Atmospheric sampling was conducted at a rural site near Egbert, about 70 km north of Toronto, Ontario, Canada from March 27 to May 8, 2003 to characterize the physical and chemical properties of the ambient aerosol in near real-time. The instrumentation included a tapered element oscillating microbalance (TEOM), an ultrafine condensation particle counter (UCPC), a scanning mobility particle sizer (SMPS), an aerodynamic particle sizer (APS), an aerosol mass spectrometer (AMS), and a particulate nitrate monitor (R&P 8400N) for aerosol measurements. Gas-phase non-methane hydrocarbon compounds (NMHCs) were measured by gas chromatograph-flame ionization detection (GC-FID). Filter samples were also collected for analysis of inorganic ions by ion chromatography (IC). Aerosol properties varied considerably depending upon meteorological conditions and airmass histories. For example, urban and industrial emissions advected from the south strongly influenced the site occasionally, resulting in higher particulate mass with the higher fractions of nitrate and organics. Cleaner northwesterly winds carried aerosols with relatively higher fractions of organics and sulfate. The AMS derived mass size distributions showed that the inorganic species in the particles with vacuum aerodynamic diameters between about 60 nm and 600 nm had mass modal vacuum aerodynamic diameters around 400–500 nm. The particulate organics often exhibited two modes at about 100 nm and 425 nm, more noticeable during fresh pollution events. The small organic mode was well correlated with gas-phase nonmethane hydrocarbons such as ethylbenzene, toluene, and propene, suggesting that the likely sources of small organic particles were combustion related emissions. The particulate nitrate exhibited a diurnal variation with higher concentrations during dark hours and minima in the afternoon. Particulate sulfate and organics showed evidence of photochemical processing with higher levels of sulfate and oxygenated organics in the afternoon. Reasonable agreement among all of the co-located measurements is found, provided the upper size limit of the AMS is considered.
INTRODUCTION
Atmospheric aerosols significantly influence air quality, the earth's radiation budget, atmospheric chemistry, cloud properties, the hydrological cycle and climate change. Ambient fine particles (aerodynamic diameters < 2.5 μ m, PM2.5) are alleged to have association with a variety of adverse health effects (CitationPope et al. 2002). Quantitative measurement of the chemical composition of atmospheric particles as a function of particle size with high temporal resolution is crucial for understanding the sources of particles and their impacts (e.g., CitationIPCC 2001; CitationMcMurry 2000; CitationSeinfeld and Pandis 1998).
Organic material constitutes 10–70% of the total fine aerosol mass in both urban and marine environments (e.g., CitationMiddlebrook et al. 1998; CitationRandles et al. 2004; CitationSaxena et al. 1995). The importance of particulate organic compounds to human health and climate has been recognized in several recent studies. The organic material internally mixed with the inorganics in the particles may either enhance or suppress the aggregate hygroscopic properties of the particles thereby altering the optical properties and their ability to become cloud condensation nuclei (CitationLohmann et al. 2004; CitationRandles et al. 2004; CitationSaxena et al. 1995). Because particulate organics comprise hundreds of different compounds that current measurement techniques are unable to characterize completely, the organic mass concentrations, composition, formation mechanisms, their fate and the potential effects are largely unknown.
During a 43-day period of the spring of 2003 (27 March–8 May), a number of continuous real-time measurements of the ambient aerosol were conducted at a rural site in Egbert, southern Ontario, about 70 km north of the city of Toronto. The sampling site, the Centre for Atmospheric Research Experiments (CARE: 44.23 N, 79.78 W; 251 m asl), is located in an agricultural area. The site experiences a wide range of pollution levels: relatively clean air arrives from northern Canada while more polluted air is advected from the urban and industrial areas of southern Ontario, and the mid-west and northeastern United States (e.g., CitationBrickell et al. 2003). This site, because of the variety of air masses and the agricultural influence, provides the opportunity to investigate the physical and chemical properties of aerosols under various conditions.
PM2.5 mass concentrations at CARE are typically higher during the summer months than in the winter months (CitationVet et al. 2001). The filter-based chemical analysis shows that the carbonaceous species (organic carbons and elemental carbon) and sulfate constitute a large fraction of PM2.5, followed by nitrate and ammonium, and soil dust as a relatively minor constituent (CitationVet et al. 2001). The fraction of nitrate to PM2.5 is higher in the wintertime aerosol and lower in summertime aerosol, whereas sulfate shows the opposite pattern (CitationBrook and Dann 1999; CitationVet et al. 2001). However, the sampling times enable an understanding based on 24-h averaging intervals and are unable to capture diurnal changes and shorter-term variations in particle size and chemistry that may reflect many processes.
These present springtime measurements, which provide more detail of the physical properties and chemical composition of the atmospheric aerosol, are used to investigate the diurnal behavior of aerosol constituents and closely examine short-term episodes. The aim is to increase our knowledge of the processes governing the aerosol mass and chemistry at a rural site in eastern North America.
INSTRUMENTATION AND METHODS
All instruments were operated concurrently in an air-conditioned building on the site. The sampling intakes were approximately 2 m above the roof and about 6 m above the ground. Ambient aerosols were sampled through a 0.8 cm ID copper tube at a flowrate of approximately 20 1 min−1 over a distance of about 7 m to various instruments.
Particle Number Concentrations and Number Size Distributions
The total particle number concentrations for diameters larger than 3 nm were measured with an ultrafine condensation particle counter (UCPC, TSI Model 3025). Particle number size distributions from 20 nm to 420 nm were measured with a scanning mobility particle sizer (SMPS, TSI Model 3070 with UCPC Model 3025). An aerodynamic particle sizer (APS, TSI Model 3321) measured the number size distributions of particles with aerodynamic diameters from 0.5 μ m to 20.5 μ m. The SMPS was operated almost continuously from April 1 to May 7 and the APS was run from April 3 to May 8. The sheath air streams in both the SMPS and APS were dried. Sampling intervals for all measurements are given in .
TABLE 1 Summary of the instrumentation deployed during sampling in Egbert
Particle Mass and Chemical Composition
The PM2.5 mass concentration is continuously monitored at CARE with a tapered element oscillating microbalance (TEOM). The TEOM measures the PM2.5 mass concentration by recording the frequency changes of an oscillating filter due to increases in the mass of the particles deposited on the filter (CitationPatashnick and Rupprecht 1991). During this study, the TEOM PM2.5 mass concentration records were averaged over every 30 min. The 24-h filter samples are collected daily (8:00 A.M. to 8:00 A.M.) and analyzed for the chemical composition of PM2.5. In addition, an Aerodyne aerosol mass spectrometer (AMS) was used to measure 15-min averaged mass size distributions and mass concentrations of non-refractory species (compounds that volatilize below 500°C) in submicron aerosols. A particle nitrate monitor (Rupprecht & Patashnick Model 8400N) was deployed for two weeks (April 17–30) to measure PM2.5 nitrate concentrations with a time resolution of 15 min. It utilizes a humidified impaction process to collect the ambient particles in an integrated collection and vaporization cell. The collected particles are flash vaporized and a chemiluminescence analyzer detects the NOx evolved from the vaporization (CitationStolzenburg and Hering 2000).
Non-Methane Hydrocarbon Compounds and Other Auxiliary Measurements
Gas phase mixing ratios of 37 non-methane hydrocarbon compounds (NMHCs) are routinely measured at CARE using a liquid nitrogen cooled glass bead pre-concentration trap and multi-column capillary gas chromatograph–flame ionization detection (GC-FID) system (CitationBrickell et al. 2003). This fully automated system measures 37 NMHCs (C2–C8) in 20-min samples at a frequency of 10–12 samples per day. The gas phase concentrations of NO, NOy, and O3 are also routinely monitored. Ammonia (NH3) is measured from citric acid impregnated filters sampled for 24 h. Meteorological data (wind direction, wind speed, temperature, relative humidity, pressure, solar irradiance and precipitation) are collected at this site every 10 min.
Aerodyne Aerosol Mass Spectrometer
The AMS measurements are central to this study. The AMS provides near real-time measurements of mass concentrations of non-refractory (NR) species in submicron particles as well as the mass distributions as a function of vacuum aerodynamic diameter (CitationJayne et al. 2000). A comprehensive description of the design of the AMS, its operation, quantification methods and calibration procedure are given elsewhere (CitationAlfarra et al. 2004; CitationAllan et al. 2003a; CitationAllan et al. 2003b; CitationJayne et al. 2000; CitationJimenez et al. 2003a). The AMS consists of three main sections: a particle sampling inlet, an aerodynamic particle sizing chamber and a particle composition analysis section. The particle sampling lens (CitationLiu et al. 1995a; CitationLiu et al. 1995b) brings nearly all submicron particles to the vacuum in a narrow collimated beam. Upon expansion into vacuum, particles acquire size-dependent velocities according to their inertia in the sizing chamber, where the particle-time-of-flight (TOF) is measured and used to determine vacuum aerodynamic diameter (Dva). In the third section, particles hit a vaporizer (∼500°C) where the non-refractory components flash vaporize, the vapor is ionized with electron impact (70 eV), and finally resultant positive ions are analyzed with a quadrupole mass spectrometer (QMA 422, Balzers, Liechtenstein).
The AMS can be operated in two modes: mass spectrum (MS) mode and particle time-of-flight (TOF) mode. The mass spectra (mass-to-charge ratio, m/z 1–300 amu) of the ensemble of particles acquired during the MS mode operation are used to obtain ambient mass concentrations of non-refractory species, without size information. In TOF mode, a set of pre-selected m/z is scanned as a function of particle TOF, from which the ensemble mass size distributions of non-refractory species as a function of vacuum aerodynamic diameter are derived. The vaporizer temperature for this AMS was set at about 500°C, which means that it was unable to detect refractory components such as sea salt, black carbon, and soil dust; however, it should be possible to see some non-refractory components internally mixed with refractory materials (CitationJimenez et al. 2003a).
The AMS particle collection efficiency (CE), i.e., the particle collection at the vaporizer, depends on the particle transmission efficiency of the critical orifice and the aerodynamic lens (EL), broadening of the particle beam as the non-spherical particles are poorly focused (ES) and the degree of “bounce” of the particles off the vaporizer (EB) (CitationJimenez 2004). The inlet and aerodynamic focusing lens has a near 100% transmission efficiency for the particles within the range of 60–600 nm vacuum aerodynamic diameters, and reduced efficiencies for the particles in the range of ∼ 20–60 nm and 600–1500 nm (CitationLiu et al. 1995a; CitationLiu et al. 1995b; CitationJayne et al. 2000; CitationZhang et al. 2002). The AMS measurements are reported as NR-PM1 i.e. the mass of the non-refractory materials in the particles with aerodynamic diameters < 1 μ m). The collection efficiency of the particles by the vaporizer (oven) is now believed to be determined largely by the degree of “bounce” of the particles off the oven (CitationJimenez 2004) related to the morphology of the particle. The non-spherical particles are more likely to bounce off the oven. The particle beam width and shape-related collection efficiency (ES) can be measured in real-time with a particle beam width probe (BWP) (CitationHuffman et al. 2005). However, the BWP was not installed in this AMS during this field campaign. The average ambient relative humidity during this campaign was 71% (Standard deviation = 21%) with a median of 76% and 25th percentile of 54%. In addition, the inorganic species (ammonium, nitrates, sulfates) dominated the particle composition. Thus, most of the particles contained some hygroscopic component, and despite some warming of the aerosol as it entered the building housing the instruments, it is believed that many of the particles were at least slightly wet when sampled. Overall, the collection efficiency is poorly understood. Laboratory aerosols of ammonium nitrate and ammonium sulfate indicate CEs of 1 and 0.2, respectively. However, the complex mixtures of nitrate, sulfate and organics found in the atmosphere do not necessarily mimic pure laboratory particles. Here, a collection efficiency of unity for all species has been used as it is the simple and fundamental assumption and the collection efficiency is not necessarily constant.
In addition to the factors mentioned previously, any change in the ionization efficiency (IE) with time introduces uncertainty in the AMS measurements. Calibrations of the absolute ionization efficiency with nearly monodisperse particles of ammonium nitrate (NH4NO3) were performed 2–3 times each week. The particles were generated by atomizing a solution of NH4NO3, drying the droplets and then size selecting the particles with a TSI Electrostatic classifier. The AMS IE was found to decrease by up to 15% between mass calibrations (2–3 day intervals); although some of this uncertainty could be related to the calibration aerosol. Thus, based on what is known about the collection efficiency, a reasonable estimate for the uncertainty in the AMS measurements if the CE is less than unity is ± 25%. The most recent information on the ionization efficiencies of other species relative to NH4NO3 was used (CitationAlfarra et al. 2004; CitationAllan et al. 2004a).
The AMS detection limits for the particle bound non-refractory species are estimated as three times the standard deviation of the 15 min (in this campaign) signals in the mass spectrum (MS) mode during periods with very low signals in each field campaign (CitationAllan et al. 2003a). The 15 min detection limits for ammonium, sulfate, nitrate and organics for this campaign are estimated to be 0.015 μ g m−3, 0.006 μ g m−3, 0.006 μ g m−3 and 0.015 μ g m−3, respectively.
Mass Estimation from the Number Size Distributions
In an effort to examine closure among some of the measurements, the particle number size distributions from the SMPS and APS were converted to mass concentrations. For this, the mobility equivalent diameter (Dm) was assumed to be equal to the volume equivalent diameter (Dv) or geometric diameter (Dp), and the mass concentration was computed assuming that the particles were spheres (dynamic shape factor, χ = 1) with a particle density (ρp) of 1.5 g cm−3 for the whole campaign. For the APS data, particles were assumed to be spheres with a particle density of 1 g cm−3. The particle densities based on the measurements of ammonium, sulfate, nitrate, and organics mass concentrations using the AMS varied between 1.25 g cm−3 to 1.66 g cm−3. The maximum uncertainty based on the variations in the relative amounts of each species in the particulate matter is estimated at ± 20% of the estimated mass concentrations. The mass concentrations from each instrument were averaged to a common interval of 1 h to perform intercomparisons between mass loadings measured with three different techniques (AMS, SMPS, and APS) that utilize different working principles. Furthermore, the number distributions measured with the SMPS and APS were transferred to the mass size distributions in vacuum aerodynamic diameter (Dva) space and compared with the AMS derived mass size distribution. The conversions were made using the following equations (CitationJimenez et al. 2003b):
OBSERVATIONS AND DISCUSSION
Local Meteorological Conditions
The average ambient temperature during the study was 5.3°C with a minimum of −11.8°C and a maximum of 28.3°C. The relative humidity varied between 19% and 100% (average = 71%). Wind speeds ranged from less than 1 m s−1 to as high as 14 m s−1 (average = 3.6 m s−1). The frequency of local wind direction was highest for winds from the northwest (270–360°), followed by the southeast (90–180°), the southwest (180°–270°) and then the northeast (0–90°). Nineteen days out of 43 days experienced either trace or moderate precipitation (9 days of rainfall, 3 days of snowfall and 7 days with a mix of both). There were trace amounts of snow on the ground until April 10. One morning (May 6) was foggy. Precipitation was observed between 8:00 A.M. and 6:00 P.M. on only 5 days. The remaining 24 days were mostly sunny, and the solar irradiance reached as high as 970 W m−2.
The 15 min averages of the total concentrations of particles measured with the UCPC varied between 300 cm−3 and 25,000 cm−3 (average = 6,500 cm−3). Variations of aerosol mass and composition are discussed in the following sections.
Mass Concentrations of the Aerosol Components
illustrates time series of AMS derived ambient mass concentrations of ammonium, nitrate, sulfate and total organics in the submicron aerosols sampled at CARE from March 27 to May 8, 2003. For the entire sampling period, the average mass concentrations of ammonium, nitrate, sulfate and the organics are 0.69 μ g m−3, 1.18 μ g m−3, 1.19 μ g m−3, and 1.73 μ g m−3, respectively. includes the summary statistics of various components during the measurement period. The minimum-maximum ranges of the percentage contribution of ammonium, nitrate, sulfate, and organics to the total mass (NR-PM1), where the total is the sum of only these four components, are 0–30%, 0–60%, 3–82%, and 14–87%, respectively, indicating that organic material constitutes a large fraction of the total fine aerosol mass.
FIG. 1 Time series of ammonium, nitrate, sulfate and organics mass concentrations of ambient aerosols measured with the AMS, together with ambient temperature (T), relative humidity (RH), wind speed (ws) and wind directions (wd) at CARE from March 27 to May 8, 2003. The chemical components are 15 min averages and the meteorological parameters are 10 min averages. The gas phase concentrations of ammonia from filter pack analysis (24 h averages) are also shown.
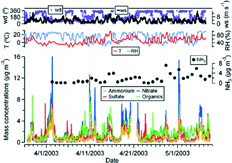
TABLE 2 Summary statistics of ambient mass concentrations (μg m−3) of chemical constituents in submicron aerosols, and aerosol acidity. The total mass is the sum of only ammonium, nitrate, sulfate and organics. Aerosol acidity is the molar ratio of ammonium to sum of two times sulfate and nitrate [NH4/(2SO4 + NO3)]. SD indicates the standard deviation
Typically, the particle composition varied considerably over a few hours. Included in are 10 min average local wind directions, wind speeds, temperatures, relative humidities and 24 h averaged gas phase ammonia (NH3) concentrations. Higher fine particle mass concentrations were observed when the winds were southerly and the wind speeds were lower. The episodes of the highest particle mass concentrations were dominated by nitrate. Higher nitrate mass concentrations occurred during dark hours, in particular early mornings with lower temperatures and higher relative humidities. Aerosol particles from the northwest were composed of mainly organics. On a few occasions (e.g., March 31, April 5, 6, and 23), sulfate dominated the northerly aerosol, but the events were short-lived. Those events correspond to the air masses that had passed over the Sudbury region (industrial area with nickel smelters noted for higher SO2 emission) to the northwest.
There is a tendency for ammonia to increase with time through the study period in correspondence with generally increasing temperatures and decreasing snow cover. Ammonia mass concentrations remained less than 1 μ g m−3 at temperatures less than 5°C. The detection limit for NH3 was 0.15 μ g m−3 (95% confidence limit). At temperatures below approximately 12°C, the gas-phase ammonia correlated positively with the particulate ammonium and nitrate mass concentrations, then the correlations became negative with the ammonia tending to be higher at higher temperatures. This may reflect a difference in the partitioning between the gas-phase ammonia and the particle-phase ammonium with more ammonia in the gas phase at higher temperatures. Ammonium correlated negatively with the sulfate for temperatures < 5°C coincident with the sulfate dominated airmasses from the northwest. The variance in NH3 may also be a result of changes in the local source strength of NH3 during the period; the ground was warming during the study period and the fields were starting to be prepared for planting. Ammonium nitrate is a reaction product of nitric acid with NH3, which is temperature dependent and influenced by relative humidity (CitationSeinfeld and Pandis 1998). Higher levels of ammonia in Egbert from agricultural activities and the cooler temperatures may favor its formation. The fog and haze droplets also appear to have influenced ammonium nitrate formation (e.g., May 6).
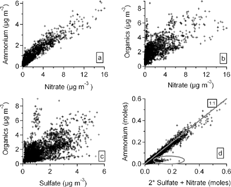
Among the mass concentrations of ammonium, nitrate, sulfate and organics, the strongest correlation is between ammonium and nitrate (correlation coefficient, r = 0.94; ). Ammonium is moderately correlated with organics (r = 0.78) and sulfate (r = 0.76). The correlation between organics and nitrate (r = 0.73) is slightly higher than that between organics and sulfate (r = 0.60). The correlation coefficient between nitrate and sulfate is 0.57. The average molar ratio of ammonium to nitrate plus sulfate (i.e., NH4 +/[2 SO4 = +NO3 −]) is 0.93 (SD = 0.28) and the median is 0.88 for the entire campaign. The smaller values of the ratio, some of them are noticeable and encircled at the bottom-left of the molar ratio plot in , were observed mostly on the days with northwesterly winds when sulfate dominated the particles. This is consistent with reduced ammonia emissions in the colder north regions during that time. Although it may be implied from this that the aerosol was more acidic when the winds were from the north, the presence of refractory ions, undetected with the AMS, and organic acids in the particles leaves some uncertainty in trying to estimate aerosol acidity from these data.
FIG. 2 Correlations among aerosol components measured with the Aerosol Mass Spectrometer (AMS). The acidity of the particles—the molar ratio of positive ions to negative ions (NH4 +/2SO4 = + NO3 −)—is also displayed (d).
Mass Distributions of Aerosol Components
illustrates the mass size distributions of ammonium, nitrate, sulfate and organic fractions of aerosol particles determined from the AMS measurements averaged over the entire sampling period. Particulate inorganic species have similar unimodal mass distributions with modal vacuum aerodynamic diameters (Dva) between 475 nm and 500 nm. Particulate organic material has two modes, a larger mode with a modal Dva close to that of the inorganics and a smaller mode peaked at about 150 nm. Most of the particle mass of the particles smaller than about 80 nm is organic. Thus, the average distribution is represented by smaller particles of mostly organics and larger particles more likely internal mixtures of organics, ammonium, sulfate and nitrate. The average distribution of species in this campaign is similar to that reported for the lower Fraser Valley, British Columbia, Canada (CitationAlfarra et al. 2004), indicative of an urban air mass that had undergone several hours of atmospheric processing. It also bears similarity to that measured at Trinidad Head, California when marine air mass influenced by continental emission was sampled (CitationAllan et al. 2004b).
FIG. 3 The average mass size distributions of ammonium, nitrate, sulfate and organics in ambient aerosols acquired by the aerosol mass spectrometer for the entire sampling period.
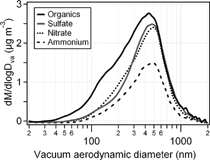
In previous urban studies, the organic mass distribution was found bimodal with a distinct smaller mode (modal Dva < 200 nm), attributed to local traffic emissions of hydrocarbon particles. The larger mode contains some oxygenated organics (CitationAlfarra et al. 2004; CitationAllan et al. 2003a; CitationDrewnick et al. 2004). As shown later, the mass loading of small mode organic particles (Dva < 200 nm) in this study has positive correlations with several gas phase NMHCs, including propene, ethylbenzene, toluene, benzene. The implication is that the small organic particles are most likely the result of primary emissions of organics from fuel combustion sources.
Although the mass distributions in provide an average picture for this springtime aerosol at CARE, they do not demonstrate the temporal variability of the aerosol mass size distributions. The mass distributions of organics and sulfate over shorter time intervals often showed significant mass below 100 nm. During the periods with higher particulate matter, often associated with southerly winds, organics showed distinct bimodal mass distribution and nitrate also tended to exhibit bimodal mass distribution. Sulfate occasionally displayed bimodal mass distribution during suspected particle nucleation events.
Intercomparisons of Measurements with Co-Located Instruments
Mass Size Distribution: AMS and SMPS + APS
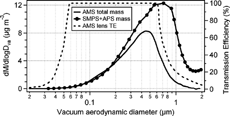
The number size distributions measured with the SMPS and APS were converted to mass size distributions as a function of vacuum aerodynamic diameter as described earlier. shows the average mass distributions of the AMS total mass concentrations and the combined estimated masses from the SMPS and APS number distribution measurements. Only those periods when all three instruments were sampling are used to produce ; hence there are slight differences between the AMS total mass distribution of and the sum of the masses in . The SMPS + APS mass distribution indicates substantially more mass above 500 nm vacuum aerodynamic diameters than the AMS (mass modal Dva of about 725 nm and 450 nm respectively). A major reason for the absence of mass in the AMS measurements at the larger particle sizes is the reduced transmission efficiency of the AMS inlet system above 600 nm (CitationAllan et al. 2003b; CitationJayne et al. 2000). The estimated curve of transmission efficiency of the inlet lens (CitationAllan et al. 2003b; CitationJayne et al. 2000) is also shown in . The measured AMS mass concentration drop-off at the larger sizes follows closely the computed AMS lens transmission efficiency. The possible presence of refractory components, such as soil dust and black carbon, is another factor that will contribute to differences between the two measurements, since this AMS does not detect those components of the particulate matter. A third factor is the morphology of the particles. Non-spherical particles or those with in an aggregate form may have a lower collection efficiency in the AMS. Differences in the particle density from the assumed 1.5 g cm−3 will introduce some uncertainty to this comparison: the maximum uncertainty based on the variations in the relative amounts of each species in the particulate matter is estimated at ± 20% of the estimated mass concentrations. Note that the AMS collection efficiency can be lower than unity for some particles.
FIG. 4 The average mass size distribution of AMS total mass (sum of ammonium, nitrate, sulfate and organics) and mass size distribution estimated from number size distributions from SMPS and APS, assuming that particles are spherical with a particle density of 1.5 g cm−3. Estimated transmission efficiencies (TE) of the AMS inlet lens as a function of particle vacuum aerodynamic diameters are also shown.
Mass Concentrations: AMS and SMPS + APS
shows the temporal variations of the hourly averaged AMS total mass concentrations (NR-PM1), the PM2.5 mass concentrations estimated from the number size distributions (SMPS + APS PM2.5), and the PM2.5 measured with the TEOM (TEOM PM2.5). The variations in the SMPS + APS PM2.5 and the AMS NR-PM1 mass concentrations are similar, but the SMPS + APS PM2.5 is higher than the AMS NR-PM1, consistent with the comparison of the size distributions in . The AMS NR-PM1 is, on average, about 61% of the SMPS + APS PM2.5 and 70% of the SMPS + APS PM1 (time series not shown). To demonstrate the role of the larger particles in the time series comparisons in , the estimated mass concentrations of particles with aerodynamic diameters larger than 0.5–2.5 μ m from the APS number concentrations are included separately. Increase in the APS mass concentrations correspond well with increases in the difference between the SMPS + APS PM2.5 and the AMS NR-PM1 mass concentrations.
FIG. 5 Intercomparison among hourly AMS NR-PM1 (AMS total mass concentrations—the sum of only ammonium, nitrate, sulfate, and organics mass concentrations), SMPS + APS PM2.5 (estimated PM2.5 mass concentrations from number distributions measured with the SMPS and APS) and TEOM PM2.5 mass concentrations. The APS mass concentrations of particles with aerodynamic diameters 0.5–2.5 μm (APS < 2.5 μm) and the AMS derived nitrate mass concentrations are also shown.
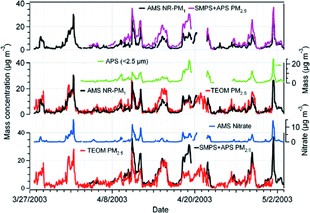
The total of the potential refractory material (calcium, magnesium, potassium, and sodium) from the ion analysis of the filter PM2.5 ranges from 0.13–4.3 μ g m−3. The total refractory material in PM2.5 was high during April 9–11 (∼ 3.75 μ g m−3), 14–15 (∼ 4.07 μ g m−3), 25–26 (3.32 μ g m−3), 27–28 (4.3 μ g m−3), and April 30–May 1 (3.42 μ g m−3), and low during April 18–20 (< 2.15 μ g m−3). Contrasted with the differences between the AMS NR-PM1 and SMPS + APS PM2.5, these levels of refractory materials, without the corresponding anion mass, may contribute to some of the deviations. The difference between AMS NR-PM1 and SMPS + APS PM2.5 is pronounced during April 18–20. In this case, the mass of the larger particles (Da = 0.5 − 2.5 μ m) reaches 24 μ g m−3, the total refractory material is less than 2.15 μ g m−3, and the AMS ammonium, nitrate and sulfate mass concentrations (24 h averages) are 35–55% lower than the filter ammonium, nitrate and sulfate. Thus, during this period, the major source of disagreement appears to be the inability of the AMS to sample the larger non-refractory particles, and a small quantity of refractory material as a minor contributor.
The difference between the SMPS + APS PM2.5 and the AMS total mass increases slightly during the afternoons when the relative humidity drops compared to the nights and early mornings. Relatively large changes in the relative humidity may influence the morphological structure of the particles. Lower relative humidity may lead to non-spherical particles that are more susceptible to bounce off the vaporizer of the AMS.
Mass Concentrations: AMS and TEOM
The AMS total mass concentrations and the TEOM PM2.5 agree reasonably well. The AMS NR-PM1 is slightly lower than the TEOM PM2.5 (on average, the AMS total mass is about 81% of TEOM PM2.5). The TEOM PM2.5 is notably higher than AMS NR-PM1 on April 14–15. Some of the difference in the averages of the TEOM and the AMS stems from increase in particles larger than 0.6 μ m, as indicated by the APS, and some is due to the presence of refractory materials. During April 14–15, the SMPS + APS PM2.5 and TEOM PM2.5 agree well (the bottom plot). In this particular case, the mass concentrations of nitrate and the APS mass (i.e., < 2.5 μ m) are relatively low. Refractory materials are relatively high, implying that a significant source of disagreement is due to materials that the AMS was unable to measure. This period coincides with lowest relative humidities (< 50%) and highest temperatures (20–28°C) that persisted over nearly 2 days. Consequently, some of the particles may have been crystalline and subject to a lower collection efficiency in the AMS. This could account for some of the difference with the TEOM measurements.
The inability of the TEOM to report PM2.5 correctly during nitrate events is noticeable. For example, during April 18–19, the TEOM PM2.5 compares well with the AMS total mass, whereas there is a significant discrepancy of the SMPS + APS PM2.5 with both the TEOM and AMS. The AMS indicates that there was a nitrate episode during April 18–19, the APS shows significant mass in the 0.5–2.5 μ m aerodynamic diameter range and the filters indicate twice the nitrate of the AMS. Thus, it appears that, in this case, the filters and the APS measured the larger nitrate-dominated particles, whereas the AMS was unable to see entire nitrate due to drop off in transmission efficiency of the larger particles. Volatilization of semivolatile species, such as ammonium nitrate, in the TEOM inlet, maintained at 30°C, may explain the reduced mass indicated by the TEOM (CitationAllen et al. 1997). Such evaporative loss can be a concern when using the TEOM to monitor PM2.5, in particular, in areas where other semivolatile species contribute significantly to the particulate matter.
Mass Concentrations: AMS and Filter Measurements
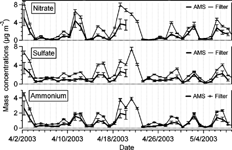
The PM2.5 bulk chemical composition (nitrate, sulfate, ammonium, chloride, sodium, calcium, magnesium and potassium) was determined by analyzing daily filter samples collected for 24 h (8:00 A.M. to 8:00 A.M.). In order to perform statistical intercomparisons with the filter data, the AMS data were averaged to a common averaging interval of 24 h (8:00 A.M. to 8:00 A.M.). The AMS did not have complete 24 h coverage on all days due to calibrations and maintenance, so only days with AMS sampling of more than 16 h are used. displays the time series of ammonium, nitrate and sulfate acquired by the two measurements. Although data from two measurements co-vary, the AMS measurement is frequently lower than filter measurements.
FIG. 6 Time series of particulate ammonium, nitrate and sulfate mass concentrations at Egbert measured with the AMS and those species from the filter pack analysis of PM2.5 (both data 24 h averages).
shows scatter plots of AMS derived ammonium, sulfate and nitrate mass concentrations with those determined from the filter analyses. The AMS measurements account for about 57% of the filter ammonium (slope = 0.57, intercept = 0.04 μ g m−3 and r2 = 0.92), 56% of the filter nitrate (slope = 0.56, intercept = −0.05 μ g m−3 and r2 = 0.92) and 50% of the filter sulfate (slope = 0.50, intercept = 0.07 μ g m−3 and r2 = 0.89). The nitrate measured by the AMS may include contributions from ammonium nitrate, sodium nitrate in road salt particles and nitrogen containing organic compounds that may not be measured in the filter analyses. Despite the lower slopes, all three comparisons correlate well. The most likely factor that contributes to the discrepancies between the measurements is the presence of larger particles that AMS samples with reduced transmission, collection and volatilization efficiencies as discussed earlier. There is also some chance of collecting particles larger than 2.5 μ m Da by the filter system due to uncertainty in size the cut-off of inlet as well as oxidation and condensation processes that may occur on the filters (CitationDrewnick et al. 2004).
FIG. 7 Intercomparison of 24 h averaged particulate ammonium, nitrate and sulfate mass concentrations acquired by the AMS and filter measurements of PM2.5.
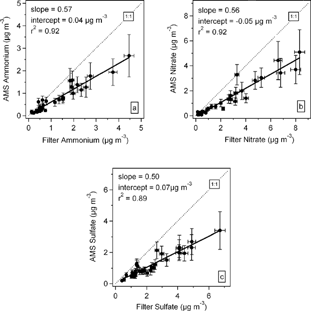
Several factors lead to uncertainties in AMS measurements and filter measurements. Those factors for the AMS measurements include the transmission efficiency of the lens within and beyond 60 nm to 600 nm size range, beam broadening, particle bounce and change in the IE with time. Therefore, based on what is known about the collection efficiency, a reasonable estimate for the uncertainty in the AMS measurements if the CE is less than unity is ± 25%. The error bars in the AMS measurements in and include these factors as well as a small contribution due to the detection limits (very small values for 24 h). The uncertainties in the filter measurements are due to flow rate change, analytical uncertainty, blank levels, volatilization, reaction and condensation on filters, and sampling of some particles larger than 2.5 μ m. A value of 5% is used to represent the uncertainty in the filter concentrations in and .
Nitrate Mass Concentrations: AMS and R&P 8400N
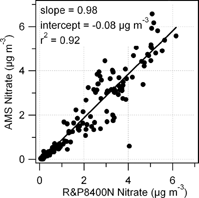
Automated semicontinuous measurements of the PM2.5 nitrate concentrations were made using a R&P nitrate monitor (R&P8400N) from 17 April to 30 April. shows the correlation between the hourly averaged AMS and R&P8400N nitrate mass concentrations. Two measurements agree well; the slope and intercept of this correlation plot are 0.98 and −0.08 μ g m−3, respectively, and r2 is 0.92.
FIG. 8 Intercomparison between particulate nitrate measured by the AMS and the R&P8400N particulate nitrate monitor (both 1 h averages).
During the two-week period the R&P8400N was used, the correlations among the AMS nitrate, R&P8400N nitrate and filter nitrate (all 24 h averages) indicate that the R&P8400N and the AMS recovered 49% and 46% of the filter nitrate, with r2 values of 99% and 98%, respectively. The collection efficiency of the impactor in the R&P8400N is > 95% for particles with aerodynamic diameters from 0.1 μ m to 0.8 μ m, the uncertainty is greater for particles > 0.8 μ m (CitationStolzenburg and Hering 2000). The evaporative loss of the species such as ammonium nitrate has been estimated to be less than 6% (CitationStolzenburg and Hering 2000). Thus, the strong agreement between the AMS and R&P8400N may be coincidental and due to similarly poor collection efficiencies of both instruments for the larger particles.
Diurnal Patterns of the Aerosol Components
displays box plots of the diurnal variations of the particulate (a) nitrate, (b) sulfate, (c) ammonium, and (d) organic mass concentrations from the AMS for the entire period. There is a diurnal pattern in the nitrate concentrations in the means, medians, upper quartiles, and 95th percentiles with higher nitrate concentrations evident during early morning hours and minima in the afternoons. Ammonium nitrate formation is favored at lower temperatures (e.g., CitationBrook and Dann 1999; CitationDrewnick et al. 2004; CitationSeinfield and Pandis 1998), which more often occurs during the nighttime. At night, the most significant mechanism that controls NH4NO3 formation is the conversion of NO3 (NO2 + O3 → NO3 + O2) to N2O5, followed by homogeneous and heterogeneous hydrolysis of N2O5 to give gaseous HNO3 and particulate phase HNO3, respectively. HNO3 then reacts with NH3 to produce NH4NO3 (CitationMcLaren et al. 2004; CitationSeinfield and Pandis 1998). Higher relative humidity will favor the hydrolysis of N2O5 on wet aerosol surfaces. The nitrate diurnal pattern is qualitatively consistent with the diurnal variations of temperature and relative humidity. Because of its relatively high vapor pressure, ammonium nitrate shows a stronger temperature dependence unlike most of other constituents, such as ammonium sulfate and most of the oxygenated organics.
FIG. 9 Diurnal patterns of (a) nitrate, (b) sulfate, (c) ammonium, and (d) organics mass concentrations along with diurnal variations of ambient temperature (T), relative humidity (RH) and solar radiation (SR). In each box, the mid-line (−) represents the median value and a marker • represents the average for each hour. The top and bottom of the box represent the upper and lower quartiles respectively. The top and bottom of the whisker represent the 95th percentile and 5th percentile respectively.
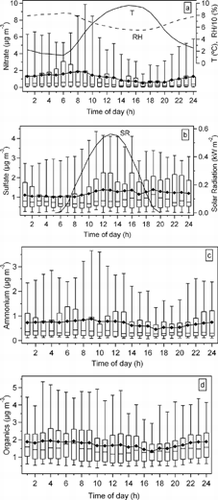
The sulfate means and upper quartiles show increased sulfate concentrations from just before noon until the late evening with minima in the morning hours (the median values peak in the late afternoon to early evening). The sulfate concentrations slightly increase with the daily increase in solar irradiance. This reflects increased photochemical production of sulfate during warm sunny afternoons (e.g., CitationDrewnick et al. 2004; CitationSeinfield and Pandis 1998). Lack of clear diurnal pattern in sulfate mass concentrations is suggestive of the regional scale production and transport of sulfates. The lack of diurnal cycle in sulfate mass concentrations also reflects the fact that sulfate, unlike nitrate, does not partition into gas-phase (CitationJimenez et al. 2003a). The different diurnal patterns of nitrate and sulfate not only indicate different formation mechanisms for the periods of higher concentrations of each, but they may also reflect a preference for the formation of NH4NO3 at lower sulfate concentrations (CitationDrewnick et al. 2004; CitationSeinfield and Pandis 1998). The diurnal pattern of ammonium tends to follow more closely that of nitrate, perhaps driven by the higher nitrate events. However, it is also influenced by sulfate around noon. The total organic concentrations are at minimum about the time that the sulfate is maximized. The organics are considered in more detail next.
Particulate Organics and Gas Phase Non-Methane Hydrocarbons
The organic material contributes, on average, the highest fraction of the submicron particle mass (). The AMS measures the mass of all particulate organics, both water-soluble and water-insoluble, that volatilize at 500–600°C. Previous aerosol studies at Egbert have referred to the “unidentified or residual mass” in the filter analysis as carbonaceous materials (CitationVet et al. 2001), and therefore the nature of particulate organics is still unknown. Organic speciation using the AMS is not possible, except identification of some functional groups. However, information about the general nature of particulate organics can be extracted by looking at the contributions of particular mass-to-charge ratios (m/z) to total organics. For example, the oxygenated organics (due to photochemistry or oxidation reactions), primarily the carboxylic acids, produce a major signal at m/z 44 (i.e., CO2 +) (CitationAlfarra et al. 2004). The m/z 43 contains contributions from both oxygenated organics, such as ketones (CH3CO+) and aldehydes (CH2CHO+), and saturated hydrocarbon compounds (C3H7 +). The m/z 57 is a fragment (C4H9 +) typical of saturated hydrocarbon compounds chiefly from combustion sources (CitationAlfarra et al. 2004; CitationAllan et al. 2003a; Citation2003b; CitationDrewnick et al. 2004). A few statistics of the fraction of particulate organics at m/z 43 (i.e., m/z 43/total organics), m/z 44 (i.e., m/z 44/total organics) and m/z 57 (i.e., m/z 57/total organics) during the entire sampling period are given in . The mass concentrations measured at m/z 44 and at m/z 57 are used here as indicators for the oxygenated particulate organics and for particulate hydrocarbons, respectively.
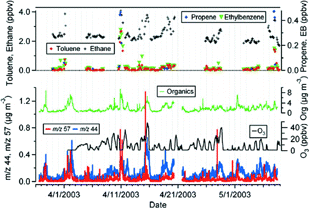
shows the time series of the mass concentrations of total particulate organics, m/z 44 and m/z 57 as well as gas phase mixing ratios of some non-methane hydrocarbon compounds (NMHCs) and O3 for the entire sampling period. Increases in total organic particulate matter coincide with increases in the various gas-phase NMHCs. Mobile emissions are the major source of atmospheric NMHCs, such as ethane, benzene, toluene, and ethylbenzene (CitationSeinfeld and Pandis 1998). The atmospheric lifetime of ethylbenzene, propene, m-xylene, toluene and ethane due to reaction with OH is approximately 2.6 h, 6 h, 7 h, 2.4 days, and 30 days, respectively, for an ambient OH concentration of 1.5 × 106 molecules cm−3 (CitationSeinfeld and Pandis 1998). There are several particulate hydrocarbon events (i.e., m/z 57) that tend to be shorter in duration than the periods with higher oxygenated organics (m/z 44) that tend to persist for several hours, sometimes 1–2 days. There is a hydrocarbon event over the night of April 10–11 with relatively high gas phase mixing ratios of several hydrocarbon compounds, including ethylbenzene, propene and toluene and very low O3. This event and others with sharp increases in ethylbenzene, propene and toluene coincide with higher m/z 57 relative to m/z 44 in the AMS time series. The April 10–11 increase of the m/z 57 mass concentration with higher NMHCs and lower O3 (most likely due to titration by NO) suggests common emission sources, such as traffic, with relatively little chemical processing. Periods with higher m/z 44 (e.g., April 8–9, 15, 28) show no peaks in short-lived NMHCs (e.g., ethylbenzene, toluene) but peaks in ethane. The April 15 event is notable because the values of m/z 44, ethane and O3 are highest during the sampling, as is the ratio of the m/z 44 mass concentrations to the total organic mass concentrations. Thus, the aerosol during this period is perhaps the most chemically processed of any during this study.
Time series of total organics, organics contributing to m/z 57 (an indicator for hydrocarbon-like organic aerosol, HOA) and m/z 44 (an indicator for oxygenated organic aerosol, OOA) reported by the AMS, and gas phase mixing ratios of some selected non-methane hydrocarbons and ozone. EB indicates ethylbenzene and Org. indicates total organics.
The organic mass loading in the small mode of the organics (< 200 nm) was occasionally significant at Egbert when urban air masses affected the site. shows positive correlations between the mass concentrations of organics in small mode (< 200 nm) and several gas phase NMHCs, pointing to fossil fuel combustion-related emission as the source of the smaller organic particles (CitationAlfarra et al. 2004; CitationAllan et al. 2003a; CitationDrewnick et al. 2004). also includes correlations between the NMHCs and m/z 57, as well as m/z 44. The strong positive correlations of the various NMHCs with m/z 57/organics suggest common sources, whereas the anti-correlations with m/z 44/organics indicate of further oxidation of hydrocarbons as the aerosol is processed.
FIG. 11 Correlations between the mass concentrations of organics in small particles (< 200 nm) and mixing ratios of the gas phase non-methane hydrocarbon compounds. The correlations between those NMHCs and particulate m/z 57/organics and m/z 44/organics are also included.

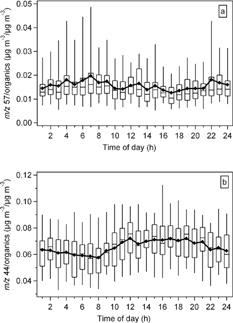
The diurnal patterns of the ratio of m/z 57/organics to total organics and the ratio of m/z 44/organics to total organics are plotted in and , respectively. The particulate hydrocarbons as represented by m/z 57/total organics exhibit a relatively weak diurnal pattern with slightly higher values during the nighttime and lower values in the afternoons. The oxygenated fraction in the organics (m/z 44/total organics) has a more pronounced and different diurnal pattern. The oxygenated fraction gradually increases from 8:00 A.M., maximizes about noon and remains elevated during the afternoon hours. Its pattern follows closely that of the solar irradiance, suggesting photochemical production of the oxygenated organics. The increase in m/z 44/total organics is also similar to that of sulfate, except that the rise in m/z 44/total organics appears to begin earlier than that of sulfate. Although the assessment of an earlier rise in m/z 44 must be considered cautiously, such a difference may indicate a more important role of the development of the boundary layer in the sulfate variation.
FIG. 12 Diurnal pattern of the (a) m/z 57/total organics—a fraction of the total particulate organics that represents hydrocarbon-like organics in the particles and (b) m/z 44/total organics—a fraction that represents oxygenated organics for the entire sampling period. The lengths of the box and whiskers are defined as in .
Case Studies
Two periods are selected for more detailed examination:
| • | Event 1 (April 10, 7:45 P.M.–April 11, 11:00 A.M.) with higher nitrate and organics, and lower sulfate. | ||||
| • | Event 2 (April 15, 10:00 A.M.–9:00 P.M.) with higher organics and sulfate, and lower nitrate. | ||||
Events 1 and 2 also mark the higher levels of particulate hydrocarbons and highest oxygenated organics, respectively.
Event 1—April 10, 7:45 P.M.–April 11, 11:00 A.M.
The average ambient temperature and relative humidity during this event were 3.5°C (min = −1.3°C, max. = 13°C) and 60% (min = 40%, max = 78%), respectively. The total particle number concentrations (> 3 nm) were in the range of 6,000–10,800 cm3. The mixing ratios of several NHMCs increased. Particulate nitrate makes the largest contribution to the fine particle mass fraction followed by organics, ammonium and very low sulfate (, ). During this and other nitrate events, the O3 concentrations drop near zero. It is likely that O3 was titrated by NO near the source and advected to the site as such. Being overnight, there was no significant production source to replenish the O3. Lower temperatures, relatively higher levels of NH3 and remarkably low concentrations of sulfate perhaps created environment favorable for NH4NO3 formation. According to , the nitrate was more coincident with the smaller organic particles, suggesting that NH4NO3 may have condensed onto pre-existing organic particles.
FIG. 13 Mass size distributions of particulate ammonium, nitrate, sulfate and organics during the two different events on (a) April 10–11 and (b) April 15 as described in the text. Mass distributions of selected key organic mass fragments are also included. Typical air mass back trajectories (red: 250 m and blue: 1000 m, and two marks along trajectory represent 24 h interval) for these events are also displayed.
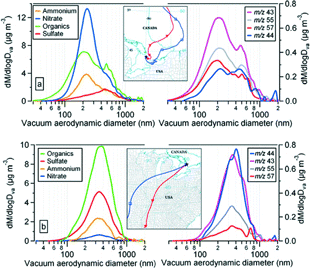
illustrates that particulate nitrate and ammonium both have dominant mass distributions at a modal vacuum aerodynamic diameter of about 225 nm. The sulfate is also unimodal peaked about 500 nm. Organics have a distinct mode at about 200 nm Dva and a second mode coincident with the sulfate peak and a secondary peak in the nitrate mass. Assuming that the available particulate ammonium neutralizes all sulfates before neutralizing nitrate it is found that there is excess nitrate in the size range between 170 nm to 350 nm, coinciding with the organic mass distribution peak. There are at least two possible explanations for this: (1) the unneutralized nitrate is due to the absorption of HNO3 or N2O5 by the aqueous particles, or (2) the excess nitrate is from organic nitrates.
The particles smaller than 100 nm appear to be mainly externally mixed organics. The presence of the high small particle mass during this event and the elevated levels of short-lived NMHCs (), indicate that the sampling site was indeed impacted by relatively recent anthropogenic emissions. also includes the back trajectories of the airmass that arrived at the site at 4:00 A.M. on April 11 at two heights (250 m and 1000 m). It manifests the anthropogenically influenced aerosol, as the airmass appears to have come from northern Canada to near Toronto and then looped to the north arriving at the Egbert site after spending significant time over urban areas. The back trajectory was computed from the HYSPLIT4 (Hybrid Single Particle Lagrangian Integrated Trajectories) model (CitationDraxler and Rolph 2000) with EDAS (Eta Data Assimilation System) data as meteorological inputs (www.arl.noaa.gov/hysplit.html).
also includes the mass distributions of some key organic fragments, m/z 44 (oxygenated organics), m/z 57, and m/z 55 (typically hydrocarbons), and m/z 43 (both). The fragment m/z 44 is bimodally distributed with a relatively higher concentration in the larger mode whereas the other three fragments are preferentially higher in the small mode. This implies that the aerosol is more oxidized in the larger mode compared with the smaller mode.
The trajectory analysis and the chemical size distribution data suggest the following explanation for the presence of the two major modes in the aerosol. There is no major local anthropogenic source, other than agricultural activities. Ammonia emission is also low in northern regions due to cold temperature. Most of the sulfate and organics in the larger mode at about 500 nm were present earlier in the history of this air parcel before it reached southern Ontario. Therefore, anthropogenic sources in the vicinity of Toronto were responsible for the addition of smaller primary organic particles and NOX that eventually, in the presence of local NH3 as well as increased relative humidity, led to relatively local formation of the nitrates in both the smaller and larger modes. Since the smaller organic aerosols were present in the urban outflow, the nitrate condensed on those particles because those particles provided a large surface area.
Event 2—April 15, 10:00 A.M.–9:00 P.M.
The ambient air was relatively warmer and drier during this event than in event 1 with an average temperature of 25.6°C (min = 18.3°C, max = 28.3°C) and an average relative humidity of 38% (min = 32%, max = 58%). The total particle number concentration varied between 7,600 and 20,000 cm−3 and the O3 concentration remained higher throughout the event. Only NMHCs with longer atmospheric lifetime (e.g., ethane, propane) were measured. The organics make the largest contribution to the submicron mass followed by sulfate and ammonium (). Nitrate is relatively low. The warmer temperatures and lower relative humidities appear to be unfavorable condition for particulate nitrate formation; the warmer temperatures on April 14 and 15 preferred the gas-phase particle-phase partitioning towards ammonia. The back trajectories show that the air mass passed over urban and industrial regions to the southwest before arriving at the site.
The particulate organics, sulfate, ammonium and nitrate were most likely internally mixed as these four constituents exhibit very similar unimodal mass distributions with modal diameters around 350 nm (). In both an absolute and relative sense, the fraction of oxygenated organics in the particles (m/z 44) is much higher than during event 1, indicating that the aerosol arriving at Egbert was much more chemically processed during this event than the previous one.
SUMMARY AND CONCLUSIONS
During the spring of 2003, secondary aerosol components (ammonium, nitrate, sulfate, and some organics) made up a large fraction of the ambient aerosol mass at a rural site about 70 km north of Toronto, Ontario. On average, the total organic material constituted the highest fraction of submicron aerosol mass, whereas nitrate fluctuated over a wider range during the sampling period. The relatively high time resolution of the instrumentation enabled characterization of the aerosol size and chemistry in detail, including examination of the diurnal patterns of the particle components and analysis of short-term events. The maximum concentration of nitrate was found in the early morning and the minimum concentration was found in the afternoon. The presence of higher concentrations of nitrate in the particles was favored by conditions of lower light, lower temperatures, higher relative humidities and air trajectories from urban regions. In contrast, sulfate and the oxygenated fraction of the organics displayed higher concentrations in the afternoon, evidence of their formation connected with the photochemical oxidation.
The mass modal vacuum aerodynamic diameter (Dva) estimated from the particle number distribution measurements made with a Scanning Mobility Particle Sizer (SMPS) and an Aerodynamic Particle Sizer (APS) was about 725 nm averaged over entire sampling period. Hence, significant concentrations of larger particles (> 0.6 μm) were present at the site. The Aerosol Mass Spectrometer (AMS) measurements showed that most of the paniculate ammonium, nitrate, sulfate and organic mass, on average, had a modal diameter of 400–500 nm Dva. Occasionally, the organic mass distribution also exhibited a second and smaller mode near 100 nm. Periods with particulate hydrocarbon mass in small particles (< 200 nm) coincided with a substantial increase in several gas-phase non-methane hydrocarbons (NMHCs), indicating a more recent impact of anthropogenic emissions. The smaller organic particles contained a higher fraction of hydrocarbons, sometimes externally mixed, while the larger organic particles were more oxygenated than the smaller ones and internally mixed with the inorganics. On at least one occasion, relatively large concentrations of nitrate were associated with the smaller organic mode.
Mass closure among the various semicontinuous measurements (AMS, TEOM PM2.5, SMPS + APS, R&P nitrate monitor) as well as time-integrated filter measurements is explained on the basis of the relative size of particles sampled by various techniques. In order to get further insights into the particulate composition at this site, black carbon measurement and analysis of the filter samples for the organics should be incorporated in the future campaigns.
Acknowledgments
This work would not have been possible without the assistance of Mr. F. Froude (manager of CARE) and the staff at CARE. We are also grateful to the Canadian Air and Precipitation Monitoring Network (CAPMoN) for providing important filter and trace gas data. Maheswar Rupakheti is grateful for the Dalhousie Graduate Fellowship from the Dalhousie University, and support from the Multiscale Air Quality Network (MAQNet) funded by the Canadian Foundation for Climate and Atmospheric Sciences (CFCAS). We also thank two anonymous reviewers for useful comments that helped improve the manuscript.
Related Research Data
REFERENCES
- Alfarra , M. R. , Coe , H. , Allan , J. D. , Bower , K. N. , Boudries , H. , Canagaratana , M. R. , Jimenez , J. L. , Jayne , J. T. , Garforth , A. A. , Li , S. and Worsnop , D. R. 2004 . Characterization of Urban and Regional Organic Aerosols in the Lower Fraser Valley Using Two Aerodyne Aerosol Mass Spectrometers . Atmos. Environ. , 38 ( l ) : 5745 – 5758 .
- Allan , J. D. , Jimenez , J. L. , Williams , P. I. , Alfarra , M. R. , Bower , K. N. , Jayne , J. T. , Coe , H. and Worsnop , D. R. 2003a . Quantitative Sampling Using an Aerodyne Aerosol Mass Spectrometer, 1, Techniques of Data Interpretation and Error Analysis . J. Geophys. Res. , 108 ( D3 ) : 4090 [CROSSREF]
- Allan , J. D. , Alfarra , M. R. , Bower , K. N. , Williams , P. I. , Gallagher , M. W. , Jimenez , J. L. , McDonald , A. G. , Nemitz , E. , Canagaratana , M. R. , Jayne , J. T. , Coe , H. and Worsnop , D. R. 2003b . Quantitative Sampling Using an Aerodyne Aerosol Mass Spectrometer, 2, Measurements of Fine Particulate Chemical Composition in Two U.K. Cities . J. Geophys. Res. , 108 ( D3 ) : 4091 [CROSSREF]
- Allan , J. D. , Coe , H. , Bower , K. N. , Alfarra , M. R. , Delia , A. E. , Jimenez , J. L. , Middlebrook , A. M. , Drewnick , F. , Onasch , T. B. , Canagaratana , M. R. , Jayne , J. T. and Worsnop , D. R. 2004a . A Generalized Method for the Extraction of Chemically Resolved Mass Spectra from Aerodyne Aerosol Mass Spectrometer Data . J. Aerosol Science , 35 ( 7 ) : 909 – 922 . [CROSSREF]
- Allan , J. D. , Bower , K. N. , Coe , H. , Boudries , H. , Jayne , J. T. , Canagaratana , M. R. , Millet , D. B. , Goldstein , A. H. , Quinn , P. K. , Weber , R. J. and Worsnop , D. R. 2004b . Submicron Aerosol Composition at Trinidad Head, CA during ITCT 2K2, Its Relationship with Gas Phase Volatile Organics Carbon and Assessment of Instrument Performance . J. Geophys. Res. , 109 ( D23 ) : D23S24 [CROSSREF]
- Allen , G. , Sioutas , C. , Koutrakis , P. , Reiss , R. , Lurmann , F. W. and Roberts , P. T. 1997 . Evaluation of The TEOM Method for Measurement of Ambient Particulate Mass in Urban Areas . J. Air & Waste Manage. Assoc. , 47 : 682 – 689 . [CSA]
- Brickell , P. C. , Bottenheim , J. W. , Froude , F. and Jiang , Z. 2003 . Continuous In-Situ NMHC Measurements in Rural Ontario, Canada. Eos. Trans. AGU 84(46), Fall Meeting . December 2003 , San Francisco. Suppl., A31D-0070
- Brook , J. and Dann , T. 1999 . Contribution of Nitrate and Carbonaceous Species to PM10 and PM2.5 Observed in Canadian Cities . J. Air & Waste Manage. Asso. , 49 : 193 – 199 . [CSA]
- Draxler , R. R. and Rolph , G. D. 2000 . “ HYSPLIT (HYbrid Single Particle Lagrangian Integrated Trajectories) Model (Version 4) ” . MD, , USA : NOAA Air Resource Laboratory, Silver Spring . http://www.arl.noaa.gov/ ready/hysplit4.html
- Drewnick , F. , Schwab , J. J. , Jayne , J. T. , Canagaratana , M. R. , Worsnop , D. R. and Demerjian , K. L. 2004 . Measurement of Ambient Aerosols During the PMTACS-NY2001 Using an Aerosol Mass Spectrometer Part I: Mass Concentrations . Aerosol Sci. Technol. , 38 ( S1 ) : 92 – 103 . [CROSSREF]
- Huffman , J. A. , Jayne , J. T. , Drewnick , F. , Aiken , A. C. , Onasch , T. , Worsnop , D. R. and Jimenez , J. L. 2005 . Design, Modeling, Optimization, and Experimental Tests of a Particle Beam Width Probe for the Aerodyne Aerosol Mass Spectrometer . Aerosol Sci. Technol. , submitted to, in press.
- Intergovernmental Panel on Climate Change (IPCC) . 2001 . “ Climate Change 2001: Scientific Basis ” . In Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change , Edited by: Houghton , J. T. Cambridge, , UK : Cambridge University Press .
- Jayne , J. T. , Leard , D. C. , Zhang , X. , Davidovits , P. , Smith , K. A. , Kolb , C. E. and Worsnop , D. R. 2000 . Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles . Aerosol Sci. Technol. , 33 : 49 – 70 . [CROSSREF]
- Jimenez , J. L. , Jayne , J. T. , Shi , Q. , Kolb , C. E. , Worsnop , D. R. , Yourshaw , I. , Seinfeld , J. H. , Flagan , R. C. , Zhang , X. , Smith , K. A. , Morris , J. and Davidovits , P. 2003a . Ambient Aerosol Sampling Using the Aerodyne Aerosol Mass Spectrometer . Geophys. Res. , 108 ( D7 ) : 8425 [CROSSREF]
- Jimenez , J. L. , Bahreini , R. , Cocker , I D. R. II , Zhuang , H. , Varutbangkul , V. , Flagan , R. C. , Seinfeld , J. H. , O'Dowd , C. D. and Hoffmann , T. 2003b . New Particle Formation from Photooxidation of Diiodomethane (CH2I2) . J. Geophys. Res. , 108 ( D10 ) : 4318 [CROSSREF]
- Jimenez , J. L. 2004 . Aerosol Mass Spectrometry Part 2: Thermal Desorption Techniques, Tutorial no. 15 . 23rd Annual Conference of American Association for Aerosol Research (AAAR) . Oct. 2004 , Atlanta, Georgia.
- Liu , P. , Ziemann , P. J. , Kittelson , D. B. and McMurry , P. H. 1995a . Generating Particle Beams of Controlled Dimensions and Divergence: I. Theory of Particle Motion in Aerodynamic Lenses and Nozzle Expansions . Aerosol Sci. Technol. , 22 : 293 – 313 .
- Liu , P. , Ziemann , P. J. , Kittelson , D. B. and McMurry , P. H. 1995b . Generating Particle Beams of Controlled Dimensions and Divergence: II. Experimental Evaluation of Particle Motion in Aerodynamic Lenses and Nozzle Expansions . Aerosol Sci. Technol. , 22 : 314 – 324 .
- Lohmann , U. , Broekhuizen , K. , Leaitch , R. , Shantz , N. and Abbatt , J. 2004 . How Efficient is Cloud Droplet Formation of Organic Aerosols? . Geophys. Res. Lett. , 31 [CSA] [CROSSREF]
- McLaren , R. , Salmon , R. A. , Liggio , J. , Hayden , K. L. , Anlauf , K. G. and Leaitch , W. R. 2004 . Nighttime Chemistry at a Rural Site in the Lower Fraser Valley . Atmos. Environ. , 38 : 5837 – 5848 . [CROSSREF]
- McMurry , P. H. 2000 . A Review of Atmospheric Aerosol Measurements . Atmos. Environ. , 34 ( 12–13 ) : 1959 – 1999 . [CROSSREF]
- Middlebrook , A. M. , Murphy , D. M. and Thomson , D. S. 1998 . Observations of Organic Material in Individual Marine Particles at Cape Grim during the First Aerosol Characterization Experiment (ACE1) . J. Geophys. Res. , 103 ( D13 ) : 16,475 – 16,483 . [CROSSREF]
- Patashnick , H. and Rupprecht , E. G. 1991 . Continuous PM10 Measurements Using the Tapered Element Oscillating Microbalance . J. Air & Waste Manage. Assoc. , 41 : 1079 – 1083 . [CSA]
- Pope , C. A. , Burnett , R. T. , Thun , M. J. , Calle , E. E. , Krewski , D. , Ito , K. and Thurston , G. D. 2002 . Lung Cancer, Cardio-Pulmonary Mortality, and Long-Term Exposure to Fine Particulate Air Pollution . J. American Medical Association , 287 ( 9 ) : 1132 – 1141 . [CROSSREF]
- Randles , C. A. , Russell , L. M. and Ramaswamy , V. 2004 . Hygroscopic and Optical Properties of Organic Sea Salt Aerosol and Consequences for Climate Forcing . J. Geophys. Res. , 31 : L16108
- Saxena , P. , Hidlemann , L. M. , McMurry , P. H. and Seinfeld , J. H. 1995 . Organics Alter Hygroscopic Behavior of Atmospheric Particles . J. Geophys. Res. , 100 ( D9 ) : 18,755 – 18,770 .
- Seinfeld , J. H. and Pandis , S. N. 1998 . Atmospheric Chemistry and Physics from Air Pollution to Climate Change , USA : John Wiley and Sons .
- Stolzenburg , M. R. and Hering , S. V. 2000 . Method for the Automated Measurement of Fine Particle Nitrate in the Atmosphere . Environ. Sci. Technol. , 34 ( 5 ) : 907 – 914 . [CSA] [CROSSREF]
- Vet , R. J. , Brook , J. R. and Dann , T. F. 2001 . “ The Nature of PM2.5 Mass, Composition, and Precursors in Canada ” . In Precursor Contributions to Ambient Fine Particulate Matter in Canada , Edited by: Brook , J. R. Meteorological Service of Canada . Environment Canada
- Zhang , X. , Smith , K. A. , Worsnop , D. R. , Jimenez , J. , Jayne , J. T. and Kolb , C. E. 2002 . A Numerical Characterization of Particle Beam Collimation by an Aerodynamic Lens-Nozzle System Part 1, an Individual Lens or Nozzle . Aerosol Sci. Technol. , 36 : 617 – 631 . [CROSSREF]