
Abstract
Objective
To assess the long-term safety of tezepelumab in Japanese patients with severe uncontrolled asthma.
Methods
This phase III, 52-week, open-label, single-arm study (NOZOMI, NCT04048343) evaluated the safety/tolerability of subcutaneous (SC) tezepelumab 210 mg every 4 weeks (Q4W) in Japanese patients aged 12–80 years with severe uncontrolled asthma using medium- to high-dose inhaled corticosteroids and at least one additional asthma controller medication, with/without oral corticosteroids. Exploratory outcomes included efficacy (asthma exacerbations, lung function, and asthma control), pharmacokinetic parameters, and immunogenicity.
Results
Among 65 patients (median age 52 years), 39 (60%) experienced 94 adverse events (AEs; predominantly nasopharyngitis [13/65]) of mild (49.2%), moderate (7.7%), or severe (3.1%) intensity. Two patients had transient injection site erythema related to tezepelumab. Four patients reported serious AEs unrelated to tezepelumab and one AE led to treatment discontinuation. AEs of special interest were infrequent and generally mild/moderate. Apart from a decrease in blood eosinophils (an expected pharmacodynamic effect), no notable trends/clinically relevant changes in hematology, clinical chemistry, or urinalysis parameters were observed. Among exploratory outcomes, tezepelumab was associated with a low annualized asthma exacerbation rate over the study period (0.11/patient-year), improved lung function (mean [standard deviation] change from baseline of 0.075 [0.226] L in pre-dose/pre-bronchodilator forced expiratory volume in 1 s), and better asthma control versus baseline (responder rate: 71.4% at Week 52).
Conclusion
Tezepelumab 210 mg SC Q4W in Japanese patients with severe uncontrolled asthma showed safety/tolerability profiles similar to international data, with low exacerbation rates and improvements in lung function and asthma control.
Introduction
Asthma is a common chronic airway disease characterized by airway inflammation that manifests as wheezing, dyspnea, and cough (Citation1). According to the World Health Organization, approximately 262 million people globally were affected by asthma in 2019 (Citation2). In Japan, the prevalence of asthma is roughly 7% (Citation3).
There are both Japanese guidelines (Citation4,Citation5) and internationally recognized criteria for the classification of “severe” asthma (Citation6–9). Asthma is considered severe if patients remain “uncontrolled” on treatment with high-dose inhaled corticosteroids (ICS) plus a second controller (and/or systemic corticosteroids), or if they need this treatment to prevent progression to “uncontrolled” asthma. Of the worldwide asthma population, 5–10% of patients are thought to have severe asthma (Citation10), and in Japan, an estimated 7–10% of asthma patients have severe asthma and 2.5% have severe uncontrolled asthma (Citation11). Patients with severe asthma are at the greatest risk of exacerbations, representing the greatest unmet medical need and economic cost among the asthmatic population today (Citation12,Citation13).
Recently, the Global Initiative for Asthma Guidelines recommended the use of biologic therapies for treatment of severe asthma (Citation1). Approved biologics in Japan include omalizumab (for severe atopic asthma), and mepolizumab, benralizumab, and dupilumab (for severe eosinophilic asthma) (Citation1). However, not all patients with severe uncontrolled asthma are eligible for treatment with currently approved biologics (Citation1), and some undergoing treatment with these agents still experience exacerbations, suggesting that they could benefit from biologics targeting different pathways.
Thymic stromal lymphopoietin (TSLP) is an epithelial cell-derived cytokine that plays a role in the pathogenesis of asthma through multiple pathways (Citation14,Citation15). Tezepelumab is a high-affinity human anti-TSLP monoclonal antibody that prevents the interaction of TSLP with its heterodimeric receptor (Citation16,Citation17), thereby decreasing levels of inflammatory biomarkers and cytokines (Citation18).
Tezepelumab has previously demonstrated efficacy with a manageable safety profile in phase II (Citation16) and III (Citation19) studies. The phase IIb PATHWAY trial (Citation16), which included both non-Japanese and Japanese patients, showed clinically meaningful reductions in exacerbations, and improved lung function and asthma control parameters with a favorable safety profile in patients with severe uncontrolled asthma treated with tezepelumab for 52 weeks. A global, phase III pivotal trial (NAVIGATOR) (Citation19) investigated the safety and efficacy of tezepelumab versus placebo in adults and adolescents with asthma and included a Japanese subpopulation. Although efficacy and safety results have been made available for the overall population in this study (Citation20), detailed data for the Japanese population have not yet been published.
The purpose of the NOZOMI study was to provide additional long-term safety data of tezepelumab to meet the regulatory requirement of 1 year of exposure to tezepelumab in at least 100 Japanese patients (Citation21). In NOZOMI, the safety of subcutaneous (SC) tezepelumab 210 mg every 4 weeks (Q4W) in Japanese patients with severe, uncontrolled asthma was evaluated. Additionally, we explored the effects of tezepelumab on asthma exacerbations, lung function, and asthma control, and evaluated its pharmacokinetics (PK) and immunogenicity.
Methods
Study design
This phase III, 52-week, open-label, single-arm study was conducted at five centers in Japan between June 2019 and June 2021 (database lock June 2021). The NOZOMI study included a screening period of 2 weeks, a treatment period of 52 weeks (on-treatment period, from baseline to Week 52), and a post-treatment follow-up period of 12 weeks (on-study period, from baseline to Week 64 or withdrawal date) (Supplementary Figure 1).
From screening and throughout the 52-week treatment period, in addition to the study drug (tezepelumab 210 mg Q4W SC), patients maintained their prescribed ICS and at least one additional asthma controller medication at the same doses as those used at screening.
This study was performed in accordance with the ethical principles of the Declaration of Helsinki, International Conference on Harmonisation Good Clinical Practice (GCP) guidelines, and other applicable local regulatory requirements relating to the conduct of clinical studies. The clinical study protocol and amendments were approved by the required Institutional Review Boards or Independent Ethics Committees, and all patients provided written informed consent prior to enrollment. The trial was registered at ClinicalTrials.gov (NCT04048343).
Owing to the COVID-19 pandemic, changes were made to the clinical study protocol to ensure the safety of participants, maintain compliance with GCP, and minimize potential risks to trial integrity. Where permitted by local health authorities, ethics committees, and health care provider guidelines, these changes included the option of home administration of tezepelumab by a qualified health care professional, and telephone and/or virtual visits to replace on-site visits.
Patients
The study population comprised patients aged between 12 and 80 years with physician-diagnosed asthma for at least 12 months and at least 3 months’ background asthma therapy of medium- or high-dose ICS (≥500 µg fluticasone propionate dry powder formulation-equivalent total daily dose; as defined in Supplementary Table 1) plus at least one additional asthma controller medication (long-acting β2-agonists [LABA], leukotriene receptor antagonists [LTRA], long-acting muscarinic antagonists [LAMA], cromones, or theophylline), with or without maintenance oral corticosteroids (OCS), which was maintained from screening and throughout the study, including the follow-up period. Additional key inclusion criteria were a history of at least one physician-diagnosed exacerbation in the year prior to screening and an Asthma Control Questionnaire-6 (ACQ-6) score ≥1.5 at screening or registration.
Table 1. Baseline demographic and clinical characteristics (safety analysis set).
Key exclusion criteria were pulmonary disease other than asthma; recent history of cancer (i.e. curative therapy completed <5 years prior to screening or <1 year for basal cell carcinoma, localized squamous cell carcinoma of the skin, or in situ carcinoma of the cervix); recent history of a clinically significant infection (i.e. requiring antibiotic/antiviral treatment completed <2 weeks prior to screening); current smoking or smoking history of ≥10 pack-years; history of chronic alcohol/drug abuse within the last year; hepatitis B, C or HIV infection; pregnancy or lactation; history of anaphylaxis following any biologic therapy; and prior participation in the current study or previous tezepelumab studies.
Study outcomes and assessments
The primary outcome was the safety and tolerability of tezepelumab based on the frequency of adverse events (AEs)/serious AEs (SAEs), and AEs of special interest (AESIs) as defined in the study protocol (opportunistic infections, helminth infections, anaphylactic or serious allergic reactions, hypersensitivity reactions, malignancy, injection site reactions, and Guillain-Barré syndrome) coded using the Medical Dictionary for Regulatory Activities v23.1, assessment of vital signs, weight, body mass index, electrocardiography, and laboratory examinations including clinical chemistry, hematology, and urinalysis parameters.
Exploratory efficacy outcome measures included: asthma exacerbations, assessed by the annualized asthma exacerbation rate (AAER) over 52 weeks; lung function, assessed by absolute change from baseline in pre-dose/pre-bronchodilator (pre-BD) forced expiratory volume in 1 s (FEV1) at 52 weeks; and asthma control, assessed by change from baseline in ACQ-6 at 52 weeks. Spirometry variables included pre-BD FEV1, forced vital capacity (FVC), and FEV1/FVC. The ACQ-6 was assessed on a 7-point scale, where 0 = totally controlled and 6 = severely uncontrolled. Mean scores of ≤0.75 indicated well-controlled asthma, scores >0.75 and <1.5 indicated partly controlled asthma, and scores ≥1.5 indicated uncontrolled asthma. Individual changes of at least 0.5 were considered clinically meaningful (Citation22). An ACQ-6 responder was defined as a patient with a decrease of at least 0.5 in the ACQ-6 score (Citation23,Citation24).
PK parameters (based on serum tezepelumab trough concentrations) and immunogenicity (incidence rate of anti-drug antibodies [ADA]) were also analyzed. ADA status (positive versus negative) at each visit was summarized for the on-study period. Descriptive statistics including the number of patients, median, range, and first and third quartiles of the actual ADA titers by visit, where possible, were provided. Samples confirmed positive for ADA were archived for possible testing for neutralizing antibodies (nAb).
Statistical analysis
Based on the number of Japanese patients randomized in PATHWAY (Citation16) and the expected number to be randomized in NAVIGATOR (Citation19), we planned to register approximately 66 Japanese patients so that 59 patients would complete the study. This was to ensure that approximately 100 Japanese patients in total would receive tezepelumab for 52 weeks across all studies.
Analyses of safety, efficacy, and ADA were performed on the safety analysis set, which comprised all patients who received at least one dose of tezepelumab. The PK analysis set comprised all patients who received tezepelumab and had at least one detectable serum concentration from a PK blood sample collected post-first dose that was assumed not to have been affected by factors such as protocol deviations.
Categorical variables were summarized using number and percent (n [%]), and continuous variables were summarized using appropriate descriptive statistics such as minimum, first quartile, median, third quartile, maximum, mean, and standard deviation (SD). AAERs were summarized along with the total number of exacerbations and the total time at risk, which was defined as the total number of patient-years minus the time during an exacerbation plus 7 days. No statistical hypothesis testing was performed. All statistical analyses were performed using SAS version 9.4 (SAS Institute, Inc., Cary, NC, USA).
Results
Patients
A total of 71 patients were recruited; of these, 65 patients from five centers in Japan were enrolled (), all prior to potential COVID-19 pandemic disruptions (i.e. from 11 March 2020). Five patients failed screening, and one patient and their legally authorized representative withdrew consent to participate. All 65 enrolled patients received at least one dose of tezepelumab and were included in the safety analysis set; 61 (93.8%) patients completed treatment, and all 65 patients completed the study. Four patients (6.2%) discontinued tezepelumab during the study, two (3.1%) by their own request, one (1.5%) due to an AE, and one (1.5%) who declined site visits due to the COVID-19 pandemic.
Figure 1. Patient disposition. aInformed consent received. bFor one patient, the legally authorized representative decided to withdraw the patient from participation because the study site could not obtain sufficient information from the patient’s primary care physician. cIncludes patients who completed treatment and the study, and those who discontinued treatment but completed study assessments. One patient who did not complete the follow-up visits was considered not to have completed the study (but to have completed treatment). dAll efficacy variables were summarized using the safety analysis set.
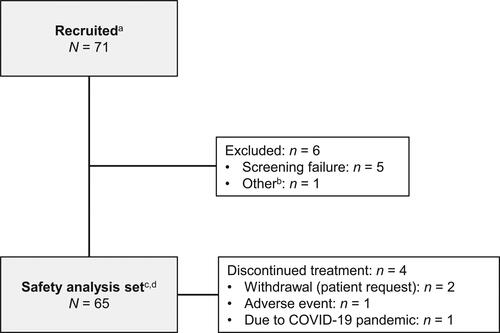
Several patients were affected by study disruptions because of the COVID-19 pandemic (Supplementary Table 2). Important protocol deviations occurred in 8/65 patients (12.3%) during the study (Supplementary Table 3); however, none raised any concerns regarding the overall conduct or quality of the study, the safety profile observed, or the interpretation of the study results.
Table 2. Baseline asthma treatments.
Table 3. Summary of adverse eventsa,b.
Baseline demographic and clinical patient characteristics are shown in . The proportions of men and women were well balanced (32/65 [49.2%] and 33/65 [50.8%] patients, respectively). Most patients in the study were aged 18 to <65 years (52/65 [80.0%]). One (1.5%) adolescent patient (aged 12 to <18 years) and 12 (18.5%) patients aged ≥65 years were also registered in the study. Other baseline asthma characteristics, including median time since diagnosis (11.00 [1.0–58.0] years), median age at diagnosis (36.0 [1–65] years), and number of exacerbations/patient in the last year (one exacerbation, n = 47 [72.3%]; two exacerbations, n = 16 [24.6%]; three exacerbations, n = 2 [3.1%]), were consistent with the eligibility criteria for the study population. The most common concomitant medical conditions at baseline were allergic rhinitis (37/65 [56.9%]), seasonal allergy (23/65 [35.4%]), and dyslipidemia (19/65 [29.2%]). Overall, 55/65 (84.6%) patients had a history of allergies.
Per the eligibility criteria, at baseline, all patients were receiving treatment with medium- or high-dose ICS (39/65 [60.0%] and 26/65 [40.0%], respectively) and at least one additional maintenance asthma controller medication (). In total, 64/65 (98.5%) patients were on LABA as additional maintenance treatment, including 36/65 (55.4%) on LABA alone and 24/65 (36.9%) on LABA + LTRA.
Exposure
The mean (SD) and median (range) treatment compliances with tezepelumab were 99.4% (2.47) and 100% (84.6–100.0), respectively. The median (range) duration of exposure was 370.0 (251–375) days, with a total of 64.8 patient-years of exposure to tezepelumab.
Primary outcome—safety
Treatment interruptions were reported for six (9.2%) patients due to AEs; two (3.1%) patients missed one dose each due to AEs of somnolence and viral gastroenteritis, and four (6.2%) patients declined site visits due to COVID-19 pandemic concerns, missing one, two, two, and four consecutive doses, respectively.
Overall, 39/65 (60.0%) patients experienced a total of 94 AEs during the on‑treatment period, the most common AE being nasopharyngitis (13/65 [20.0%]) (). The maximum reported intensity of any AE was mild in 32/65 (49.2%) patients, moderate in 5/65 (7.7%), and severe in 2/65 (3.1%) during the on-treatment period.
Two (3.1%) patients had AEs that were assessed as causally related to the study drug by the investigator (mild AEs of injection site erythema that resolved without treatment). Four (6.2%) patients reported one SAE each during the on‑treatment period, and all were assessed as unrelated to the study drug by the investigator; these were atrial fibrillation (severe), viral gastroenteritis (moderate), lung abscess (severe), and tonsillitis (moderate), which were reported in one (1.5%) patient each. The SAE of lung abscess led to tezepelumab discontinuation, and the SAE of atrial fibrillation remained unresolved at the end of the follow-up period. There were no AEs with a fatal outcome.
AESIs reported during the on-treatment period are described in Supplementary Table 4. Severe infections were reported in nine (13.8%) patients during the on-treatment period and included herpes zoster and oral herpes (two [3.1%] patients each), and viral gastroenteritis, genital herpes simplex, influenza, lung abscess, and tonsillitis (one [1.5%] patient each). None of these events were assessed by the investigator as causally related to tezepelumab. Injection site reactions occurred in two patients (3.1%) and were classed as mild, non-serious AEs of injection site erythema on the abdominal wall, which were assessed by the investigator to be causally related to tezepelumab administration. During the study, no AESIs of opportunistic infection, helminth infection, anaphylactic reaction, hypersensitivity, malignancy, or Guillain–Barré syndrome were reported, and no patient had confirmed immune complex disease. The male patient had a mild, non-serious AE of drug-induced liver injury on Day 171, but this was attributed to bepridil hydrochloride monohydrate that was used to treat an AE of atrial fibrillation and was assessed by the investigator to be unrelated to tezepelumab.
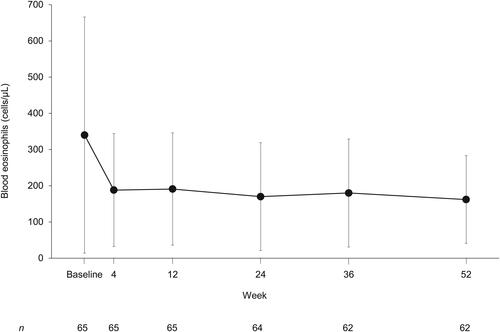
The change in blood eosinophils over time is shown in . The mean (SD) decrease from baseline (340 [326] cells/μL) to Week 52 (162 [121] cells/μL) was 181 (291) cells/μL, in line with the expected pharmacodynamic effect of tezepelumab. No notable trends or clinically meaningful shifts in the values of other hematology parameters or clinical chemistry parameters were observed over time.
Figure 2. Blood eosinophil levels over time for patients receiving tezepelumab (safety analysis set, on-study). Data are shown as mean (standard deviation).
A few shifts from negative or trace urinalysis parameters to positive values were observed; however, no clinically important abnormalities or clinically significant trends were identified during the study. No notable trends in changes to vital signs, electrocardiography, weight, or body mass index were observed over time.
Exploratory outcomes—efficacy, pharmacokinetics, and immunogenicity
The estimated AAER was 0.11/patient-year; 3/65 (4.6%) patients had one exacerbation each, and 2/65 (3.1%) patients had two exacerbations each during the planned treatment period. All five patients with exacerbations were treated with systemic corticosteroids, and one patient required an emergency room visit. No exacerbations led to hospitalization.
A numerical increase in mean (SD) pre‑BD FEV1 from baseline (2.257 [0.744] L) to Week 52 (2.329 [0.711] L) was observed, with a mean (SD) change from baseline of 0.075 (0.226) L. Of the 63 patients included in the summary analysis of ACQ‑6 at Week 52, most (45/63 [71.4%]) were responders, i.e. they had decreases of ≥0.5 in ACQ‑6 scores (Supplementary Table 5). Higher proportions of patients had well-controlled asthma (ACQ-6 score ≤0.75) and partly controlled asthma (ACQ-6 score >0.75 to <1.5) at Week 52 than at baseline: 23/63 (36.5%) and 23/63 (36.5%) versus 1/65 (1.5%) and 9/65 (13.8%) patients, respectively.
All 65 patients received tezepelumab and had at least one detectable tezepelumab serum concentration from a sample collected post-first dose. The arithmetic mean (SD) serum concentrations of tezepelumab 210 mg administered SC Q4W were similar at Week 24 (25.5 [8.61] μg/mL) and Week 52 (27.0 [8.11] μg/mL), demonstrating that steady state was achieved by Week 24, the first PK sampling time point (Supplementary Table 6).
Based on the immunogenicity analysis, 3/65 (4.6%) patients were ADA-positive: 2/65 (3.1%) patients were positive at baseline only, and 1/65 (1.5%) was ADA-positive at baseline and at follow-up Week 64 (Supplementary Table 7). None of these ADA-positive cases were considered treatment‑emergent, and no patient was nAb-positive at any time during the study.
Discussion
This study demonstrated the safety of tezepelumab over 52 weeks in Japanese patients with inadequately controlled severe asthma. No new safety concerns were identified with the administration of tezepelumab 210 mg Q4W in a study population considered to be representative of the intended target population. Importantly, approximately 60% of patients in our study had baseline blood eosinophil counts <300 cells/μL, so our results confirm that tezepelumab is generally safe and well-tolerated in patients with the non-eosinophilic/non-type 2 asthma phenotype.
Human TSLP is involved in a wide range of inflammatory reactions (Citation25), and its blockage by tezepelumab did not result in any new safety findings (Citation19) in the present study. The most commonly reported AEs with an incidence >3% were nasopharyngitis, pharyngitis, back pain, herpes zoster, and upper respiratory tract inflammation, and most AEs were classified as mild or moderate. AESIs were reported in 11/65 patients, of which three events were considered serious (lung abscess, which led to discontinuation of tezepelumab; viral gastroenteritis; and tonsillitis), but were not judged to be treatment-related. No immune complex disease, anaphylactic reactions, or hypersensitivity occurred during the study. The decreases in mean blood eosinophil counts are an expected pharmacodynamic effect of tezepelumab (Citation16,Citation19,Citation26) and not a safety concern. No other notable trends or clinically meaningful shifts in the values of hematology, clinical chemistry, or urinalysis parameters were observed. No patients had alanine aminotransferase, aspartate aminotransferase, or total bilirubin elevations that fulfilled Hy’s Law criteria for potential drug-induced liver injury.
The overall safety profile of tezepelumab in the present study was similar to that of the international NAVIGATOR trial: 60.0% of patients in NOZOMI experienced an AE versus 77.1% of those in the tezepelumab arm of NAVIGATOR (Citation19). The rates of common AEs were also comparable between NOZOMI and NAVIGATOR (tezepelumab arm): nasopharyngitis, 20.0% and 21.4%; pharyngitis, 6.2% and 3.2%; back pain, 4.6% and 4.0%; and herpes zoster, 4.6% and 2.5%, respectively (Citation19, AstraZeneca data on file).
Although all patients in the present study had presented at least one exacerbation in the past year, the estimated AAER in NOZOMI was 0.11/patient-year, and 5/65 (7.7%) patients had a total of seven asthma exacerbations during the planned treatment period, reflecting a considerable reduction in exacerbations during 52 weeks of treatment. All exacerbations were managed with systemic corticosteroids; one patient required an emergency room visit but recovered without the need for hospitalization. Numerical improvements were observed in pre‑BD FEV1 (mean change from baseline of 0.075 L at Week 52), although the change was smaller than that observed in the previous phase III NAVIGATOR trial (Citation19). Baseline FEV1 was higher in our study population than in the NAVIGATOR population, which may have contributed to this discrepancy, as there was less room for improvement in patients in our study. Overall, 71.4% of patients (45/63) had decreases of ≥0.5 in their ACQ‑6 scores and were classified as responders at Week 52, and the mean change in the ACQ-6 score from baseline was −0.98. The change in ACQ-6 score was also smaller than that of the NAVIGATOR trial, and similar to the pre-BD FEV1 results; this could be because patients in our study had lower baseline ACQ-6 scores. Finally, in the present study, more patients had well-controlled or partly controlled asthma at Week 52 compared with baseline.
Serum concentrations of tezepelumab were similar at Weeks 24 and 52, suggesting that steady state was achieved by Week 24 (first post‑baseline PK sampling time point). This is consistent with results from the subset of Japanese patients in the NAVIGATOR study (AstraZeneca, data on file).
The results of this study in Japanese patients with inadequately controlled severe asthma are consistent with findings in other populations. The multinational, 52-week phase IIb PATHWAY study showed that compared with placebo, tezepelumab 210 mg Q4W significantly reduced exacerbations by up to 71%, and had a similar safety profile (Citation16). In the NAVIGATOR study, which evaluated the effect of tezepelumab in 1059 patients with a range of severe uncontrolled asthma phenotypes, including those with low blood eosinophil counts, the frequency and types of AEs reported with tezepelumab did not differ significantly from those seen with placebo (Citation19). Additionally, compared with placebo, patients with severe uncontrolled asthma who received tezepelumab had fewer asthma exacerbations and better lung function, asthma control, and health-related quality of life.
This study has several limitations. As only Japanese patients were recruited, the findings may not be generalizable to other populations. Furthermore, we were only able to recruit a single adolescent and a small number of patients aged ≥65 years, so results in these age groups should be interpreted with caution. There was no blinding in this open-label study, and no comparator was assessed. Nevertheless, the study met the aim of accruing additional long-term safety data following tezepelumab treatment, and also provided additional information regarding the impact of tezepelumab on exacerbations and other asthma-related symptoms.
Conclusions
No new safety concerns were detected during 52-week treatment with tezepelumab 210 mg SC Q4W in Japanese patients with severe uncontrolled asthma; thus, tezepelumab was considered safe and tolerable. Additionally, AESIs were infrequent and generally mild or moderate. Japanese patients with severe uncontrolled asthma in our cohort appeared to have low immunogenicity to tezepelumab. Exploratory efficacy findings suggest that Japanese patients benefited from tezepelumab, based on the reduced number of exacerbations and improvement in pre‑BD FEV1 and ACQ-6 scores.
Authors’ contributions
Hiroshi Okada, Tatsuro Tokiyo, Nobuya Hayashi, Mami Takikawa, Gene Colice, and Gun Almqvist conceptualized the study and developed the methodology. Nobuya Hayashi and Gene Colice conducted the formal analysis. Masaharu Shinkai, Motohiro Ebisawa, Yasushi Fukushima, and Satomi Takeuchi coordinated project administration and resources. Mami Takikawa, Gene Colice, and Gun Almqvist were responsible for supervision and oversight of the study. All authors participated in conducting the research or data/evidence collection, critical review of the manuscript, and approval of the final version. The study sponsor (AstraZeneca) was involved in the study design, data collection, analysis/interpretation of data, and review of the manuscript.
Supplemental Material
Download PDF (252.1 KB)Acknowledgements
The authors thank the participants and their families, the study investigators, the clinical site staff, the NOZOMI study team, and all other persons involved in this study. Additionally, the authors wish to acknowledge Dr Keyra Martinez Dunn and Nila Bhana, MSc, of Edanz, Japan, for providing medical writing support, which was funded by AstraZeneca, Japan, through EMC K.K., Japan, in accordance with Good Publication Practice guidelines (https://www.ismpp.org/gpp3).
Data sharing statement
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
Declaration of interest
Masaharu Shinkai, Motohiro Ebisawa, Yasushi Fukushima, and Satomi Takeuchi report receipt of funding from AstraZeneca related to the current study. Hiroshi Okada, Tatsuro Tokiyo, Nobuya Hayashi, Mami Takikawa, Gene Colice, and Gun Almqvist are employees of AstraZeneca and hold stock and/or stock options in AstraZeneca.
Funding
This research was funded by AstraZeneca and Amgen.
References
- Global Initiative for Asthma (GINA). Global strategy for asthma management and prevention (2021 update). Available from: https://ginasthma.org/wp-content/uploads/2021/05/GINA-Main-Report-2021-V2-WMS.pdf.
- World Health Organization. Key facts – asthma. Available from: https://www.who.int/news-room/fact-sheets/detail/asthma.
- Ichinose M, Sugiura H, Nagase H, Yamaguchi M, Inoue H, Sagara H, Tamaoki J, Tohda Y, Munakata M, Yamauchi K, Japanese Society of Allergology, et al. Japanese guidelines for adult asthma 2017. Allergol Int. 2017;66(2):163–189. doi:10.1016/j.alit.2016.12.005.
- Japanese Society of Allergology. Asthma prevention and management guideline. Kyowakikaku, Tokyo, Japan; 2021 (in Japanese).
- Yoshihara S. The Japanese Respiratory Society Guidelines 2019 for refractory asthma in children. Arerugi. 2021;70(2):95–99 (in Japanese). doi:10.15036/arerugi.70.95.
- Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, Sterk PJ, Adcock IM, Bateman ED, Bel EH, Bleecker ER, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J. 2014;43(2):343–373. doi:10.1183/09031936.00202013.
- Holguin F, Cardet JC, Chung KF, Diver S, Ferreira DS, Fitzpatrick A, Gaga M, Kellermeyer L, Khurana S, Knight S, et al. Management of severe asthma: a European Respiratory Society/American Thoracic Society guideline. Eur Respir J. 2020;55(1):1900588. doi:10.1183/13993003.00588-2019.
- Jarjour NN, Erzurum SC, Bleecker ER, Calhoun WJ, Castro M, Comhair SA, Chung KF, Curran-Everett D, Dweik RA, Fain SB, NHLBI Severe Asthma Research Program (SARP), et al. Severe asthma: lessons learned from the National Heart, Lung, and Blood Institute Severe Asthma Research Program. Am J Respir Crit Care Med. 2012;185(4):356–362. doi:10.1164/rccm.201107-1317PP.
- GINA. Difficult-to-treat & severe asthma in adolescents and adult patients: diagnosis and management. A GINA pocket guide for health professionals, V2.0 April 2019. Available from: https://ginasthma.org/wp-content/uploads/2019/04/GINA-Severe-asthma-Pocket-Guide-v2.0-wms-1.pdf.
- Hekking PW, Wener RR, Amelink M, Zwinderman AH, Bouvy ML, Bel EH. The prevalence of severe refractory asthma. J Allergy Clin Immunol. 2015;135(4):896–902. doi:10.1016/j.jaci.2014.08.042.
- Nagase H. Severe asthma in Japan. Allergol Int. 2019;68(2):167–171. doi:10.1016/j.alit.2019.02.004.
- Côté A, Godbout K, Boulet LP. The management of severe asthma in 2020. Biochem Pharmacol. 2020;179:114112. doi:10.1016/j.bcp.2020.114112.
- Jansson SA, Backman H, Andersson M, Telg G, Lindberg A, Stridsman C, Lundbäck B, Rönmark E. Severe asthma is related to high societal costs and decreased health related quality of life. Respir Med. 2020;162:105860. doi:10.1016/j.rmed.2019.105860.
- Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, Gilliet M, Ho S, Antonenko S, Lauerma A, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3(7):673–680. doi:10.1038/ni805.
- Allakhverdi Z, Comeau MR, Jessup HK, Yoon BR, Brewer A, Chartier S, Paquette N, Ziegler SF, Sarfati M, Delespesse G. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med. 2007;204(2):253–258. doi:10.1084/jem.20062211.
- Corren J, Parnes JR, Wang L, Mo M, Roseti SL, Griffiths JM, van der Merwe R. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. 2017;377(10):936–946. doi:10.1056/NEJMoa1704064.
- Gauvreau GM, O’Byrne PM, Boulet LP, Wang Y, Cockcroft D, Bigler J, FitzGerald JM, Boedigheimer M, Davis BE, Dias C, et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N Engl J Med. 2014;370(22):2102–2110. doi:10.1056/NEJMoa1402895.
- Pham T, Ren P, Parnes JR, Griffiths JM. Tezepelumab reduces multiple key inflammatory biomarkers in patients with severe, uncontrolled asthma in the phase 2b PATHWAY study. AJRCCM. 2019;199:A2677.
- Menzies-Gow A, Corren J, Bourdin A, Chupp G, Israel E, Wechsler ME, Brightling CE, Griffiths JM, Hellqvist Å, Bowen K, et al. Tezepelumab in adults and adolescents with severe, uncontrolled asthma. N Engl J Med. 2021;384(19):1800–1809. doi:10.1056/NEJMoa2034975.
- Menzies-Gow A, Corren J, Bourdin A, Chupp G, Israel E, Griffiths J, Hellqvist Å, Bowen K, Kaur P, Almqvist G, et al. Efficacy and safety of tezepelumab in adults and adolescents with severe, uncontrolled asthma: results from the phase 3 NAVIGATOR study. J Allergy Clin Immunol. 2021;147(2 suppl):AB249. doi:10.1016/j.jaci.2020.12.050.
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: the extent of population exposure to assess clinical safety for drugs intended for long-term treatment of non-life-threatening conditions. 1994. Available from: https://www.pmda.go.jp/files/000156791.pdf.
- American Thoracic Society. Sleep related questionnaires. Available from: https://www.thoracic.org/members/assemblies/assemblies/srn/questionaires/acq.php.
- Juniper EF, O’Byrne PM, Guyatt G, Ferrie P, King D. Development and validation of a questionnaire to measure asthma control. Eur Respir J. 1999;14(4):902–907. doi:10.1034/j.1399-3003.1999.14d29.x.
- Juniper EF, Svensson K, Mörk AC, Ståhl E. Measurement properties and interpretation of three shortened versions of the asthma control questionnaire. Respir Med. 2005;99(5):553–558. doi:10.1016/j.rmed.2004.10.008.
- Gauvreau GM, Sehmi R, Ambrose CS, Griffiths JM. Thymic stromal lymphopoietin: its role and potential as a therapeutic target in asthma. Expert Opin Ther Targets. 2020;24(8):777–792. doi:10.1080/14728222.2020.1783242.
- Diver S, Khalfaoui L, Emson C, Wenzel SE, Menzies-Gow A, Wechsler ME, Johnston J, Molfino N, Parnes JR, Megally A, et al. Effect of tezepelumab on airway inflammatory cells, remodelling, and hyperresponsiveness in patients with moderate-to-severe uncontrolled asthma (CASCADE): a double-blind, randomized, placebo-controlled, phase 2 trial. Lancet Respir Med. 2021;9(11):1299–1312. doi:10.1016/S2213-2600(21)00226-5.