Abstract
An aerosol nitrogen analyzer has been developed for determination of total nitrogen (TN) content in aerosols collected on a filter substrate. It uses rapid thermoevolution of nitrogenous species at 800°C in a 2.5% O2/97.5% He carrier gas. Evolved nitrogen is oxidized to nitrogen oxides on a manganese dioxide catalyst, converted to nitrogen monoxide on a molybdenum catalyst, and quantified with a chemiluminescence detector. The analyzer is built upon components of two commercially available instruments, a thermal aerosol carbon analyzer and a chemiluminescent NO x analyzer. This system is able to provide fast (3 min per sample) and highly sensitive (a detection limit of 26 ng N) TN measurements for aerosol samples of a small size (∼1× 1 cm2) without any pretreatment. When the aerosol nitrogen analyzer was properly calibrated by ammonium sulfate, excellent agreement was obtained between TN measurements of twelve aerosol samples by the aerosol nitrogen analyzer and those obtained from an elemental analyzer. This instrument provides a useful and convenient tool for characterization of the organic nitrogen (ON) pool in atmospheric aerosols. The ON concentrations in the twelve aerosol samples, calculated from the difference between TN and the sum of the inorganic nitrogen species determined by ion chromatographic analysis, were found comparable in magnitude to particulate nitrate-N. The contribution of ON to TN ranged from 1.7% to 54% for the limited set of test samples. While twelve samples are insufficient to draw general conclusions about the relative distribution of ambient aerosol nitrogen species, they have demonstrated the applicability of the new method to ambient aerosol characterization and exposed a potential limitation in deriving at ON concentrations by taking a sometime small difference between two large numbers (i.e., TN and inorganic nitrogen). Differentiation of organic and inorganic nitrogen using thermal methods is also explored and found not to be feasible.
INTRODUCTION
Atmospheric particles can be an important source of fixed nitrogen in the environment because of their typically high nitrogen content (CitationFinlayson-Pitts and Pitts 2000; CitationSeinfeld and Pandis 1998). A large number of N-containing species have been identified to exist in ambient aerosols. To date, most studies have focused on inorganic nitrogen (IN) species. The IN species mainly are ammonium (NH4 +), nitrate (NO3 –), and nitrite (NO2 –) (CitationSeinfeld and Pandis 1998). They are most commonly determined by ion chromatography (IC) or flow injection analysis (CitationLuke and Valigura 1997). These measurement techniques are established, and they have been well tested by researchers in this field. Organic nitrogen (ON) appears to be a ubiquitous and significant component in the atmospheric nitrogen budget (CitationCornell et al. 1995), but it is much less studied, partly due to a lack of simple analytical methods. ON is usually determined as the difference between total nitrogen (TN) and IN. As such, TN measurement techniques provide indirect solutions for ON determination.
Several methods for the determination of TN in aqueous solutions are currently in widespread use. They include persulfate oxidation, UV photo-oxidation, and high-temperature oxidation or catalytic oxidation (CitationBronk et al. 2000). These methods fall into two categories, wet methods and combustion-based methods. In the first category, ON species are converted into IN ions either by the use of chemical oxidants such as persulfate or by UV photolysis with or without addition of oxidants (CitationBronk et al. 2000; CitationSolorzano and Sharp 1980). The wet methods have the drawback of being laborious and often suffer high and variable nitrogen blank levels caused by the oxidization agents (CitationCornell and Jickells 1999). In the second category, the methods involve oxidation of nitrogenous species by high-temperature oxidation or catalytic oxidation, followed by detection of the N gas products (CitationSuzuki et al. 1985; CitationSugimura and Suzuki 1988; CitationHansell 1993; CitationAmmann et al. 2000; CitationAlvarez-Salgado and Miller 1998). These combustion-based methods analyze the entire sample content without sample preparation. They have the advantages of being simple and rapid and require a smaller amount of the sample, although the instrumentation is less readily available.
Historically, the methods in both categories have been developed for the analysis of aqueous samples, particularly seawater matrices. Their applications to atmospheric samples are limited to rainwater and water extracts of aerosol samples (CitationCornell et al. 1995; CitationCornel and Jickells 1999; CitationZhang and Anastasio 2001; CitationZhang et al. 2002). Direct analysis of solid matrices, such as aerosol-laden filters, is not accommodated by methods in either category. On the other hand, combustion-based methods are routinely used in the direct analysis of the carbon content in aerosols (e.g., CitationChow et al. 1993; CitationBirch and Cary 1996). These methods involve evolution of carbonaceous materials through thermal decomposition, volatilization, and oxidation under programmed heating followed by catalytic oxidation to CO2 for detection. A limited number of studies have explored the combustion-based approach for the determination of nitrogen in aerosols. Dod and his colleagues have demonstrated the feasibility of combining thermal evolution techniques and chemiluminescence detection for the measurement of aerosol nitrogen (CitationDod and Novakov 1982; CitationDod et al. 1984; CitationDaisey and Gundel 1991), but no detailed method characterization and validation is available in their reports. More recently, Stolzenburg and his coworkers (CitationStolzenburg and Hering 2000; CitationStolzenburg et al. 2003) reported an automated method for one group of aerosol nitrogenous compounds, i.e., nitrates. Their method uses flash vaporization and selectively converts nitrate compounds in the aerosols to nitrogen oxides for subsequent chemiluminescent detection. We describe here a simple aerosol TN analysis system, called the aerosol nitrogen analyzer (ANA) hereafter, which employs flash heating of aerosol-laden filters in a 2.5% O2/97.5% He carrier gas with subsequent catalytic conversion to nitrogen monoxide for chemiluminescent detection. The system is built upon components of two commercially available instruments, a combustion-based aerosol carbon analyzer (Sunset Laboratory, Forest Grove, OR, USA) and a chemiluminescent NOx analyzer (Model 200A, API, San Diego, CA, USA). Presented below are details about the instrument and characterization of the instrument's performance. Also shown are example data of aerosol TN and ON measurements made with the instrument.
EXPERIMENTAL SECTION
Apparatus
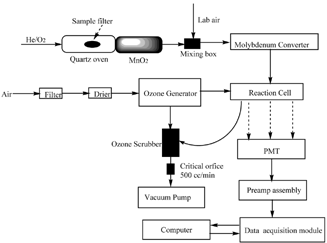
The aerosol nitrogen analyzer is designed for determination of microgram levels of aerosol nitrogen. A block diagram of the instrument is shown in . The major components of this apparatus are the following:
| 1. | The quartz tube (0.5 cm o.d., 12 cm in length) is divided into two zones, the front oven zone and the back oven zone, each 4 cm in length and wrapped in separate 20-gauge nichrome wires. The front oven accepts a rectangular filter (1–1.5 cm2) cut from an aerosol-laden quartz filter. The back oven houses the manganese dioxide (MnO2) catalyst. A filter cut is first placed in the front oven, after which the system is closed and flushed with a 2.5% O2/97.5% He mixture at 100 mL/min at room temperature. During analysis, the temperature of the front oven is programmed to allow thermal evolution of the aerosol materials into the carrier gas stream. The back oven temperature is fixed at a preset, programmable value, which is 870°C for all ensuing analyses. The MnO2 catalyst in the back oven oxidizes the nitrogen-containing gases that have evolved from the front oven to nitrogen oxides (e.g., NO, NO2). After the heating zones, the O2/He mixture gas mix with a separate air stream supplied at ∼400 mL/min in the mixing box. | ||||
| 2. | The molybdenum converter is a stainless steel cartridge containing molybdenum chips heated to 315°C. The converter reduces various forms of higher nitrogen oxides (e.g., NO2, N2O5) to nitric oxide (NO) when the sample stream is routed through the heated molybdenum. | ||||
| 3. | The ozone generator subsystem consists of a 47 mm particulate filter, a permeation drier, and a generator module. Ozone is needed for the chemiluminscent detection of NO as described below. Excessive ozone is destroyed in a catalytic ozone scrubber. | ||||
| 4. | The detector module consists of a reaction cell, a photo multiplier tube (PMT), and a preamplifier assembly. In the reaction cell, ozone mixes with the NO gas generated after the carrier gas passes through the Mo converter to produce electronically excited NO2 molecules. The excited NO2 molecules emit chemiluminescent light when they drop to a lower energy level. The chemiluminescent light intensity is directly proportional to the NO concentration. Parts of a commercial chemiluminescence NOx analyzer are modified to provide components 2–4. | ||||
| 5. | The data acquisition module (Microlink 751, Biodata Limited, Manchester, UK) collects the analog voltage signal from the preamplifier assembly at a frequency of 0.2 Hz and sends the data to the computer for display. The microprocessor in the NOx analyzer has to be bypassed to achieve this high frequency. | ||||
FIG. 1 Block diagram of the aerosol nitrogen analyzer.
Elemental Analyzer
A PE2400 series II CHNS/O analyzer (Perkin Elemer, Shelton, CT, USA) was used in its CHN mode to provide independent N measurements for evaluating the accuracy of the measurements by the ANA. This elemental analyzer uses a combustion method to convert the measured elements (C, H, and N) to simple gases, i.e., CO2, H2O, and N2. These gases are then quantified as a function of thermal conductivity. A known standard, acetanilide (C8H9NO), is first analyzed to calibrate the analyzer. The resulting calibration factor is then used to determine the samples. A filter cut sized 1× 1 cm is placed on a tin disk, folded, and rolled into a cylinder. The cylinder is then dropped into the analyzer for elemental analysis. The elemental analyzer has a detection limit of 0.68 μgN.
Reagents and Aerosol Materials
The standard nitrogenous compounds for testing include ammonium chloride, potassium nitrate, sodium nitrite, ammonium nitrate, ammonium sulfate, ammonium hydrogen sulfate, methylamine, urea, and a mixture of 20 individual amino acids. Ammonium hydrogen sulfate was made by mixing ammonium sulfate and sulfuric acid in stoichiometric proportions. The amino acid mixture was purchased from Waters (Milford, MA) and all the other standards were from Acros (Geel, Belgium). All chemicals were reagent grade or better and used without further purification.
Aerosol samples were collected on quartz fiber filters (Pall Gelman, Ann Arbor, MI, USA) from three locations, Jeju Island, S. Korea, Hong Kong, and Nanjing, China. All quartz filters were prebaked in a muffle furnace at 550°C overnight before use. The Jeju samples were collected onto 20 cm quartz filters at a flow rate of 0.50 m3/min using a high-volume particle trap impactor/denuder sampler with an impactor inlet to remove particles of larger than 5 μm (CitationMader et al. 2001). The Hong Kong and the Nanjing samples were collected onto 20 × 25 cm quartz filters at a flow rate of 1.13 m3/min using a PM2.5 high-volume sampler (Andersen Instruments, Smyrna, GA, USA). All of the samples were collected under ambient temperature and relative humidity. No attempts were made to remove gaseous nitric acid upstream the filters.
Determination of Organic and Inorganic Nitrogen
Concentrations of the IN species in aerosols, NH4 +, NO3 −, and NO2 −, were determined by injecting water extracts of the aerosol samples into an ion chromatograph (DX500, Dionex, Sunnyvale, CA, USA). Each water extract was obtained by extracting 2–4 pieces of filter strips sized in 1× 1.45 cm with two portions of water (2 ml each) in a sonication bath, each extraction for 15 min. The two extractions were then passed through a same syringe filter and combined in a 5.0 volumetric flask. An additional 1 ml of water was then passed through the same syringe filter and combined with the previous two extractions. The final volume of the combined water extracts was made to be 5.0 ml. An IonPac CG12 guard column (4× 50 mm), an IonPac CS12 analytical column (4× 250 mm), and an ED40 electrochemical detector were used in the cation analysis with isocratic elution at a flow rate of 1 mL/min. The eluent was 20 mM of methanesulfonic acid. In the anion analysis, an Ionpac AG11 guard column (4× 50 mm), an IonPac AS11 analytical column (4× 250 mm), and a CD20 conductivity detector were used. The eluent was NaOH with a programmed gradient from 0.2 to 5 mM, and the flow rate was 2 mL/min. The sampling loop was 50 μL in both cation and anion analyses. ON concentrations were calculated as the difference between TN and the sum of the three IN species.
RESULTS AND DISCUSSION
Calibration, Precision, and Detection Limit
Prior to the commencement of an analysis, the front oven temperature is maintained at ca 50°C due to its proximity to the back oven, which is maintained at 870°C at all times. Once the filter sample is placed in the oven and the system is closed, the front oven temperature is set to 800°C and the analysis starts. shows the evolved peaks for the various tested standard compounds and an aerosol sample. The standard nitrogenous compounds vary in the N oxidation states and chemical structures. It is noted that they evolve at slightly different times and appear in different peak shapes.
FIG. 2 N thermoevolution peaks of a few standard nitrogen species and an aerosol sample using a fast thermoevolution temperature program. Analysis conditions: Front oven, 800°C; MnO2 catalyst oven, 870°C; Mo converter, 315°C; Carrier gas flow 500 mL/min.
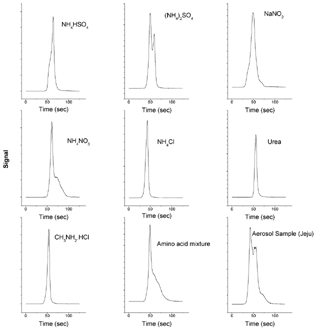
Ammonium chloride, methylamine, and urea appear as single symmetric peaks; ammonium sulfate appears as two overlapping peaks; the other tested standards show either leading or tailing peaks. Regardless of the peak shapes, the peak area is proportional to the amount of N present for each individual standard, suggesting that the conversion of N species to NO and the subsequent detection of NO is quantitative. As illustrated in , the evolution of N is completed in less than 3 min. This demonstrates that the analyzer is capable of rapid analysis of aerosol N through thermal evolution.
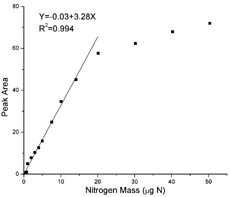
Ammonium sulfate is chosen to calibrate the aerosol nitrogen analyzer on the grounds that it is a known major nitrogen species in atmospheric aerosols (CitationSeinfeld and Pandis 1998). The detector calibration factor, called the K-factor hereafter, is defined as the ratio of the peak area of the chemiluminescence signal and the spiked (NH4)2SO4–N amount in μ gN. shows an example response curve of TN in the range of 0.5–50 μ gN, introduced into the ANA by syringe injection of known amounts of an (NH4)2SO4 aqueous solution onto prebaked quartz filter strips. The detector response is linear up to 15 μ gN. The K-factor is determined from the linear regression of the measurements below 15 μgN. The detection limit of the analyzer, calculated from 3SDb/K-factor, is estimated to be 0.026 μgN, where SDb is the standard deviation of replicate measurements of blanks. The relative standard deviation (RSD) for 4–5 replicate analyses of the standard samples is typically less than 5%, even at N levels as low as 0.1 μ g N.
FIG. 3 Response curve of the aerosol nitrogen analyzer to the amount of N in ammonium sulfate. The linear curve is generated using calibration standards of 0.5–15 μgN.
Validation
For assessment of the compound dependence of the instrument response, a number of standard N compounds of known amounts were analyzed to determine their recoveries. They are listed in . They were selected for analysis because of their established atmospheric presence. The characterization of the molecular composition of ON is still far from being comprehensive and complete. Available data indicate that urea, aliphatic amines, and amino acids are present in atmospheric particulate matter (CitationZhang and Anastasio 2001; CitationZhang et al. 2002; CitationGorzelska and Galloway 1990; CitationGorzelska et al. 1997; CitationCornell et al. 1998). The above-mentioned ON species, together with the few IN compounds, cover the full range of oxidation states of nitrogen (from −3 to +5) and represent a variety of chemical structures. The recoveries of these compounds were determined to range from 80–129% (). A unity recovery for a given compound indicates that the instrument response for this compound is the same as the instrument response for the calibration compound (NH4)2SO4. Six out of nine compounds had their recoveries deviate less than 6% from unity, demonstrating the robustness of the oxidation catalyst and the Mo converter. The remaining three compounds, NH4Cl, KNO3, and KNO2, exhibited deviations of –20%, 30%, and –15%, respectively. The recovery, and thereby the instrument response, is apparently dependent on both the N oxidation state and its chemical surroundings in the individual compound (e.g., NH4Cl versus (NH4)2SO4). It is conceivable that both factors could affect the resulting composition of the nitrogen gas products and thereby the oxidation efficiency of the MnO2 catalyst and subsequently the efficiency of the Mo converter.
TABLE 1 Recoveries of a few nitrogenous standards as measured by the aerosol nitrogen analyzer
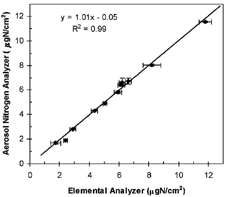
The nitrogen pool in real aerosol particulate matter is poorly understood. In addition, the chemical composition of aerosols varies in space and time. Without knowledge of the detailed molecular composition, it is impractical to test every N species possibly present in the aerosols. In addition, it is unknown if the presence of numerous other chemicals alters thermal evolution behaviors and products of a given N compound. Consequently, it is difficult to assess the impact of the few N species that have moderate deviations from the reference compound (NH4)2SO4on the measurement accuracy of the TN content. The alternative approach is to assess the extent of the agreement between measurements made by the ANA and a different technique. Elemental analyzers, which have been routinely used to determine the elemental composition of C, H, and N in chemical compounds for decades (CitationSkoog et al. 1998), provide such an alternative technique for TN measurement. In this connection, we have compared the TN measurements of twelve ambient aerosol samples by the aerosol nitrogen analyzer and those by the Perkin Elemer elemental analyzer. The aerosol samples collected from different locations in the East Asian region were chosen to capture aerosols of different chemical composition. The results in μ gN per cm2 of filter area are listed in and depicted in . The linear regression line in has a slope and an intercept not statistically different from unity and zero, respectively, by a t test at the 95% confidence level (CitationMiller and Miller 1993). The excellent agreement between the two techniques demonstrate that despite the complex and variable chemical matrix, the ANA can provide accurate N measurements using (NH4)2SO4 as the instrument calibration compound.
FIG. 4 Comparison of total nitrogen measurements by the aerosol nitrogen analyzer and the elemental analyzer.
TN, ON, and IN Concentrations of Ambient Aerosol Samples
The TN air concentrations of the twelve ambient aerosol samples range from 0.76–5.83 μgN/m3 (). The IN species, NH4 +, NO3 −, and NO2 − in these samples, are determined by IC analysis and have a method detection limit of 2.1, 1.2, and 0.12 μ M, respectively. Nitrite is below its detection limit in all samples, that is, it is less than 0.7 ngN/m3 in air concentrations. The ON concentrations range from 0.04–0.87 μ gN/m3, comparable to the particulate NO3 −-N in magnitude in each individual sample (). The organic nitrogen pool accounts for 1.7–54% of the total aerosol nitrogen, ranging from being negligible to substantial. This limited data set indicates that the ON concentrations in ambient aerosols could have considerable variation and that ON could be a significant part of aerosol nitrogen loading. The results also reveal an inherit problem with the approach of taking a small difference (i.e, ON) between two large numbers (i.e., TN and IN) (CitationHuebert and Charlson 2000). Large ON measurement uncertainties are observed in the Hong Kong samples, which have TN and IN of similar magnitudes. In particular, the presence of ON in two of the samples cannot be verified due to the measurement uncertainties. Despite this shortcoming, the ANA offers a convenient tool for assessing the abundance of aerosol ON.
TABLE 2 TN, ON, and IN concentrations in twelve ambient aerosol samples
Feasibility of Differentiation of ON and IN by Thermal Methods
CitationDod et al. (1984) suggested the possibility of programming the thermal evolution temperature to differentiate ON and IN. They reported that under a linear temperature program, the amount of N evolved before 300°C in one ambient sample was roughly equal to the sum of NH4 + and NO3 –, as determined by X-ray photoelectron spectroscopy. In addition, after a sequential extraction of the filter with benzene and a methanol:chloroform mixture that aimed to remove ON, they observed that little of the residue N evolved at temperature higher than 300°C. On the basis of these observations, they suggested that the N that evolved after 300°C was linked to ON. However, it is noted that the N evolution peaks were far from being well resolved in the vicinity of 300°C for the real aerosol samples shown in their paper. Moreover, water-soluble ON species (e.g., amino acids) would have been part of the residue N on the filter after the extractions by organic solvent, and they evolve together with the IN species before 300°C. In summary, there is insufficient evidence to support the use of 300°C as an adequate benchmark for quantification of ON and IN.
We have examined the thermal evolution characteristics of nitrogenous materials on the ANA. The temperature program employed consists of two temperature steps, 300°C and 800°C, with a residence time of 120 s at each temperature step. The residence time includes the time spent in raising the temperature from the previous value to the set value. There is a 60 s cooling period between the two temperature steps, during which the heating source is switched off and a fan is turned on. The purpose of this cooling period is to ensure that the first evolution peak signal returns to the baseline before the next temperature step. shows the N thermograms of a few IN and ON standards of known presence in atmospheric aerosols. On one hand, an ON compound could have part (e.g., amino acids) or all (e.g., urea, methylamine) of its N moiety evolve before or at 300°C. On the other hand, (NH4)2SO4 and NH4NO3 have a second peak that evolves after 300°C, and all of KNO3 appears as a single peak that evolves well after the 300°C temperature step. It is obvious that there is not a clear-cut temperature that differentiates ON from IN. In other words, ON and IN species cannot be distinguished on the basis of their thermal volatility.
FIG. 5 N thermograms of selected organic and inorganic nitrogen species and two aerosol samples using a two-step temperature program. Front oven temperature program: initially 300°C for 120 s, cooling for 60 s, 800°C for 120 s.
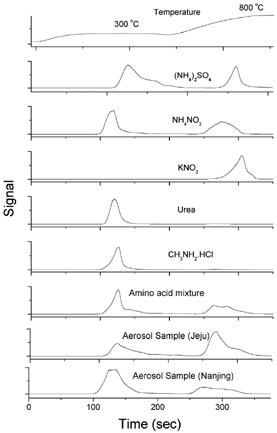
also shows two example N thermograms of ambient aerosol samples, one sample from Jeju Island, South Korea and the other sample from Nanjing, China. The thermograms appear as two broad but resolved peaks corresponding to the two temperature steps. For the Jeju sample, the ratio between the second and the first peaks is 1.65, much greater than its ON:IN ratio of 0.38. For the Nanjing sample, the ratio between the second and the first peaks is 0.42, close to its ON:IN ratio of 0.40 (). Again, this demonstrates that contrary to Dod et al.'s suggestion, the two resolved peaks that evolved before/at and after the 300°C temperature step cannot be conclusively assigned to IN and ON, respectively. Any close match is more likely than not a fortuitous result. If one considers the complex and unknown nature of aerosol N composition, it is unlikely to resolve the various N species into separate ON and IN peaks by fine-tuning the temperature program. As a result, we conclude that it is not feasible to differentiate IN and ON using thermal methods.
CONCLUSIONS
We have demonstrated that an aerosol nitrogen analyzer can be constructed using components of two commercially available instruments, an aerosol carbon analyzer and a NOx chemiluminescent analyzer. The aerosol nitrogen analyzer is capable of fast and highly sensitive determination of aerosol total nitrogen at submicrogram levels. The TN measurement allows the determination of total organic N and therefore the quantification of relative contribution of inorganic and organic N species to the total fixed N in aerosols. The ON measurement also makes it possible to evaluate the relative contribution of individual ON compounds to the total organic N, and to determine whether there are major unidentified ON compounds. This instrument provides a useful and convenient tool for characterization of the organic nitrogen pool in atmospheric aerosols.
Acknowledgments
This work was supported by the Research Grants Council of Hong Kong, China (DAG 00/01.SC14 and N_HKUST612/01). The authors thank Eric C. H. Wan and Hong Yang for collecting the aerosol samples, Jinhui Xu for providing the IC analysis, and the Marine Coastal Lab at HKUST for allowing us to use its elemental analyzer.
Notes
a A unity recovery indicates that a given compound has the same instrument response as the calibration compound (NH4)2SO4.
b A mixture of 20 individual amino acids.
REFERENCES
- Alvarez-Salgado , X. A. and Miller , A. E. J. 1998 . Simultaneous Determination of Dissolved Organic Carbon and Total Dissolved Nitrogen in Seawater by High Temperature Catalytic Oxidation: Condition for Precise Shipboard Measurements . Mar. Chem. , 62 : 325 – 333 .
- Ammann , A. A. , Rüttimann , T. B. and Bürgi , F. 2000 . Simultaneous Determination of TOC and TNB in Surface and Wastewater by Optimized High Temperature Catalytic Combustion . Water Res. , 34 : 3573 – 3579 .
- Birch , M. E. and Cary , R. A. 1996 . Elemental Carbon-Based Method for Monitoring Occupational Exposures to Particulate Diesel Exhaust . Aerosol Sci. Technol. , 25 : 221 – 241 .
- Bronk , D. A. , Lomas , M. W. , Gilbert , P. M. , Schukert , K. J. and Sanderson , M. P. 2000 . Total Dissolved Nitrogen Analysis: Comparisons Between the Persulfate, UV and High Temperature Oxidation Methods . Mar. Chem. , 69 : 163 – 178 .
- Chow , J. C. , Watson , J. G. , Pritchett , L. C. , Pierson , W. R. , Frazier , C. A. and Purcell , R. G. 1993 . The DRI Thermal Optical Reflectance Carbon Analysis System—Description, Evaluation and Applications in United States Air Quality Studies . Atmos. Environ. , 27A : 1185 – 1201 .
- Cornell , S. E. and Jickells , T. D. 1999 . Water-Soluble Organic Nitrogen in Atmospheric Aerosol: A Comparison of UV and Persulfate Oxidation Methods . Atmos. Environ. , 33 : 833 – 840 .
- Cornell , S. E. , Jickells , T. D. and Thornton , C. A. 1998 . Urea in Rainwater and Atmospheric Aerosol . Atmos. Environ. , 32 : 1903 – 1910 .
- Cornell , S. , Rendell , A. and Jickells , T. 1995 . Atmospheric Input of Dissolved Organic Nitrogen to the Oceans . Nature , 376 : 243 – 246 .
- Daisey , J. M. and Gundel , L. A. 1991 . Tracing the Sources of Indoor Aerosols Using Gas-Analysis . Aerosol Sci. Technol. , 14 : 25 – 32 .
- Dod , R. L. , Gundel , L. A. , Benner , W. H. and Novakov , T. 1984 . Non-Ammonium Reduced Nitrogen Species in Atmospheric Aerosol Particles . Sci. Total Environ. , 36 : 277 – 282 .
- Dod , R. L. and Novakov , T. 1982 . “ Application of Thermal Analysis and Photoelectron Spectroscopy for the Characterization of Particulate Matter ” . In Industrial Applications of Surface Analysis , Edited by: Casper , L. A. and Powell , C. J. pp. 397 – 409 . Washington, DC : American Chemical Society . ACS Symposium Series 199
- Finlayson-Pitts , B. J. and Pitts , J. N. Jr. 2000 . Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Application , San Diego, CA : Academic Press .
- Gorzelska , K. and Galloway , J. N. 1990 . Amine Nitrogen in the Atmospheric Environment over the North Atlantic Ocean . Global Biogeochem. Cycles. , 4 : 309 – 333 .
- Gorzelska , K. , Scudlark , J. R. and Keene , W. C. 1997 . “ Dissolved Organic Nitrogen in the Atmospheric Environment ” . In Atmospheric Deposition of Contaminants to the Great Lakes and Coastal Waters , Edited by: Baker , J. E. pp. 379 – 392 . Pensacola, FL : SETAC Press .
- Hansell , D. A. 1993 . Results and Observations from the Measurement of DOC and DON in the Southern California Bight Using Oxidation by High Temperature Combustion Technique . Mar. Chem. , 41 : 195 – 202 .
- Huebert , B. J. and Charlson , R. J. 2000 . Uncertainties in Data on Organic Aerosols . Tellus , 52B : 1249 – 1255 .
- Luke , W. T. and Valigura , R. A. 1997 . “ Methodologies to Estimate the Air-surface Exchange of Atmospheric Nitrogen Compounds ” . In Atmospheric Deposition of Contaminants to the Great Lakes and Coastal Waters , Edited by: Baker , J. E. pp. 415 – 429 . Pensacola, FL : SETAC Press .
- Mader , B. T. , Flagan , R. C. and Seinfeld , J. H. 2001 . Sampling Atmospheric Carbonaceous Aerosols Using a Particle Trap Impactor/Denuder Sampler . Environ. Sci. Technol. , 35 : 4857 – 4867 .
- Miller , J. C. and Miller , J. N. 1993 . Statistics for Analytical Chemistry , 3rd ed. , pp. 120 – 124 . West Sussex, , UK : Ellis Horwood and Prentice Hall .
- Seinfeld , J. H. and Pandis , S. N. 1998 . Atmospheric Chemistry and Physics , New York : John Wiley & Sons .
- Skoog , D. A. , Holler , F. J. and Nieman , T. A. 1998 . Principles of Instrumental Analysis , pp. 844 – 845 . Chicago : Harcourt Brace & Co. .
- Solorzano , L. and Sharp , J. H. 1980 . Determination of Total Dissolved Nitrogen in Natural Waters . Limnol. Oceanogr. , 25 : 751 – 754 .
- Stolzenburg , M. R. , Dutcher , D. D. , Kirby , B. W. and Hering , S. V. 2003 . Automated Measurement of the Size and Concentration of Airborne Particulate Nitrate . Aerosol Sci. Technol. , 37 : 537 – 546 .
- Stolzenburg , M. R. and Hering , S. V. 2000 . Method for the Automated Measurement of Fine Particle Nitrate in the Atmosphere . Environ. Sci. Technol. , 34 : 907 – 914 .
- Sugimura , Y. and Suzuki , Y. 1988 . A High Temperature Catalytic Oxidation Method on Non-Volatile Dissolved Organic Carbon in Seawater by Direct Injection of Liquid Samples . Mar. Chem. , 24 : 105 – 131 .
- Suzuki , Y. , Suginura , Y. and Itoh , T. 1985 . A Catalytic Oxidation Method for the Determination of Total Nitrogen Dissolved in Seawater . Mar. Chem. , 16 : 83 – 97 .
- Zhang , Q. and Anastasio , C. 2001 . Chemistry of Fog Waters in California's Central Valley—Part 3: Concentrations and Speciation of Organic and Inorganic Nitrogen . Atmos. Environ. , 35 : 5629 – 5643 .
- Zhang , Q. , Anastasio , C. and Jimenez-Cruz , M. 2002 . Water-soluble Organic Nitrogen in Atmospheric Fine Particles (PM2.5) from Northern California . J. Geophy. Res. , 107 : 4112