of RV in asthma development into early adulthood (Citation10,Citation11).
As seen in these studies, there is a distinct difference between the manifestations of RSV versus RV in pediatric patients. RSV and RV are both risk factors for viral induced wheezing and may result in the development of asthma. RSV bronchiolitis within the first year of life seems to result in asthma development into early adulthood, while RV results in asthma development into early adolescence. RV-induced wheezing seems to be more likely to occur if the patient has had allergic sensitization prior to the onset of RV infection; whereas, RSV-induced wheezing is more associated with atopic characteristics after the onset of viral disease.
The connection between viral disease, allergic sensitization, and asthma has been shown; however, there are several discussions as to the mechanism(s) behind this connection (Citation12). There is an antiviral immune response that involves cytokines and immune cell types such as neutrophils and dendritic cells (Citation12). The rest of this review will discuss the mechanisms involved in RSV and RV infections including the inflammatory profile and cell types involved.
Mechanisms
The antiviral immune response between RSV and RV infections differs in terms of the mechanism at play: the difference lies not only in the virus but also in the inflammatory profile, including specific cytokines and cell types such as T cells, neutrophils, and dendritic cells (see ). This difference in mechanism can lead to different manifestations of post-viral airway disease, as well as association with atopic disease. Based on several studies discussed earlier, asthma can be a manifestation of both RSV and RV viral disease. One hypothesis, known as the Th2 hypothesis, explains that asthma development involves the helper T cell (Th) response; the two Th responses that are the most common involve Th1 responses and Th2 responses (Citation13). The Th1 response involves the production of IL-2 and IFNγ while the Th2 response involves production of IL-4, IL-5, IL-6, and IL-13 (Citation3). The “Th2 hypothesis” states that, in asthma, the Th2 response is up-regulated while the Th1 response is downregulated (Citation13). This hypothesis as well as variations among mechanisms and research between RSV and RV will be explored below.
Figure 1. Comparison of rhinovirus (RV) and respiratory syncytial virus (RSV) in development of post-viral wheezing. RSV drives post-viral wheezing in those without preexisting atopy through an immune response characterized by decreased CCL5 and increased IL13, IL8, and cysteinyl leukotrienes (CysLTs). This is in contrast to RV, which drives post-viral wheezing in those with preexisting atopy, and through an immune response characterized by increased CCL5, and decreased type I and II IFN.
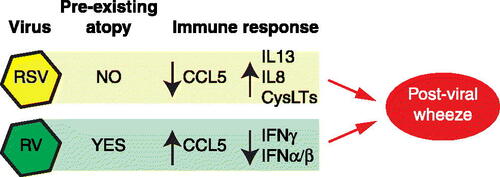
Respiratory syncytial virus infections
The potential mechanism of RSV and the immune pathway involved has been studied by using Sendai virus (SeV) in murine models; SeV is genetically related to RSV so the mechanism of disease with SeV could be representative of that seen with RSV. When mice are infected with Sendai virus (SeV), airway epithelial cells release type I interferons (IFNα/β), which, although having an antiviral effect, can often lead to epithelial cell death, with the dead epithelial cells being eventually engulfed by macrophages (Citation13). The chemokine, CCL5 (RANTES) protects macrophages from virus-induced death, effectively allowing them to participate in phagocytic viral clearance (Citation14). In propagating acute post-viral disease, CD49d + neutrophils are recruited to the airway, where they are able to induce expression of the high affinity IgE receptor, FcεRI, on lung conventional dendritic cells (DC) (Citation13,Citation15). Next, anti-SeV IgE is produced, which binds to the DC expressed FcεRI, and then can be crosslinked by SeV, leading to release of the chemokine CCL28 (Citation13). CCL28 attracts Th2 cells that then produce IL-13 (Citation16). IL-13 is involved in the propagation of chronic post-viral airway disease (Citation13,Citation17). However, Th2 IL-13 production is not required in the SeV model as IL-13 can be produced by iNKT (invariant natural killer T cells) and then by a subset of activated macrophages (Citation13,Citation17). Macrophage activation requires iNKT production of IL-13, and when macrophages are activated by IL-13 (through IL-13Rα), they produce more IL-13 (Citation17). IL-13 acts on the airway to drive mucous cell metaplasia and airway hyperreactivity (Citation17).
Severe RSV disease could be due to a variety of mechanisms, but one potential mechanism is an IgE type I hypersensitivity reaction resulting in mast cell release of factors leading to inflammation and potential bronchospasm (Citation3). As with Cheung’s study mentioned above, in mice there is evidence of SeV causing an increase in antiviral IgE that binds to the high affinity IgE receptor (FcεRI) on the lung DCs (Citation15). An increase in IgE is associated with patients with severe RSV disease (Citation16,Citation18,Citation19). Antiviral IgE seems to be present in higher concentrations in RSV patients with RSV-related wheeze than those without as well as in patients with RSV bronchiolitis (Citation16,Citation18,Citation19). The presence of antiviral-IgE during respiratory viral infection further supports the development of atopic characteristics as a result of the respiratory viral disease. In addition to the type I interferon response (mentioned above), the type II interferon response is very important to the antiviral immune response and can be interconnected with the total IgE response. IgE production is up-regulated by IL-4 and down-regulated by IFNγ (type II IFN) (Citation20). Some studies hypothesize that an ineffective IFNγ response could lead to more severe progression of RSV disease, and that this ineffective IFNγ response could lead to a higher level of IgE potentially resulting in a more robust type I hypersensitivity reaction (Citation20). As RSV can cause an ineffective IFNγ response, this is supported with findings of increased levels of IgE in the nasopharyngeal secretions of patients with RSV, which potentially could explain the onset of allergic sensitization post-RSV infection (Citation16).
IFNγ is an important component of the antiviral immune response and has effects beyond just those on the production of IgE. In fact, reduced IFNγ levels are posited to result in delayed viral clearance (Citation20). Considering IFNγ dependent viral clearance, an important component of the inflammatory profile are the cells that produce IFNγ. Natural killer (NK) cells and CD8 T cells play a role in viral clearance and the antiviral immune response in murine models (Citation20). Severe RSV disease involves decreased CD8 levels, which along with reduced IFNγ levels, and reduced viral clearance (Citation20). Decreased CD8 levels, compared to CD4 levels, have also been detected in the lungs of RSV patients (Citation21). Therefore, an impairment in the IFNγ immune response, as seen with severe RSV, can result in a more severe immune response and potentially drive post-viral allergic sensitization.
Cellular release of cytokines is a largely studied component of RSV-related post-viral disease; some of these cytokines include CCL5, IL-8, IL-6, and IL-10. Sheeran’s study finds epithelial cell produced CCL5 in the nasal wash (NW) and tracheal aspirate (TA) of patients with RSV (Citation14). Furthermore, Sheeran finds that non-intubated patients had higher levels of CCL5 than intubated patients; specifically, they find that TA CCL5 inversely correlated with RSV disease severity (Citation22). This suggests that low CCL5 levels may have a role in propagating the severity of RSV disease.
Aside from disease severity, CCL5 has been found to be a regulator of allergic inflammation further suggesting the connection between RSV and post-viral allergic disease/airway disease. In addition to CCL5, high levels of IL-8, produced by Th1 cells, have been found in the nasal secretions and TA of RSV patients (Citation14,Citation15). IL-8 is a chemo-attractant for neutrophils to the lung (Citation22). Neutrophils produce a significant portion of inflammatory mediators and IL-8’s role in bringing them to the lung could explain why several studies have shown neutrophils were the predominant cell type in the nasopharyngeal aspirate (NPA) and broncho-alveolar lavage (BAL) of infants infected with RSV (Citation21–23). Neutrophils were found in the upper and lower respiratory tracts of these infants infected with RSV and were noted to produce large amounts of inflammatory products resulting in significant airway inflammation (Citation21). IL-8, while produced by Th1 cells, has also been shown to be produced during RSV infection by alveolar macrophages and airway epithelial cells (Citation20,Citation21). As mentioned earlier, a specific subset of neutrophils, the CD49d expressing cells, seems to play a large role in the respiratory viral response in mice. As Cheung’s study demonstrates, the CD49d + neutrophils induce the expression of dendritic cell FcεRI, which, when bound IgE is crosslinked, leads to the production of CCL28, a Th2 chemoattractant (Citation15). Fullmer’s study finds that RSV infection in mice leads to the release of CysLT (cysteinyl leukotrienes) (Citation24). Furthermore, Cheung, et al, finds that CysLT1 signaling led to an accumulation of the CD49d + neutrophils in the airways of mice (Citation15). Neutrophils, particularly the CD49d + subset, point to a potential important role in the antiviral immune response during an RSV infection. Sigua, et al, further explores the CD49d + neutrophils’ role in atopy and found that these neutrophils were present at higher concentrations in human atopic patients’ nasal lavage and peripheral blood (Citation25). With Sigua’s study suggesting that the CD49d + neutrophils have a role in atopy, the prevalence of this subset of neutrophils during RSV infection could further provide mechanistic possibilities in the development of allergic disease after viral infection (Citation25).
Like CCL5 and IL-8, Sheeran also finds increased levels of IL-10 and IL-6, produced by Th2 cells, in the NW and TA of RSV patients (Citation22). Both IL-10 and IL-6, according to Sheeran, play a role in inflammatory response in the respiratory tract with RSV infection, supporting the up-regulated Th2 hypothesis involved in asthma (Citation22). Together, both Th1 (increased levels of IL-8) and Th2 (increased levels of IL-6 and IL-10) responses are at play in Sheeran’s examination of the RSV inflammatory profile. While IL-8 does not specifically attract one subset of neutrophil, RSV is associated with elevated lung CysLTs, which could lead to an increase in CD49d + neutrophils (Citation15,Citation22). The increased CD49d + neutrophils in the airways could then upregulate the FcεRI receptor on DC, which could propagate the Th2 response (Citation15). So, in this way a Th1 response (IL-8) could be tied to the subsequent development of a Th2 response.
Rhinovirus infections
Like most viral immune responses, the antiviral response in RV involves a type I interferon response. However, RV infected asthmatic patients have both reduced IFNβ and IFNα responses (Citation26). In primary bronchial epithelial cells (PBEC) isolated from asthmatic donors, there is reduced baseline IFNβ expression in response to viral infection. Further, plasmacytoid dendritic cells (pDCs) from asthmatic individuals have demonstrated reduced ability to produce IFNα ex vivo in response to a viral infection (Citation27,Citation28). This proposes the idea that asthmatic individuals have a reduced type I interferon response in viral infections. According to Cakebread, RV is recognized after replication as dsDNA, which essentially activates interferon release factor (IRF) that drives the release of IFNβ (Citation27). IFNβ activates the STAT (signal transducer and activator of transcription proteins) pathway which prompts the transcription of antiviral genes (Citation27). Giving exogenous sources of IFNβ to PBEC’s from asthmatic patients reduced viral replication (Citation27). They find that giving just IFNβ to PBECs from asthmatic and non-asthmatic patients resulted in no significant up-regulation of CCL5 or IL-6 in either (Citation27). However, they found that when an IFNβ pretreatment was given to the cells prior to an RV infection, there was decreased production of CCL5 and IL-6 in PBECs of asthmatic patients, but not in PBECs from non-asthmatic patients (Citation27). Further, without pretreatment there were increased levels of CCL5 in both groups with RV infection (Citation27). Thus, in asthmatic bronchial epithelial cells (PBECs), there is a disrupted innate immune response involving reduced type I interferon signaling, which leads to a more severe viral disease (Citation29). This suggests that in atopic individuals, the inflammatory cytokines CCL5 and IL-6 could be responsible for the increased severity of viral disease in these patients as a result of an impaired type I interferon response (Citation27). In fact, patients with preexisting asthma (i.e. prior to RV infection) have reduced IFNβ expression in PBECs, which as mentioned above could correlate with a more severe RV infection (Citation13,Citation27). These studies support the idea that preexisting atopy in patients who are infected with RV could result in more severe disease. Unlike RSV, where the virus increases the risk of wheeze and/or atopy post-viral infection in those who are not initially atopic, allergic sensitization prior to viral disease seems to increase the risk of wheezing with RV infection. The requirement for preexisting atopy may demonstrate the need for an impaired antiviral response (present as a result of the atopic condition) to translate RV infection into a wheezing illness (Citation8). Further emphasizing the role of the type I interferon response in the antiviral immune response, Gill finds reduced IFNα production in the plasmacytoid dendritic cells (pDCs) of asthmatic individuals. During viral infections, pDCs play an important role in releasing IFNα (Citation28). In humans, pDCs express the high affinity IgE receptor, FcεRI, and atopic patients (such as those with asthma) have elevated IgE levels (Citation28). Using pDCs from asthmatic and non-asthmatic patients, Gill finds that pDC from patients with asthma have evidence of increased cross-linking of FcεRI, which correlated with reduced IFNα production from the pDCs in vitro as well as reduced toll-like receptor 7 (TLR7) expression. Stimulating pDCs with TLR7 agonists increased IFNα production from pDCs 0.27 Thus, this further suggests that an impaired type I interferon response in patients with allergic disease could be associated more with severe viral disease, and that producing IgE (i.e. being atopic) may be required to fully depress the type I IFN response (Citation8).
Aside from the type I interferon response, Jackson argues (similar to Aberle’s comments on RSV) that children with more susceptibility to allergic sensitization have lower IFNγ responses, and that a decreased IFNγ response could associate with early childhood post-viral wheeze (Citation8,Citation20). The inflammatory profile, particularly the interferon response, is strongly associated with RV infection and an impaired interferon response due to previous allergic sensitization. The impaired interferon response in RV leads to a reduced viral clearance which may result in a more severe viral disease outcome and consequent post-viral wheeze.
Cell types involved in RV infection are less commonly studied than cell types in RSV; nonetheless, similar to RSV, cell types do appear to play an important role in the development of viral disease with RV infection. RV specifically targets and binds to respiratory epithelial cells causing airway inflammation, airway remodeling, and eventually post-viral airway disease (Citation28). Preexisting allergic inflammation can impair the respiratory epithelial cell barrier making it more susceptible to RV infection, primarily through goblet cells (Citation8). Goblet cells are abundant in airways of individuals with allergic disease and are especially vulnerable to RV infection, suggesting that atopic individuals have a higher ability to be infected than non-atopic individuals (Citation8). Further studies on the cellular components of the anti-RV immune response are needed, especially with respect to how these responses differ in atopic and non-atopic individuals.
Conclusion
Respiratory tract viruses are a common contributor to the onset of wheezing, a marker for asthma development post-viral disease; RSV and RV are two of the most common viruses associated with development of wheezing (Citation6). RSV is associated more with the incidence of post- viral atopic disease in patients without prior atopic disease, while RV has a higher risk of post- viral disease when the patient has atopic disease before the incident RV infection (Citation10). Though the mechanism through which RSV imparts a risk of post-viral wheezing is not clearly delineated, mouse models have identified possible pathways to translate a viral infection into post-viral atopic disease. In murine models, SeV infection results in a well-demarcated pathway involving CysLTs, CD49d + PMNs, FcεRI on lung cDCs, anti-SeV IgE, CCL28, and IL-13 producing Th2 cells (Citation15). The IL-13 produced in the airways can promote mucous cell metaplasia and airway hyper-reactivity resulting in allergic inflammation and wheeze (Citation17). On the other hand, RV infections in individuals with impaired type I and type II interferon responses may drive development of post-viral wheeze (Citation26,Citation29). Atopic individuals appear to have an impaired both type I and type II interferon response, which may explain the need for atopy to be present prior to RV infection for post-viral wheeze to develop after infection (Citation8).
Understanding the mechanisms behind both the antiviral immune response to RSV and RV and the link with atopic disease could help to direct treatment options, and possibly even primary prevention strategies. If there are particular mechanisms in RSV and RV that result in post-viral wheeze, these cytokines or cell types could be targeted to prevent the onset of post- viral wheeze and the potential complication of long-term asthma. Moreover, recent studies have exploring a variety of pathogenic mechanisms involved in the development of asthma. Holtzman, et al, explored which mechanisms control common viral infections and how these infections eventually cause long-term disease (Citation30). Several mechanisms of pathogenesis are discussed including the role of epithelial immune cells and goblet cell hyperplasia (Citation30). They propose the “hit and run hypothesis” where development of an asthmatic phenotype occurs after an infection in the appropriate genetic background (Citation30). Indeed there is a great need for research looking at potential endotypes, phenotypes, and genotypes that associate with development of asthma (and recurrent wheezing) after a viral insult (see ). Some research has been published trying to understand the genetic basis of asthma and its multitude of endotypes, with a focus on cytokine production and immune cell response, but in general these studies fail to account for initial viral insults (Citation31). Certain alleles of the gene orosomucoid-like 3 (ORMDL3), however, have been associated with an increased risk of RV induced wheeze (Citation32). In this specific case it appears that these alleles drive increased ICAM-1 (the receptor for RV), and that may then lead to increased post-RV wheezing (Citation32). Less is known about other potential genetic risks, such as those from dual specificity phosphatase 22 (DUSP22), Ikaros family zinc finger protein 3 (IKZF3), and runt-related transcription factor 1 (RUNX1), where other factors such as exposure or environment beyond just presence of a risk allele influence the development of asthma (Citation33). Future research is needed to better understand the genetic (and epigenetic) phenotypes involved in viral induced asthma, as well as to target the variety of mechanisms involved and develop preventative strategies with the goal of suppressing or even preventing the onset of post-viral wheeze.
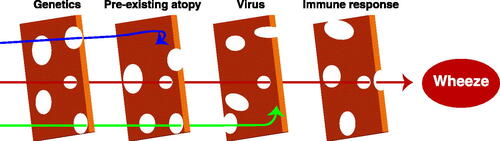
Figure 2. Development of post-viral wheezing involves multiple aligned risks. Post-viral wheezing likely requires the appropriate genetics, presence (or absence) of atopy, an appropriate virus (RV with atopy, RSV in the absence of atopy), and a subsequent specific immune response. Like a “Swiss cheese” model, having only one or two of these risks, will likely not end up leading to post-viral wheeze (blue and green arrows). Only when all of the “holes” are aligned will the subject go on to develop post-viral wheezing and asthma.
Conflict of interest statement
MHG has been a medical advisory board participant for Aimmune, DBV, GlaxoSmithKline, and Genzyme, and has stock options for Invirsa. MHG is an Associate Editor at the Annals of Allergy, Asthma, and Immunology, Director and Treasurer of the American Board of Allergy and Immunology, Chair of the Medical Scientific Council and Board Member of the Asthma and Allergy Foundation of America, and Member of the Scientific Advisory Committee of the American Lung Association. DN reports no potential conflicts of interest.
Additional information
Funding
References
- Ferrante G, La Grutta S. The burden of pediatric asthma. Front Pediatr. 2018;6:186. doi:https://doi.org/10.3389/fped.2018.00186.
- Busse WW, Lemanske RF, Gern JE. Role of viral respiratory infections in asthma and asthma exacerbations. Lancet. 2010;376(9743):826–834. doi:https://doi.org/10.1016/S0140-6736(10)61380-3.
- Mosmann T, Coffman R. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7(1):145–173. doi:https://doi.org/10.1146/annurev.iy.07.040189.001045.
- Mikhail I, Grayson MH. Asthma and viral infections: an intricate relationship. Ann Allergy Asthma Immunol. 2019;123(4):352–358. doi:https://doi.org/10.1016/j.anai.2019.06.020.
- Hussain SA, Mejias A, Ramilo O, Peeples ME, Grayson MH. Post-viral atopic airway disease: pathogenesis and potential avenues for intervention. Expert Rev Clin Immunol. 2019;15(1):49–58. doi:https://doi.org/10.1080/1744666X.2019.1541737.
- Turunen R, Koistinen A, Vuorinen T, Arku B, Söderlund-Venermo M, Ruuskanen O, Jartti T. The first wheezing episode: respiratory virus etiology, atopic characteristics, and illness severity. Pediatr Allergy Immunol. 2014;25(8):796–803. doi:https://doi.org/10.1111/pai.12318.
- Midulla F, Pierangeli A, Cangiano G, Bonci E, Salvadei S, Scagnolari C, Moretti C, Antonelli G, Ferro V, Papoff P, et al. Rhinovirus bronchiolitis and recurrent wheezing: 1-year follow-up. Eur Respir J. 2012;39(2):396–402. doi:https://doi.org/10.1183/09031936.00188210.
- Jackson DJ, Evans MD, Gangnon RE, Tisler CJ, Pappas TE, Lee W-M, Gern JE, Lemanske RF. Evidence for a causal relationship between allergic sensitization and rhinovirus wheezing in early life. Am J Respir Crit Care Med. 2012;185(3):281–285. doi:https://doi.org/10.1164/rccm.201104-0660OC.
- Rubner FJ, Jackson DJ, Evans MD, Gangnon RE, Tisler CJ, Pappas TE, Gern JE, Lemanske RF. Early life rhinovirus wheezing, allergic sensitization, and asthma risk at adolescence. J Allergy Clin Immunol. 2017;139(2):501–507. doi:https://doi.org/10.1016/j.jaci.2016.03.049.
- Sigurs N, Gustafsson PM, Bjarnason R, Lundberg F, Schmidt S, Sigurbergsson F, Kjellman B. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am J Respir Crit Care Med. 2005;171(2):137–141. doi:https://doi.org/10.1164/rccm.200406-730OC.
- Sigurs N, Aljassim F, Kjellman B, Robinson PD, Sigurbergsson F, Bjarnason R, Gustafsson PM. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax. 2010;65(12):1045–1052. doi:https://doi.org/10.1136/thx.2009.121582.
- Cheung DS, Grayson MH. Role of viruses in the development of atopic disease in pediatric patients. Curr Allergy Asthma Rep. 2012;12(6):613–620. doi:https://doi.org/10.1007/s11882-012-0295-y.
- Holtzman MJ. Asthma as a chronic disease of the innate and adaptive immune systems responding to viruses and allergens. J Clin Invest. 2012;122(8):2741–2748. doi:https://doi.org/10.1172/JCI60325.
- Tyner JW, Kim EY, Ide K, Pelletier MR, Roswit WT, Morton JD, Battaile JT, Patel AC, Patterson GA, Castro M, et al. Blocking airway mucous cell metaplasia by inhibiting EGFR antiapoptosis and IL-13 transdifferentiation signals. J Clin Invest. 2006;116(2):309–321. doi:https://doi.org/10.1172/JCI25167.
- Cheung DS, Sigua JA, Simpson PM, Yan K, Hussain S-RA, Santoro JL, Buell EJ, Hunter DA, Rohlfing M, Patadia D, et al. Cysteinyl leukotriene receptor 1 expression identifies a subset of neutrophils during the antiviral response that contributes to postviral atopic airway disease. J Allergy Clin Immunol. 2018;142(4):1206–1217.e5. doi:https://doi.org/10.1016/j.jaci.2017.11.026.
- Welliver R, Wong D, Sun M, Middleton E, Vaughan R, Ogra P. The development of respiratory syncytial virus-specific IgE and the release of histamine in nasopharyngeal secretions after infection. N Engl J Med. 1981;305(15):841–846. doi:https://doi.org/10.1056/NEJM198110083051501.
- Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, Benoit LA, Byers DE, Alevy Y, Tucker J, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. 2008;14(6):633–640. doi:https://doi.org/10.1038/nm1770.
- Bui RH, Molinaro GA, Kettering JD, Heiner DC, Imagawa DT, St Geme JW. Virus-specific IgE and IgG4 antibodies in serum of children infected with respiratory syncytial virus. J Pediatr. 1987;110(1):87–90. doi:https://doi.org/10.1016/S0022-3476(87)80295-0.
- Russi JC, Delfraro A, Borthagaray MD, Velazquez B, García-Barreno B, Hortal M. Evaluation of immunoglobulin E-specific antibodies and viral antigens in nasopharyngeal secretions of children with respiratory syncytial virus infections. J Clin Microbiol. 1993;31(4):819–823. doi:https://doi.org/10.1128/JCM.31.4.819-823.1993.
- Aberle JH, Aberle SW, Dworzak MN, Mandl CW, Rebhandl W, Vollnhofer G, Kundi M, Popow-Kraupp T. Reduced interferon-gamma expression in peripheral blood mononuclear cells of infants with severe respiratory syncytial virus disease. Am J Respir Crit Care Med. 1999;160(4):1263–1268. doi:https://doi.org/10.1164/ajrccm.160.4.9812025.
- Everard ML, Swarbrick A, Wrightham M, McIntyre J, Dunkley C, James PD, Sewell HF, Milner AD. Analysis of cells obtained by bronchial lavage of infants with respiratory syncytial virus infection. Arch Dis Child. 1994;71(5):428–432. doi:https://doi.org/10.1136/adc.71.5.428.
- Sheeran P, Jafri H, Carubelli C, Saavedra J, Johnson C, Krisher K, Sanchez P, Ramilo O. Elevated cytokine concentrations in the nasopharyngeal and tracheal secretions of children with respiratory syncytial virus disease. Pediatr Infect Dis J. 1999;18(2):115–122. doi:https://doi.org/10.1097/00006454-199902000-00007.
- Pitrez PMC, Brennan S, Sly PD. Inflammatory profile in nasal secretions of infants hospitalized with acute lower airway tract infections. Respirology. 2005;10(3):365–370. doi:https://doi.org/10.1111/j.1440-1843.2005.00721.x.
- Fullmer JJ, Khan AM, Elidemir O, Chiappetta C, Stark JM, Colasurdo GN. Role of cysteinyl leukotrienes in airway inflammation and responsiveness following RSV infection in BALB/c mice. Pediatr Allergy Immunol. 2005;16(7):593–601. doi:https://doi.org/10.1111/j.1399-3038.2005.00248.x.
- Sigua JA, Buelow B, Cheung DS, Buell E, Hunter D, Klancnik M, Grayson MH. CD49d-expressing neutrophils differentiate atopic from nonatopic individuals. J Allergy Clin Immunol. 2014;133(3):901–904.e5. doi:https://doi.org/10.1016/j.jaci.2013.09.035.
- Sykes A, Edwards MR, Macintyre J, del Rosario A, Bakhsoliani E, Trujillo-Torralbo M-B, Kon OM, Mallia P, McHale M, Johnston SL. Rhinovirus 16-induced IFN-α and IFN-β are deficient in bronchoalveolar lavage cells in asthmatic patients. J Allergy Clin Immunol. 2012;129(6):1506–1514.e6. doi:https://doi.org/10.1016/j.jaci.2012.03.044.
- Cakebread JA, Xu Y, Grainge C, Kehagia V, Howarth PH, Holgate ST, Davies DE. Exogenous IFN-β has antiviral and anti-inflammatory properties in primary bronchial epithelial cells from asthmatic subjects exposed to rhinovirus. J Allergy Clin Immunol. 2011;127(5):1148–1154.e9. doi:https://doi.org/10.1016/j.jaci.2011.01.023.
- Gill MA, Bajwa G, George TA, Dong CC, Dougherty II, Jiang N, Gan VN, Gruchalla RS. Counterregulation between the FcεRI pathway and antiviral responses in human plasmacytoid dendritic Cells. J Immunol. 2010;184(11):5999–6006. doi:https://doi.org/10.4049/jimmunol.0901194.
- Wark PAB, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, Holgate ST, Davies DE. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201(6):937–947. doi:https://doi.org/10.1084/jem.20041901.
- Holtzman MJ, Kim EY, Lo MS, Tyner JW, Shornick LP, Sumino KC, Zhang Y. Defining and adjusting divergent host responses to viral infection. Immunol Res. 2005;32(1-3):123–141. doi:https://doi.org/10.1385/IR:32:1-3:123.
- Kuruvilla ME, Lee FE, Lee GB. Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol. 2019;56(2):219–233. doi:https://doi.org/10.1007/s12016-018-8712-1.
- Zhang Y, Willis-Owen SAG, Spiegel S, Lloyd CM, Moffatt MF, Cookson WOCM. The ORMDL3 asthma gene regulates ICAM1 and Has multiple effects on cellular inflammation. Am J Respir Crit Care Med. 2019;199(4):478–488. doi:https://doi.org/10.1164/rccm.201803-0438OC.
- Rathod A, Duan J, Zhang H, Holloway JW, Ewart S, Arshad SH, Karmaus W. Interweaving between genetic and epigenetic studies on childhood asthma. Genet Epigenet. 2020;13:251686572092339. doi:https://doi.org/10.1177/2516865720923395.