Abstract
Cibacron blue is a potent inhibitor of 3-HBA-6-hydroxylase at a concentration < 1 μM. Kinetic analyses revealed that at a concentration below 0.5 μM the dye behaves as an uncompetitive inhibitor with respect to 3-HBA and competes with NADH for the same site on the enzyme. The alteration of the near-UV CD spectrum and quenching of the emission fluorescence of the enzyme by cibacron blue indicates a significant alteration in the environment of aromatic amino acid residues due to a stacking interaction and subtle conformatiodnal changes in the enzyme. The concentration-dependent quenching of the intrinsic fluorescence of the enzyme by cibacron blue was employed to determine the binding parameters such as association constant (Ka) and stoichiometry (r) for the enzyme-dye complex.
Introduction
The flavin containing enzyme, 3-HBA-6-hydroxylase is an external flavin monooxygenase that is inducible in Pseudomonads Citation1-3, Micrococcus sp [Citation4] and Klebsiella pneumoniae [Citation5,Citation6] by the presence of 3-HBA (3-hydroxybenzoate). The flavin monooxygenase catalyzes para hydroxylation of 3-hydroxybenzoate (3-HBA) to 3,6-dihydroxybenzoate (gentisate) and requires NADH or NADPH as the external reductant for its hydroxylating activity. In an earlier paper we reported the facile purification of a NADH-dependant 3-HBA-6-hydroxylase to homogeneity by employing blue sepharose affinity chromatography [Citation4]. We demonstrated that this flavin containing enzyme has an obligatory requirement for 3-HBA to bind to the blue sepharose matrix. This unique interaction results in the formation of a ternary complex consisting of enzyme, 3-HBA and blue sepharose that can be dissociated by depletion or total absence of 3-HBA or by addition of NADH. The activity profile of this enzyme in the presence of increasing concentrations of cibacron blue F3GA, the chemical component of blue sepharose, reveals that the dye is a potent inhibitor causing 50% reduction in the rate of catalysis at a concentration as low as 0.4 μM [Citation4]. The above described preferential interaction of the polyaromatic dye has led to the present report of its effect on the enzyme in detail. This paper reports kinetic and optical spectroscopic studies that were conducted to characterize the nature of cibacron blue inhibition, binding constants for the interaction of the enzyme with the dye and finally the effects of 3-HBA and urea on the stability of the enzyme-dye complex. The significance of such a study is to evaluate the sensitivity of cibacron blue to subtle conformational changes and perturbations in the structure of 3-HBA-6-hydroxylase during its interactions with cofactors and substrate. Also such a study is of interest because of the ability of the dye to mimic the conformation of enzyme bound cofactors such as NADH and other aromatic substrates.
Materials and methods
The following chemicals were purchased from Sigma chemical company: FAD, NADH, and urea. 3-HBA was purchased from Fluka. All the other chemicals used were of analytical reagent grade and locally available. Purification of the enzyme and polarographic activity measurements were done according to the methods described in our earlier paper [Citation4]. Buffer A is 50 mM KH2PO4–NaOH buffer, pH 8.2.
Effect of cibacron blue on the activity of 3-HBA-6-hydroxylase as determined by fluorimetric assay
Aliquots of enzyme (5 μg/ml) in buffer-A were incubated with increasing concentrations (0.1–3.0 μM) of cibacron blue for 10 min in a cuvette at 30°C following which 0.1 nmole of FAD and 500 nmoles of 3-HBA were added. The reaction was initiated by the addition of 500 nmoles of NADH and appearance of the product, gentisate was monitored by following the increase in the intensity of emission fluorescence at 440 nm (excitation at 335 nm) as a function of time. The control experiment was performed in the absence of cibacron blue. Fluorimetric measurements were done using a Hitachi 650 fluorescence spectrophotometer in the ratio mode using slit widths of 5 nm for excitation and emission. Polarographic activity measurements were done according to the methods described in our earlier paper [Citation4].
Nature of inhibition of 3-HBA-6-hydroxylase activity by cibacron blue
Initial velocity studies were done using varying concentrations of 3-HBA (40–1000 μM) or NADH (20–600 μM) at different fixed concentrations (0.05–0.4 μM) of cibacron blue. The concentrations of other components in the assay mixtures were kept constant and activities were measured polarographically according to the methods described earlier [Citation4]. In order to study the nature of inhibition of enzyme activity by cibacron blue with respect to 3-HBA and NADH the results were plotted according to Lineweaver and Burk and analyzed according to Cleland [Citation7,Citation8]. The Ki value (inhibition constant) for inhibition of enzyme activity by cibacron blue with respect to NADH was obtained from a Dixon plot [Citation9].
Effect of cibacron blue on the fluorescence spectrum of 3-HBA-6-hydroxylase
Aliquots containing 25 μg/ml of 3-HBA-6-hydroxylase in its native form or after treatment with 1 M urea or 1 μM FAD or 60 μM 3-HBA were titrated individually with incremental concentrations (0.1–0.85 μM) of cibacron blue in a cuvette. The urea-treated enzyme was pre-incubated for 1 h while the enzyme solutions containing FAD or 3-HBA were pre-incubated for 10 min prior to commencement of titration with cibacron blue. The emission fluorescence spectra as well as the emission fluorescence intensity at 335 nm (λexc = 285 nm) were recorded for all solutions after subtraction from the appropriate blanks. Equivalent concentrations of cibacron blue dissolved in 1 ml of buffer-A alone or in 1 M urea or 1 μM FAD or 60 μM 3-HBA solutions prepared in 1 ml of buffer-A were used as the corresponding blanks. It was not necessary to apply correction factor for dilution since the increase in volume due to the addition of cibacron blue dye was less than 5%. The quenching of the emission fluorescence of the enzyme by incremental concentrations of cibacron blue was analyzed according to the method of Ward and data applied to graphical plots described by Stinson and Holbrook to determine association constants (Ka) and stoichiometry (r) for the enzyme-dye complex [Citation10,Citation11]. The details of the analysis method are described in the following section.
Analysis of binding parameters for the interaction of cibacron blue with 3-HBA-6-hydroxylase
Consider a situation wherein the acceptor, E (enzyme) has p sites for the binding of ligand, X (cibacron blue). The equilibrium for the interaction can be described by Equation (1).
For the above equation the equilibrium constant for association (Ka) can be represented by Equation (2).
Assuming that the interaction of each ligand (dye) molecule with the enzyme causes a constant incremental change in the quantum yield of the intrinsic emission fluorescence of the protein we can represent this change by Equation (3).
In Equation (3), F represents the fluorescence intensity of the purified enzyme in the presence of a specific concentration of dye and F0 is the fluorescence of an equivalent concentration of enzyme in the absence of dye. The maximum fluorescence change observed when all dye binding sites on the enzyme are occupied is represented as ΔFmax. Since it is not possible to experimentally saturate the ligand binding sites of the enzyme due to reasons such as inner filter effect at high concentrations of the dye, ΔFmax was obtained by extrapolation, as described below. A fixed concentration of enzyme was titrated with increasing concentrations of dye. As the concentration of dye increases, the fraction of ligand bound to protein also increases proportionately which in turn causes incremental quenching of the intrinsic fluorescence of the enzyme. Since the total concentration of cibacron blue, [XT], used was much greater than the total protein concentration, [ET], the free dye concentration [XF] can be equated with the total ligand concentration, [XT], i.e. [XF] = [XT]. An extrapolated double reciprocal plot of 1/ΔF versus 1/[XT] yields the y-axis intercept, the ΔFmax. In this plot, as [XT] approaches ∞, 1/[XT] approaches zero and correspondingly, 1/ΔF approaches 1/ΔFmax. Since it is assumed that a constant change in the protein fluorescence accompanies ligand binding, one can experimentally determine the fraction of total binding sites, θ on protein that is occupied for a chosen concentration of dye by applying Equation (4).
The θ values obtained for a selected range of dye concentration can be applied to a graphical method described by Stinson and Holbrook [Citation11] to calculate r (stoichiometry) and Ka (association constant). Equation (5) describing this plot is based on the assumption of a homogenous and non-cooperative mode of ligand binding.
If θ = 1 then Equation (5) is modified to:
From Equations (5) and (6), plots of 1/(1 − θ) versus [XT]/θ will be linear with a slope, Ka, and x-axis intercept of [XT]/θ = r [ET]. Thus if [ET] is known, r can be calculated for the interaction of dye with the enzyme.
Effect of cibacron blue on the CD spectrum of 3-HBA-6-hydroxylase
The enzyme was pre-incubated with 2 μM cibacron blue for 30 min and the far-UV and near-UV CD spectra were recorded after subtracting from the blank solution containing an equivalent concentration of the dye. The concentrations of the enzyme used were 50 μg/ml and 75 μg/ml for the far-UV and near-UV regions, respectively.
Results
Effect of cibacron blue on the activity of 3-HBA-6-hydroxylase analyzed by fluorimeteric assay
(a) and (b) depict the profiles of inhibition of 3-HBA-6-hydroxylase activity with increasing concentrations of cibacron blue as measured by fluorimetric and polarographic assay methods, respectively. The dye is a potent inhibitor of monooxygenase activity causing 50% reduction in the velocity at concentration as low as 0.5 μM. It may be worth mentioning that the inhibition curves obtained by fluorimetric () and polarographic () methods run almost parallel to each other thereby confirming the consistency of the two assay methods used.
Figure 1 Effect of cibacron blue on the initial velocities of 3-HBA-6-hydroxylase; a) activity measured by fluorimetric assay. b) activity measured by polarographic method.
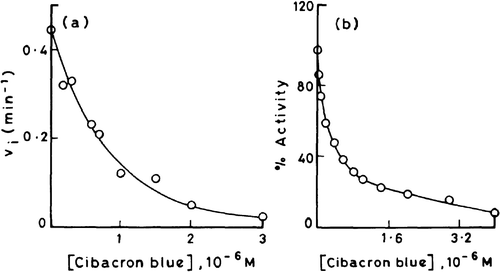
Steady state kinetic analysis of the inhibition of 3-HBA-6-hydroxylase activity by cibacron blue
Nature of inhibition with respect to 3-HBA
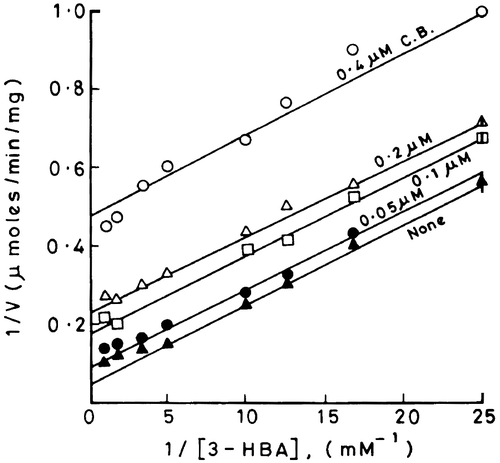
The Lineweaver-Burk plots of 1/V versus 1/[3-HBA] at different fixed concentrations of cibacron blue are shown in . It is evident from these plots that there is a change in the intercept value which increases with increasing concentration of the dye. However the slopes remain unaffected in the presence of the dye. Such observations if analyzed according to Cleland suggest that cibacron blue and 3-HBA bind to two different states of the enzyme that are not reversibly linked to each other [Citation8]. This type of inhibition may be classified as uncompetitive; for example the substrate binds to E, the native form of the enzyme and the dye binds only to ES, the enzyme-substrate complex. Since 3-HBA was used at sub-saturating concentrations, the reversibility between the two forms of enzyme, namely, E and ES is probably disrupted by saturating concentrations of NADH or molecular oxygen resulting in an uncompetitive mode of inhibition of the hydroxylase by cibacron blue with respect to 3-HBA. However the order of binding of cofactor and oxygen needs investigations.
Figure 2 Double reciprocal plots for demonstrating the nature of inhibition of 3-HBA-6-hydroxylase activity by cibacron blue with respect to 3-HBA.
Nature of inhibition with respect to NADH
The competitive nature of inhibition of hydroxylase by cibacron blue with respect to NADH was established from the double reciprocal plots of 1/V versus 1/[NADH] at different fixed concentrations of cibacron blue wherein the intercept remains constant and slope values increase with increasing concentrations of cibacron blue (plots not shown). Based on a Dixon plot (figure not shown) of 1/V versus [cibacron blue] at different fixed concentrations of NADH, a Ki value of 0.056 μM was obtained for the interaction of cibacron blue with enzyme [Citation9].
Fluorescence spectroscopic studies
The intrinsic emission fluorescence of the enzyme is increasingly quenched with increasing concentrations of cibacron blue (). However the quenching is not accompanied by shifting of the emission fluorescence spectral peak. Since the highest concentration of cibacron blue used had an optical density of less than 0.05, the inner filter effect may be neglected. Therefore one can attribute the concentration-dependant quenching of the fluorescence to the formation of a ground state complex between the enzyme and dye. The plots of 1/ΔF versus 1/[cibacron blue] are shown in (see section on “Analysis of binding parameters” for description of this plot) for the native enzyme, enzyme complexed with 3-HBA or FAD and urea denatured enzyme. The maximum fluorescence change (ΔFmax) that can occur when all the binding sites on enzyme molecules are occupied by the dye were obtained from the Y axis intercept of these double reciprocal plots for various situations. A ratio of ΔF/ΔFmax, which corresponds to the fraction of total binding sites occupied by the ligand (θ), were calculated and applied to Stinson and Holbrook plots as shown in [Citation11]. These plots are linear suggesting that the binding of cibacron blue to enzyme molecules is homogenous and non-cooperative. The intercept and slope values of each of these plots give r, the binding stoichiometry and Ka, the association constant value, respectively for the formation of enzyme-cibacron blue complex under different conditions (). The following salient features emerge from the Table:
Cibacron blue binds to the native enzyme with a stoichiometry of ∼ 1:1 which is not significantly affected by the presence of 3-HBA, FAD and urea.
The association constant value is not significantly altered by the presence of FAD or 3-HBA. However, we noticed a ten-fold reduction in the association constant in the presence of 1 M urea.
Figure 3 Profile of quenching of intrinsic fluorescence (emission maximum at 335 nm) of 3-HBA-6- hydroxylase by cibacron blue.
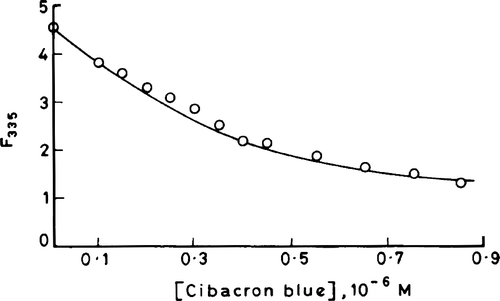
Figure 4 Double reciprocal plots for the observed changes in enzyme fluorescence with increasing concentrations of cibacron blue using native enzyme, FAD-enzyme, 3-HBA-enzyme and urea denatured enzyme.
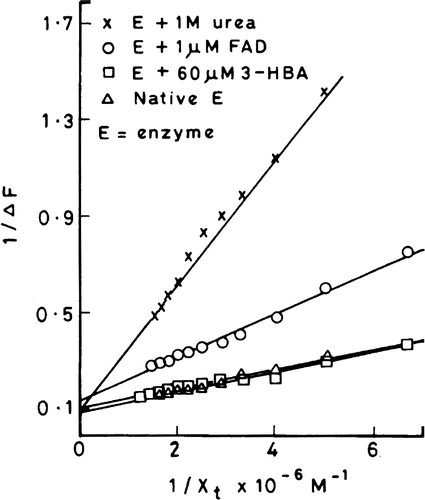
Figure 5 Stinson and Holbrook plots for the quenching of enzyme fluorescence with increasing concentrations of cibacron blue using native enzyme, 3-HBA-enzyme, FAD-enzyme and urea denatured enzyme.
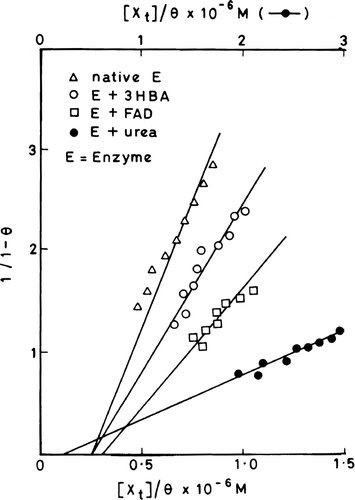
Table I. Binding parameters, stoichiometry (r) and association constant (Ka), for the enzyme–cibacron blue interaction. The binding parameters were calculated from the equation: 1/(1−θ) Ka=[XT]/θ−r[ET].
Effect of cibacron blue on the CD spectrum of enzyme
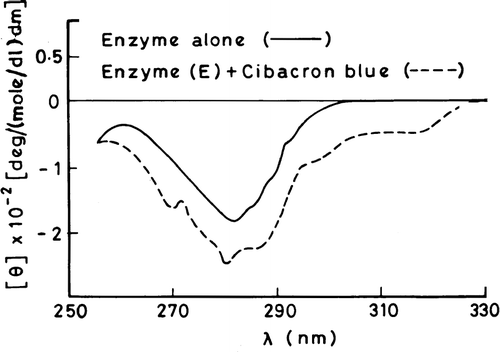
A comparison of the CD spectra of the native and cibacron blue treated enzyme did not reveal significant differences in the far-UV region, probably indicating that the dye does not induce gross conformational changes in the backbone conformation of the enzyme (data not shown). The near-UV CD spectra of the enzyme in the presence and absence of cibacron blue are compared in . The intensity of the negative CD band is significantly increased with a small shift in the negative peak by about 3 nm in the presence of cibacron blue. This is accompanied by the appearance of small bands at 270 and 280 nm with simultaneous loss of fine vibronic structures in the region of 285–295 nm that leads to a pronounced shoulder band at 285 nm. In addition there is induction of a new negative CD band between 300–325 nm.
Figure 6 Near-UV CD spectral profiles of 3-HBA-6-hydroxylase in the absence and presence of cibacron blue.
Discussion
Cibacron blue is a versatile dye that is capable of binding to a wide variety of proteins through ionic and hydrophobic forces. The dye has been used as a probe to obtain information about dinucleotide binding domains, forces stabilizing the interaction of coenzymes, conformational changes in wild type enzymes and characterization of mutant enzymes that have undergone change in their overall conformation Citation12-18. Immobilized cibacron blue is known to have high binding affinity for NADH/FAD sites in enzymes such as lysozyme and been used extensively for purification of proteins [Citation19].
Kinetic and optical spectroscopic analyses of the interaction of cibacron blue with 3-HBA-6-hydroxylase suggest that the dye is a useful conformational probe. The dye acts as a competitive inhibitor with respect to NADH and uncompetitive with respect to 3-HBA. This observation in conjunction with our earlier observation wherein 3-HBA-6-hydroxylase exhibits 3-HBA dependent binding to blue-sepharose matrix and elution using NADH probably suggests that the dye mimics NADH and binds to the cofactor-binding site in the hydroxylase. The dye/NADH binding site on the enzyme is accessible to blue-sepharose matrix following binding of 3-HBA. There are several reports suggesting the structural similarity of cibacron blue to heterocyclic naturally occuring compounds such as NADH, ATP and tetrahydrafolate due to which the dye exhibits selective competition with these coenzyme and cofactor binding sites Citation13Citation14Citation20-26. The inhibition constant, (Ki value = 0.06 μM) obtained for 3-HBA-6-hydroxylase by us is relatively lower as compared to the value reported for p-HBA hydroxylase [Citation22]. Thus the dye appears to exhibit high affinity for 3-HBA-6-hydroxylase that is accompanied by drastic inhibition of its catalytic activity. However, how closely the bound dye mimics the bound coenzyme (NADH) is unknown.
The reduction in the intensity of the intrinsic fluorescence of 3-HBA-6-hydroxylase by cibacron blue may be ascribed to stacking interactions and formation of a charge transfer complex between aromatic amino acid residues (such as the indole ring of tryptophan) and aromatic rings of the dye at the binding site. An alternative explanation is the induction of a local conformational change in the enzyme that leads to the exposure of tryptophan residues to solvent thereby reducing the quantum yield of emission fluorescence. Quenching of protein fluorescence by cibacron blue has been also reported for various proteins and enzymes such as p-hydroxybenzoate hydroxylase from Pseudomonas fluorescens and pyruvate kinase from Neurospora crassa [Citation10,Citation22,Citation27]. The concentration-dependent quenching of the intrinsic fluorescence of the enzyme by cibacron blue provided a convenient method for the quantification of binding parameters under various conditions. The dye does not seem to interact significantly with the FAD or 3-HBA sites of the enzyme since the affinity constants as well as stoichiometry is not affected by their absence or presence. On the other hand, reduction in the binding affinity of dye for urea treated enzyme might indicate the importance of hydrogen bonding forces in maintaining the dye binding site in the enzyme and/or facilitating their interaction with each other.
The kinetic analysis of the inhibition of 3-HBA-6-hydroxylase revealed that at concentrations below 0.4 μM cibacron blue behaves as an uncompetitive inhibitor with respect to 3-HBA; for example the dye preferentially binds to the ES complex. On the other hand the equilibrium studies based on quenching of the intrinsic fluorescence of enzyme suggests that the dye is capable of forming a complex with native enzyme even in the absence of 3-HBA. Also the Ka value for the enzyme-dye interaction does not seem to change significantly in the presence of 3-HBA. This discrepancy between the kinetic and equilibrium studies may be explained on the basis of a dominant effect of the ESI complex on the catalytic activity as compared to the EI complex under steady-state conditions that is attained in the kinetic experiments.
The increased intensity of the negative peak and induction of a new band in the 300–325 nm region in the near-UV CD spectrum of the enzyme in the presence of cibacron blue may be attributed to the exciton splitting due to a stacking interaction between aromatic acid residue(s) of the enzyme and aromatic rings in the dye [Citation28,Citation29]. The observed changes probably reflect subtle perturbations in the conformation of hydroxylase that is accompanied by changes in the environment of aromatic amino acid residues of the enzyme due to the binding of dye.
In conclusion, our studies suggest that in addition to its usefulness in affinity chromatography, cibacron blue may be also used as a convenient monitor to assess subtle perturbations in structure of 3-HBA-6-hydroxylase during its interaction with molecules such as NADH or 3-HBA.
Acknowledgements
The financial support given by the Indian Institute of Science, Bangalore, India is greatly appreciated. We also acknowledge S. L. Nagraj for his excellent technical assistance.
References
- Groseclose EE, Ribbons DW. Biochem Biophys Res Commun 1973; 55: 897–903
- Wang L-H, Hamzah RY, Yu Y, Tu S-C. Biochemistry 1987; 26: 1099–1104
- Yu Y, Wang L-H, Tu S-C. Biochemistry 1987; 26: 1105–1110
- Sumathi R, Rajasekharan R, Vaidyanathan CS. Arch Biochem Biophys 1990; 278: 21–25
- Jones DCN, Cooper RA. Arch Microbiol 1990; 154: 489–495
- Suarez M, Ferrer E, Garrido-Pertierra A, Martin M. FEMS Microbiol Lett 1995; 126: 283–290
- Lineweaver H, Burk D. J Amer Chem Soc 1934; 56: 658–666
- Cleland WW. The enzymes3rd ed., PD Boyer. Academic press, New York, USA 1970; 2: 1–65
- Dixon M. Biochem J 1953; 55: 170–171
- Ward LD. Meth Enzymol 1985; 117: 400–414
- Stinson RA, Holbrook JJ. Biochem J 1973; 131: 719–728
- Thompson ST, Cass KH, Stellwagen E. Proc Natl Acad Sci 1975; 72: 669–672
- Thompson ST, Stellwagen E. Proc Natl Acad Sci 1976; 73: 361–365
- Stellwagen E. Acc Chem Res 1977; 10: 92–98
- Stellwagen E, Baker B. Nature 1976; 261: 719–720
- Einarsson R, Eklund H, Zeppezauer E, Boiwe T, Branden CI. Eur J Biochem 1974; 49: 41–47
- Federici MM, Chock PB, Stadtman ER. Biochemistry 1985; 24: 647–660
- Yang M, Kirley TL. Biochemistry 2004; 43: 9185–9194
- He LZ, Sun Y. Bioprocess Biosyst Eng 2002; 25: 155–164
- Ryan LD, Vestling CS. Arch Biochem Biophys 1974; 160: 279–284
- Watson DH, Harvey MJ, Dean PD. Biochem J 1978; 173: 591–596
- Muller F, van Berkel WJH, Steennis PJ. Biochem Int 1983; 7: 115–122
- Apps DK, Gleed CD. Biochem J 1976; 159: 441–443
- Ashton AR, Polya GM. Biochem J 1978; 175: 501–506
- Lepo JE, Stacey C, Wyss O, Tabita FR. Biochim Biophys Acta 1979; 568: 428–436
- Ramesh KS, Appaji Rao N. Biochem J 1980; 187: 249–252
- Kapoor M, O'Brien MD. Can J Microbiol 1980; 26: 613–621
- Edwards RA, Woody RW. Biochem Biophys Res Commun 1977; 79: 470–476
- Edwards RA, Woody RW. Biochemistry 1979; 18: 5197–5204