The Aerodyne Aerosol Mass Spectrometer (AMS) provides size-resolved chemical composition of non-refractory (vaporized at 600°C under vacuum) submicron aerosols with a time resolution of the order of minutes. Ambient measurements were performed in Tokyo between February 2003 and February 2004. We present intercomparisons of the AMS with a Particle-Into-Liquid Sampler combined with an Ion Chromatography analyzer (PILS-IC) and a Sunset Laboratory semi-continuous thermal-optical carbon analyzer. The temperature of the AMS inlet manifold was maintained at > 10 ˆ C above the ambient dew point to dry particles in the sample air (relative humidity (RH) in the inlet < 53%). Assuming a particle collection efficiency of 0.5 for the AMS, the mass concentrations of inorganic species (nitrate, sulfate, chloride, and ammonium) measured by the AMS agree with those measured by the PILS-IC to within 26%. The mass concentrations of organic compounds measured by the AMS correlate well with organic carbon (OC) mass measured by the Sunset Laboratory carbon analyzer (r 2 = 0.67–0.83). Assuming the same collection efficiency of 0.5 for the AMS organics, the linear regression slope is found to be 1.8 in summer and 1.6 in fall. These values are consistent with expected ratios of organic matter (OM) to OC in urban air.
INTRODUCTION
The importance of aerosols in the atmosphere has been widely recognized, as they have significant impacts both on local/regional air pollution and global climate. Recent advances in aerosol measurement techniques have greatly improved our current understanding of aerosol physics and chemistry in the atmosphere (e.g., CitationMcMurry 2000). In particular, Aerodyne Research has developed a mass spectrometric analyzer for real-time measurements of aerosols, which is referred to as the Aerosol Mass Spectrometer (AMS) (CitationJayne et al. 2000; CitationJimenez et al. 2003a). The AMS provides size-resolved chemical composition of non-refractory (vaporized within ∼ 1 s at 600°C under high vacuum) submicron aerosols, with an integration time of the order of seconds/minutes. The AMS is not designed to detect refractory components such as sea salt, mineral dust, and elemental carbon under typical operating conditions. The performance of the AMS has been evaluated by previous studies based on intercomparisons with other aerosol measurements (CitationAllan et al. 2003b; CitationBoudries et al. 2004; CitationDrewnick et al. 2003; CitationJimenez et al. 2003a; CitationMiddlebrook et al. 2003; CitationHogrefe et al. 2004; CitationSchneider et al. 2004; CitationTopping et al. 2004; CitationZhang et al. 2005a). CitationDrewnick et al. (2003) discussed the intercomparison with semi-continuous sulfate analyzers and found that the particle collection efficiency (CE) for those species can be approximated as 0.5 (i.e., correction by a factor of ∼ 2). More recently, CitationAllan et al. (2004a) have investigated the effects of relative humidity (RH) on CE during the Intercontinental Transport and Chemical Transformation 2002 (ITCT-2K2). They showed that the CE value depends on the RH of the sample air, with an approximate step function between CE ∼ 0.5 at low humidity and CE ∼ 1 at high humidity. These studies have provided useful information for understanding the characteristics of the AMS for ambient measurements. However, there are still some issues that require further investigation: e.g., variability in the particle collection efficiency, intercomparison of organic concentrations, and stability of the overall instrument performance.
We conducted ground-based measurements of aerosols in Tokyo (35°39′N, 139°40′E) using an AMS between February 2003 and February 2004. The observatory building, on the University of Tokyo campus, is located near the center of the city. The AMS measurements are compared to other semi-continuous aerosol measurements carried out in parallel by our group: a Particle-Into-Liquid Sampler combined with an Ion Chromatography analyzer (PILS-IC) (CitationWeber et al. 2001; CitationOrsini et al. 2003) and a semi-continuous thermal-optical carbon aerosol analyzer manufactured by Sunset Laboratory, Inc. (CitationBae et al. 2004). The data obtained in May 2003, July 2003, and October 2003 (with PM1 inlets for the PILS-IC and Sunset Lab carbon analyzer) are used for the intercomparison (). The major purpose of this paper is to evaluate the performance of the AMS based on routine calibrations and intercomparison with those independent measurements.
TABLE 1 Summary of intercomparison periods
AERODYNE AEROSOL MASS SPECTROMETER (AMS)
Instrument Configuration for Ambient Sampling
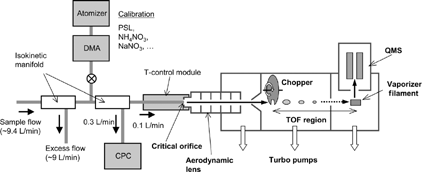
The principles of operation and details of the AMS have been presented in previous publications (e.g., CitationJayne et al. 2000; CitationJimenez et al. 2003a; CitationAllan et al. 2003a). A brief description of the performance of our AMS is given in this section. shows a schematic diagram of the AMS and ambient sampling system used in this study. The AMS consists of a particle sampling inlet, a particle time-of-flight (PTOF) chamber, and a vaporizer/ionizer that is interfaced to a quadrupole mass spectrometer (QMS). The sample air is aspirated through a critical orifice (diameter 100 μ m) that maintains a sample flow rate of 1.4 cm3 s−1. A narrow particle beam is generated by an aerodynamic lens that consists of a series of apertures embedded in a 1/2-inch outer diameter (1 cm inner diameter) stainless steel tube (CitationLiu et al. 1995; CitationZhang et al. 2002, Citation2004). The particles are accelerated in the supersonic expansion of gas molecules into a vacuum at the end of the lens, resulting in size-dependent velocities of the particles. A vaporizer (typically heated at 600°C) and a filament with a relative electrical potential of 70 eV are placed at the end of the TOF chamber, yielding a flash vaporization of the particles followed by electron impact ionization of the evolved gas molecules. The ions for a particular mass-to-charge (m/z) ratio are detected using the QMS (QMA 410, Balzers, Liechtenstein). The integration time employed for this study was 10 min.
FIG. 1 Schematic diagram of the Aerodyne Aerosol Mass Spectrometer (AMS) and the sampling system for ambient measurements.
The AMS is operated in two modes: mass spectrum (MS) mode and PTOF mode. The PTOF mode is a size distribution measurement mode for selected m/z settings of the QMS (CitationJimenez et al. 2003a; CitationAllan et al. 2003a). The MS mode is a continuous mass spectrum mode (m/z = 1−300) without size information. In the MS mode, a mechanical chopper is moved between two positions typically every 4 s to measure total signal (beam open position) and background signal (beam blocked position). The signal from ambient aerosols is determined by subtracting the background signal from the total signal.
The particle diameter measured by the AMS, i.e., vacuum aerodynamic diameter (d va ), is defined as d va = (ρ p /χ v ρ 0)d ve , where ρ p is the particle density, ρ 0 is the unit density (= 1 g cm−3), χ v is the dynamic shape factor in the free molecular regime, and d ve is the volume-equivalent diameter (CitationJimenez et al. 2003b; CitationDeCarlo et al. 2004). The aerodynamic lens allows particle transmission efficiencies of ∼ 100% for d va = 50−600 nm, with some transmission for d va > 1 μ m, which is roughly similar to the commonly used PM1 cutoff. Therefore, the aerosols measured by the AMS are referred to as non-refractory PM1 (NR − PM1) aerosol concentrations.
The sample air for the AMS was aspirated from the rooftop of the observatory building (∼ 18 m above ground level). The sampling line from the rooftop to the observation room is about 6 meters long and made of 1/2-inch outer diameter stainless steel tubing. The flow rate was maintained at ∼ 9.4 L/min, using an external pump and critical orifice (). A PM2.5 cyclone (URG Corp., USA) was used for the inlet to remove coarse particles, although the actual size-cut of the AMS is determined by the more restrictive transmission characteristics of its aerodynamic lens, as mentioned earlier. Isokinetic manifolds were used at each junction where the flow rate and the inner diameter of the tubing were changed.
The effect of the AMS inlet temperature/humidity on particle collection efficiency has been discussed by CitationAllan et al. (2004a). These authors found that mass concentrations of particulate water (H2O) increased and particle collection efficiencies for inorganic species were improved as the inlet temperature approached the ambient dew point. They suggested that the particle collection efficiency could be approximated as 0.5 when the inlet temperature was more than 10°C higher than ambient dew point (i.e., dry conditions). Based on their findings, the inlet temperature of our AMS was controlled to dry particles in the sample air (). The inlet temperature was manually determined depending on the ambient dew point to keep the RH value in the inlet manifold below ∼ 50% (> 10°C above ambient dew point). The mass loadings of particulate water detected by the AMS were nearly zero at these low RH values. The residence time of sample air in the inlet manifold was ∼ 1.5 s. Here we assume that evaporation of particulate materials due to this artificial temperature change was significant only for water vapor at this short residence time. However, it is possible that some evaporation of highly volatile constituents may have occurred in the inlet manifold. This could be a source of systematic uncertainties for our AMS measurements.
Data Processing
Details of the data processing and analysis procedures have been described in previous publications (CitationJimenez et al. 2003a; CitationAllan et al. 2003a, Citation2004b). The chemical composition of the particles can be determined based on standard mass spectrometric analysis (CitationMcLafferty and Turecek 1993), although the fragmentation patterns of the AMS are generally shifted to smaller m/z because of its higher vaporization temperature compared to gas chromatography mass spectrometry (CitationAlfarra 2004). The mass concentration of “organics” is defined as the sum of all fragments after subtracting all the identified signals originating from ambient gas molecules, inorganic compounds, and instrumental artifacts such as sodium ions (Na+) from surface ionization on the vaporizer (CitationAllan et al. 2004b).
The quantification procedure for deriving AMS mass concentrations was first described by CitationJimenez et al. (2003a), and later revised by CitationAlfarra et al. (2004). The mass concentration of species i (C i : μ g m− 3) is calculated using the following equation,
Ionization Efficiency
Ionization efficiency for nitrate, IE NO 3 , was routinely determined by supplying mono-disperse particles from a calibration unit () and measuring the ion signals in the AMS. The calibration unit consists of a Collison atomizer (Model 3076, TSI, Inc., USA), a diffusion dryer, and a scanning mobility particle sizer (SMPS) (Model 3936, TSI, Inc., USA). The SMPS consists of a differential mobility analyzer (DMA, Model 3081, TSI, Inc., USA) and a condensation particle counter (CPC, Model 3022, TSI, Inc., USA). The DMA sizing was calibrated using NIST-traceable polystyrene latex (PSL) particles (Duke Scientific, Inc., USA). The mobility diameter indicated by the SMPS was found to be 2.5% smaller than the nominal diameter of the PSL particles. Pure, dry ammonium nitrate (NH4NO3) particles with a mobility diameter of 359 nm (corresponding to the 350-nm setting of the SMPS) were used for the calibration. In the calibration procedure, we can exclude larger diameter particles of equivalent mobility diameter (multiply charged particles) because they are clearly resolved in aerodynamic diameter space (CitationJayne et al. 2000).
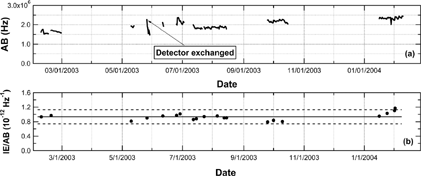
The AMS signal at m/z = 28 (N2 +), which is referred to as the airbeam (AB), is used as an internal standard for correcting for changes in the ionization efficiency in between IE NO 3 calibrations (CitationAllan et al. 2003a; CitationJimenez et al. 2003a). The AB in our instrument is typically 2 MHz under normal operating conditions. It has been found that the ratio of IE NO 3 to AB depends (a) on the pressure in the TOF chamber (∼ 10−5 torr), due to changes in the fraction of airbeam molecules scattered by background molecules with pressure, and (b) on the detailed positioning of the vaporizer and filament relative to other surfaces in the ionization region (J. Jayne et al., unpublished data). Ideally, the IE NO 3 /AB ratio remains constant as long as these factors do not change. shows a time series of AB signals and IE NO 3 /AB ratios between February 2003 and February 2004. In , the AB signal data represent either ambient measurements or laboratory experiments, while the IE NO 3 /AB ratio data represent periods when routine calibrations were performed. There are some large gaps in the AB signal data because the AMS was operated mainly during each intensive measurement period (typically of ∼ 2-week duration every 2–3 months). In between the intensive measurement periods, the instrument was not sampling ambient air (i.e., inlet valve was closed) and was mostly under high-vacuum condition due to continuous operation of the pumps. The average (± 2σ) of the IE NO 3 /AB ratios was found to be 0.93 (± 0.19) × 10−12 Hz−1. The 2σ variability range corresponds to 20% of the average.
FIG. 2 Time series of (a) the air beam (AB) signal and (b) the ratio of the ionization efficiency for nitrate (IE NO 3 ) to the AB signal. The AB data are 12-h averages. The solid line and dashed line in b indicate the average and 2σ variability range of the IE NO 3 /AB ratios, respectively.
Factors Affecting Absolute Accuracy
As described in the previous section, the AB signal is used for the correction of the ionization efficiency, based on the assumption that the IE NO 3 /AB ratios do not significantly change between IE NO 3 calibrations. Therefore, an upper limit of the uncertainty in the AB correction can be evaluated as the relative difference between the consecutive IE NO 3 /AB ratios. The differences are found to be smaller than 10% during each intensive measurement period (excluding the differences across the large temporal gaps).
The uncertainty in the actual mass of the calibration particles affects the accuracy of the IE NO 3 determined from the routine calibration procedure. It comes from (1) uncertainty in the DMA sizing, (2) uncertainty in the dynamic shape factor of the calibration particles (e.g., CitationDeCarlo et al. 2004; CitationZiemann et al. 1995), and (3) possible evaporative loss of the calibration particles between the DMA and AMS. The uncertainties of the measurements corresponding to the above three factors are referred to as ϵ1, ϵ2, and ϵ3, respectively. ϵ1 is estimated to be ∼ 8% (i.e., from 1.025 cubed), based on the PSL calibration. ϵ2 is estimated to be of the order of 6% for 350-nm NH4NO3 particles, based on the theoretical calculations performed by CitationDeCarlo et al. (2004). Here we assume that the calibration particles have no internal voids. ϵ3 can be estimated by comparing the vacuum aerodynamic diameter with the mobility diameter. The average (± 2σ) vacuum aerodynamic diameter of the calibration particles measured by the AMS was 497 ± 15 nm between February 2003 and February 2004, without a systematic dependence on room temperature (20°–29°C). On the other hand, the vacuum aerodynamic diameter estimated from the mobility diameter was 494 nm, which agreed with the measured aerodynamic diameter to within 0.6%. The conversion from the mobility diameter to the vacuum aerodynamic diameter is based on the formula given by CitationDeCarlo et al. (2004). This result suggests that the ϵ3 was negligibly small as compared to ϵ1 and ϵ2 in the case of this experiment. The total uncertainty due to the uncertainties in the IE NO 3 /AB ratios (10%), PSL calibration (8%), and mass of the calibration particle (6%) is estimated to be ∼ 14%.
The relative ionization efficiency, RIE i , was determined based on various laboratory experiments. In this study, we used the previously determined RIE i values as given by CitationAlfarra et al. (2004): i.e., 1.1 for nitrate, 1.15 for sulfate, 1.3 for chloride, 3.5 for ammonium, and 1.4 for organics. It should be noted that RIE NO 3 is not equal to unity. The standard AMS calibration procedure determines IE NO 3 using only the signals at m/z 30 (NO+) and 46 (NO2 +), which account for ∼ 90% of the total fragments from nitrate. Therefore, the effect from the other nitrate fragments that are not used for the calibration, but are used in the field data analysis, is incorporated in RIE NO 3 . For inorganic species, the uncertainties in the RIE i values are small. For organic compounds, the RIE i values can vary with functional groups (e.g., hydrocarbons, oxygenated organics, etc.) (CitationJimenez et al. 2003a). Without a priori information on the chemical compositions of organics, however, we assume a constant RIE i of 1.4 for organics. This assumption could be a source of uncertainty in determining the mass concentrations of organics.
The particle collection efficiency, CE i (typically between 0.5 and 1), has been experimentally determined in previous studies by comparison to other aerosol instruments such as PILS-IC. As mentioned earlier, it has been suggested that CE i can be approximated as 0.5 under dry conditions (CitationAllan et al. 2004a). Strictly speaking, the terminology “collection efficiency” might be somewhat ambiguous because it could be a combined effect of particle focusing in the aerodynamic lens and particle bounce from the vaporizer (T. Onasch et al., unpublished data). Intense work is ongoing in the AMS community for identifying and correcting the physical processes leading to CE i < 1 in some cases.
In addition, the approximation of the AMS size-cut to the classical PM1 size-cut may not be appropriate when a significant fraction of ambient PM1 mass lies between 600 nm and 1 μ m in vacuum aerodynamic diameter. Although this effect may not be distinguishable from particle collection efficiency in ambient data, we describe them separately because the physical processes are different. The overall accuracy of the AMS measurements, including the uncertainties in IE NO 3 , RIE i , CE i , and AMS size-cut, is difficult to evaluate because some of these uncertainties depend on the physical and chemical properties of aerosols in ambient air.
Limits of Detection
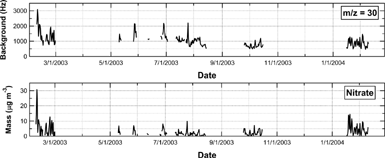
The background signals, which are routinely measured at the beam-blocked position, control the noise for most of the important m/z values used in the AMS. The background signals are due to the ionization of gases and vapors outgassing from the walls of the vacuum chamber or any other components in the chamber. shows the time series of the background signal at m/z 30 (NO+), which is mainly a nitrate fragment. The background signal was normalized using the AB signal to account for the temporal variation in the sensitivity of the instrument. Mass loadings of nitrate in ambient air are also plotted in . It can be seen that variations in the background signals showed positive correlation with the nitrate mass loadings (e.g., r2 = 0.84 for February 2003 and r2 = 0.69 for June–August 2003). This result suggests that nitrate particles originating from ambient air were the important source of the background variability at m/z 30.
FIG. 3 Time series of the background signal at m/z = 30 (NO+) in the AMS mass spectra and nitrate mass loadings in ambient air. The data are 12-h averages.
The limit of detection (LOD) can be estimated as the concentration needed to produce a signal-to-noise ratio (SNR) of 3 above the dominant source of noise due to the background signal (CitationAllan et al. 2003a). The LOD can also be measured directly by placing a particle filter in front of the AMS and recording the mass concentration (CitationZhang et al. 2005a). In this case, the LOD is defined as 3 times the standard deviation of the recorded mass concentration. The LOD values (1-h average) estimated using the above two methods are compared in . The data obtained in May 2003 were used for this calculation. The average mass concentrations of nitrate, sulfate, chloride, ammonium, and organics through the filter were below the ion-counting based LODs, indicating that there was no significant artifact or leak in the sampling line. The LOD values from ion counting noise show reasonable agreement with those from the filter measurement, although the former tends to be lower than the latter. This tendency is understandable because the filter measurement includes various factors other than ion counting noise. Hereafter, we use the LOD values from the filter measurements to describe the performance of the instrument.
TABLE 2 Limits of Detection (LODs) for AMS measurements (1-h average)
PILS-IC AND EC/OC ANALYZER
PILS-IC
The PILS-IC provides near-real-time measurements of water-soluble inorganic ions such as NO3 −, SO4 2 −, Cl−, NH4 +, Na+, Ca2 +, and K+. The configuration of the PILS-IC used in this study is basically the same as that described by CitationOrsini et al. (2003). The PILS produces supersaturated water vapor by adding steam to the sample air. The particles in the sample air grow into droplets (diameter > 1 μ m) large enough to be collected by an impactor. The collected droplets are introduced into an IC by adding a continuous liquid water flow. The sampling interval employed for this study was typically ∼ 15 min.
Recent laboratory experiments have indicated a loss of ammonium (NH4 +) in the PILS likely due to losses in liquid sample lines and volatility losses from the hot droplets. NH4 + is estimated to be underestimated by 15 ± 3% (CitationMa 2004), based on a number of comparisons, which include: comparisons from airborne and ground-based studies between the PILS-IC and integrated filters and micro-orifice impactors; comparisons of ambient measurements to a mist chamber, which does not involve heating of the collecting water; and laboratory measurements comparing molar ratios of sodium sulfate (Na2SO4) and ammonium sulfate ((NH4)2SO4). For sulfate and nitrate, PILS measurements have been compared to other methods, and the agreement was generally found to be within 20% (CitationMa 2004; CitationMa et al. 2003; CitationOrsini et al. 2003; CitationWeber et al. 2003).
For the PILS-IC used in this study, the NH4 + loss was evaluated by supplying pure NH4NO3 particles into the PILS-IC and was found to be 20%. The NH4 + concentrations presented in this paper were corrected using our experimental correction factor of 1.25 (= 1/0.8). The LOD for nitrate was estimated to be 0.01 μ g/m3 (15-min average). A PM1 cyclone (URG Corp., USA) was used for the PILS inlet between April and July 2003. The dataset obtained during this period is used for the intercomparison with the AMS measurements. The inlet was then switched to PM10 starting in August 2003 in order to investigate the interaction between anthropogenic and sea-salt aerosols in this region.
EC/OC Analyzer
The mass concentrations of organic carbon (OC) and elemental carbon (EC) were measured using a Sunset Laboratory semi-continuous EC/OC analyzer (CitationBae et al. 2004). Here we focus on OC measurements only because the AMS does not measure EC. Ambient particles are collected on a quartz filter and then analyzed based on the thermal-optical-transmittance method. The temperature protocol proposed by the National Institute for Occupational Safety and Health (NIOSH) (CitationBirch and Cary 1996) was used in this study. A comparison with the temperature protocol proposed by the Interagency Monitoring of Protected Visual Environments (IMPROVE) of the Desert Research Institute was performed. Overall, the mass concentrations of OC based on the NIOSH protocol were found to be higher by ∼ 13% than those based on the IMPROVE protocol, under typical ambient conditions in Tokyo. The difference can be attributed to higher maximum temperature in the helium atmosphere for the NIOSH protocol than the IMPROVE protocol and provides an indication of the degree of systematic uncertainty in the OC measurements in Tokyo.
The temperature of the quartz filter during the sampling period (45 min of every 1-h cycle) can be an important factor affecting the gas-particle equilibrium of organic compounds. The quartz filter temperature (i.e., room temperature) was ∼ 29°C in July 2003 and ∼ 24°C in October 2003. These values are close to ambient temperature for each period (), suggesting that the perturbation of the gas-particle equilibrium due to the temperature difference between ambient air and the quartz filter was small during the intercomparison period.
Volatile organic compounds (VOCs) are the most critical factor affecting the background level of OC. A carbon denuder provided by the Sunset Laboratory, Inc. was used to reduce the effects of volatile organic compounds (VOCs) on the OC measurements. Because the VOC removal efficiency of the denuder could vary with time, we periodically checked the background level of OC by placing a particle filter in front of the denuder. The LOD for the OC measurements was estimated to be 1 μ g m−3 (1-h average) based on the background measurements. A PM1 cyclone (URG Corp., USA) was used for the EC/OC inlet for the intercomparison periods.
INTERCOMPARISONS
AMS versus PILS-IC
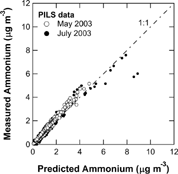
Intercomparisons of AMS with PM1 PILS-IC were carried out on May 10–12 and July 23–30 of 2003 (). Before discussing the intercomparison results, we need to describe the ion balance of ambient aerosols during these periods using the PILS-IC data only. The PILS-IC data showed that cations in the PM1 mode were dominated by NH4 +. We observed only a small amount of Na+ (< 0.2 μ g/m3) in the PM1 mode. This may originate from sea salt aerosols because the RCAST observatory is located at ∼ 10 km from a coastal area (Tokyo Bay). We estimated the concentrations of non-sea salt (nss) Cl− assuming that all of the observed Na+ originated from sodium chloride (NaCl) in sea salt. The fraction of nss-Cl− was found to be ∼ 90% of the total Cl− in the PM1 mode for both the May and July periods. It should be noted that the nss-Cl− concentration estimated above is a lower limit because Cl− from sea salt could be partially/fully depleted by reaction between NaCl and nitric acid (HNO3). presents a scatter plot of measured and predicted NH4 + derived from the PM1 PILS measurements in May and July 2003. The predicted NH4 + was determined assuming that measured anions (SO4 2 −, NO3 −, and nss-Cl−) were fully neutralized by NH4 +. Except for a few outlying data points with higher mass loadings (July) indicative of acidic particles, the measured NH4 + agreed well with the predicted NH4 +. This result indicates that these anions were mostly in the form of NH4NO3, (NH4)2SO4, and ammonium chloride (NH4Cl), which can be efficiently detected by the AMS at its standard vaporizer temperature of 600°C.
FIG. 4 Scatter plot of measured and predicted NH4 + derived from the PM1 PILS-IC measurements in May (open circles) and July (solid circles) 2003. The data are 1-h averages. The predicted NH4 + was determined assuming that measured anions (SO4 2 −, NO3 −, and nss-Cl−) were fully neutralized by NH4 +. The dot-dashed line indicates 1:1 correspondence line.
shows scatter plots of AMS versus PM1 PILS measurements in May and July 2003. Note that CE = 0.5 is assumed for all of the AMS inorganic species. summarizes the fit parameters that were determined by a standard linear regression procedure. For the May dataset, the difference between AMS and PILS-IC was found to be 12%, 24%, 16%, and 26% for nitrate, sulfate, chloride, and ammonium, respectively, where the difference is defined as (slope – 1) × 100%. For the July dataset, the difference was −19%, −10%, −9%, and −5% for nitrate, sulfate, chloride, and ammonium, respectively. The AMS measurements tend to be slightly higher than the PILS-IC measurements in May, and vice versa in July, although the cause of this systematic difference has not been identified. What is clear from this intercomparison is that the assumption of CE = 0.5 yields an agreement between AMS and PILS-IC to within 26% or better for nitrate, sulfate, chloride, and ammonium.
FIG. 5 Scatter plots of AMS versus PM1 PILS measurements in May (open circles) and July (solid circles) 2003. The data are 1-h averages. The dashed and solid lines represent the linear regression lines for May and July 2003, respectively. Note that CE = 0.5 is assumed for all of the AMS inorganic species. The dot-dashed line indicates 1:1 correspondence line.
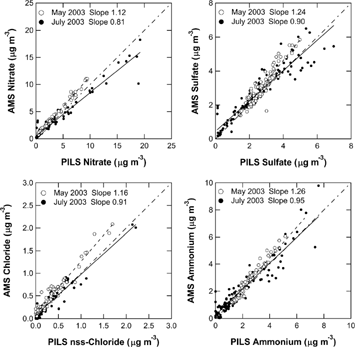
TABLE 3 Summary of intercomparison results
Previous studies also showed that the AMS measurements agreed with PILS-IC or other semi-continuous measurements to within 25% or better when CE = 0.5 was assumed (CitationDrewnick et al. 2003; CitationAllan et al. 2004a; CitationZhang et al. 2005a). However, these previous studies mostly focused on sulfate and ammonium because the mass loadings of nitrate and chloride were not high enough in many cases. The new finding obtained in this study is that the agreement between AMS and PILS-IC for nitrate and chloride is as good as sulfate and ammonium.
AMS versus EC/OC Analyzer
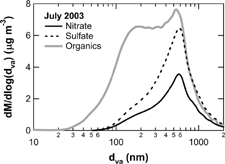
Intercomparison of OMAMS and OC measurements was made in July and October of 2003 (), where OMAMS represents the mass concentration of organic matter detected by the AMS. shows average size distributions of nitrate, sulfate, and organics for July 2003. In general, the size distribution of organics in Tokyo (versus d va ) was bimodal in all seasons (Takegawa et al. manuscript in preparation), as is often the case for AMS measurements in urban areas (e.g., CitationJimenez et al. 2003a; CitationAllan et al. 2003b; CitationZhang et al. 2005a). Analysis of the size distributions of the individual m/z sampled suggests that the small mode (d va < 200 nm) was dominated by aliphatic or aromatic hydrocarbons that are the major constituents of combustion-generated primary organic aerosols and that the accumulation mode (d va > 200 nm) was dominated by oxygenated organic compounds that are the major constituents of secondary organic aerosols. The size distributions suggest that the former is externally mixed with inorganic species and the latter is internally mixed with inorganic species (e.g., CitationAllan et al. 2003b; CitationZhang et al. 2005a). Therefore, the AMS particle collection efficiency of 0.5 is a reasonable assumption for the accumulation-mode organics. In this analysis we assume a constant collection efficiency of 0.5 for the small-mode organics as well as the accumulation-mode organics.
FIG. 6 Monthly average size distribution of nitrate (solid line), sulfate (dashed line), and organics (shaded line) for July 2003.
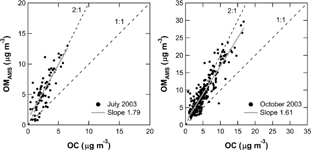
shows scatter plots of OMAMS versus OC mass concentrations in July and October 2003. In general, the mass concentrations of OMAMS were systematically higher than those of OC for almost the entire dataset, which is expected since organics contain several elements other than carbon (H, O, N, etc.). The linear regression slope was found to be 1.8 for July 2003 and 1.6 for October 2003. CitationTurpin and Lim (2001) summarized the ratio of OM to OC for various cases. The ratio of molecular weight to carbon weight is 1.2 for n-alkanes (C23–C34) and 1.0–1.1 for polycyclic aromatic hydrocarbons (PAHs), while it reaches as high as 1.7–3.8 for aliphatic dicarboxylic acids (C2–C9). They suggested that the OM/OC ratio could be approximated as 1.6 ± 0.2 for urban aerosols and 2.1 ± 0.2 for aged (non-urban) aerosols. More recently, CitationRussell (2003) estimated OM/OC ratios of 1.2–1.6 for aerosol samples collected over northeastern Asia and the Caribbean. The average OMAMS/OC ratios of 1.6–1.8 (i.e., linear regression slope) obtained in this study were consistent with the OM/OC ratios from CitationTurpin and Lim (2001) but slightly larger than those from CitationRussell (2003). The only previous published comparison between OMAMS and OC in urban air found an average OMAMS/OC ratio of 1.69 ± 0.03 during fall in Pittsburgh (CitationZhang et al. 2005a). This value is also consistent with the OM/OC ratio proposed by CitationTurpin and Lim (2001). However, Zhang et al. assumed an average collection efficiency of 0.7 for total organics (CE = 1 for small-mode organics and CE = 0.5 for accumulation-mode organics), based on laboratory experiments performed by CitationSlowik et al. (2004).
FIG. 7 Scatter plots of AMS organics (OMAMS) and OC mass concentrations in July and October 2003. The data are 1-h averages. The shaded line represents the linear regression line. The dashed lines indicate 1:1 and 2:1 correspondence lines.
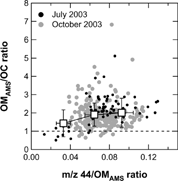
We investigate the relationship between the OMAMS/OC ratios and the degree of photochemical processing for the individual data points. Previous AMS studies have shown that signals at m/z 44 are dominated by COO+, which typically originates from oxygenated organics such as dicarboxylic acids (CitationAllan et al. 2004a; CitationAlfarra et al. 2004; CitationZhang et al. 2005b). Therefore, the m/z 44/OMAMS ratio can be used as a diagnostic for photochemical processing (oxidation) of organic aerosols: i.e., higher ratios are generally associated with more aged air masses. Note that m/z 44 represents the equivalent mass concentration of organic compounds represented by the signal intensity at this peak (CitationZhang et al. 2005b). shows the OMAMS/OC ratio as a function of the m/z 44/OMAMS ratio. Data with mass loadings below the LOD of either instrument were excluded. In this analysis, the observed data points were classified as follows: (a) less-processed organics, i.e., m/z 44/OMAMS < 0.04; (b) moderately processed organics, i.e., 0.04 < m/z 44/OMAMS < 0.08; (c) highly processed organics, i.e., 0.08 < m/z 44/OMAMS. The threshold numbers were arbitrarily chosen to span the dynamic range of m/z 44/OMAMS ratios observed in Tokyo. The average OMAMS/OC ratios (± standard deviation) were calculated to be 1.4 ± 0.7, 1.9 ± 0.6, and 2.0 ± 0.8 for less, moderately, and highly processed organics, respectively. Although the scatter in the data is considerable, the OMAMS/OC ratios tend to increase with m/z 44/OMAMS ratio. This tendency can be attributed to the increase in the elements other than carbon in organic aerosols with photochemical processing.
FIG. 8 OMAMS/OC ratios as a function of m/z 44/OMAMS ratio. The data are 1-h averages. The open squares and bars represent the average values and standard deviations for each m/z 44/OMAMS ratio bin, respectively.
SUMMARY AND CONCLUSIONS
We conducted ground-based measurements of submicron aerosols in Tokyo between February 2003 and February 2004 (typically ∼ 2-week intensive measurement period every 2–3 months) using an Aerodyne AMS. The temperature of the inlet manifold was controlled at > 10°C above ambient dew point to dry particles in the sample air (RH in the inlet < 53%). The inorganic aerosols measured by the AMS were compared to those measured by a PILS-IC, assuming that the AMS particle collection efficiency is 0.5 for those species. The mass concentrations of nitrate, sulfate, chloride, and ammonium measured by the AMS agreed with those measured by the PILS to within 26% or better. The organic mass concentrations measured by the AMS were also compared to OC mass concentrations measured by the Sunset Lab semi-continuous EC/OC analyzer, assuming the same particle collection efficiency. The average OMAMS/OC ratio was found to be 1.8 and 1.6 for July and October 2003, respectively. Those values were consistent with the OM/OC ratios of 1.6–2.1 reported by CitationTurpin and Lim (2001). Consequently, the evaluation of AMS performance presented in this paper is important not only for the interpretation of aerosol data obtained in Tokyo (Takegawa et al. manuscript in preparation) but also for other AMS experiments conducted by other investigators.
Acknowledgments
This study was funded by the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT) and the Japanese Science and Technology Agency (JST).
Notes
a Average ± standard deviation. Values in parentheses represent minimum and maximum values.
b Particle-Into-Liquid Sampler combined with Ion Chromatography (PILS-IC).
c Semi-continuous thermal-optical carbon analyzer manufactured by the Sunset Laboratory, Inc.
d N/A: Not available.
a The LOD (Ion counting) is defined as the concentration needed to produce a signal-to-noise ratio (SNR) of 3 above the dominant source of noise due to background ions from the mass spectrometer.
b The LOD (Filter) is defined as 3 times the standard deviation of the mass concentration measured by placing a particle filter in front of the AMS.
a Linear regression fit parameters of AMS versus PILS-IC or OC correlation. Errors are 95% confidence intervals.
Related Research Data
REFERENCES
- Alfarra , M. R. 2004 . Insights into Atmospheric Organic Aerosols using an Aerosol Mass Spectrometer Ph.D. thesis , UK : The University of Manchester .
- Alfarra , M. R. , Coe , H. Allan , J. D. 2004 . Characteristics of Urban and Rural Organic Particulate in the Lower Fraser Valley using Two Aerodyne Aerosol Mass Spectrometers . Atmos. Environ. , 38 : 5745 – 5758 .
- Allan , J. D. , Jimenez , J. L. Williams , P. I. 2003a . Quantitative Sampling using an Aerodyne Aerosol Mass Spectrometer, 1, Techniques of Data Interpretation and Error Analysis . J. Geophys. Res. , 108 : 4090 [CROSSREF]
- Allan , J. D. , Alfarra , M. R. Bower , K. N. 2003b . Quantitative Sampling using an Aerodyne Aerosol Mass Spectrometer, 2, Measurements of Fine Particulate Chemical Composition in Two U.K. Cities . J. Geophys. Res. , 108 : 4091 [CROSSREF]
- Allan , J. D. , Bower , K. N. Coe , H. 2004a . Submicron Aerosol Composition at Trinidad Head, CA during ITCT 2K2, Its Relationship with Gas Phase Volatile Organic Carbon and Assessment of Instrument Performance . J. Geophys. Res. , 109 : D23S24 – 0 . [CROSSREF]
- Allan , J. D. , Delia , A. E. Coe , H. 2004b . A Generalised Method for the Extraction of Chemically Resolved Mass Spectra from Aerodyne Aerosol Mass Spectrometer Data. (Technical Note) . J. Aerosol Sci. , 35 : 909 – 922 . [CROSSREF]
- Bae , M. S. , Schauer , J. J. , DeMinter , J. T. , Turner , J. R. , Smith , D. and Cary , R. A. 2004 . Validation of a Semi-Continuous Instrument for Elemental Carbon and Organic Carbon using a Thermal-Optical Method . Atmos. Environ. , 38 : 2885 – 2893 . [CROSSREF]
- Birch , M. E. and Cary , R. A. 1996 . Elemental Carbon-Based Method for Monitoring Occupational Exposures to Particulate Diesel Exhaust . Aerosol Sci. Technol. , 25 ( 3 ) : 221 – 241 .
- Boudries , H. , Canagaratna , M. R. Jayne , J. T. 2004 . Chemical and Physical Processes Controlling the Distribution of Aerosols in the Lower Fraser Valley, Canada, during the PACIFIC 2001 Field Campaign . Atmos. Environ. , 38 : 5759 – 5774 . [CROSSREF]
- DeCarlo , P. , Slowik , J. G. , Worsnop , D. R. , Davidovits , P. and Jimenez , J. L. 2004 . Particle Morphology and Density Characterization by Combined Mobility and Aerodynamic Diameter Measurements. Part 1: Theory . Aerosol Sci. Technol. , 38 : 1185 – 1205 . [CROSSREF]
- Drewnick , F. , Schwab , J. J. Hogrefe , O. 2003 . Intercomparison and Evaluation of Four Semi-Continuous PM2.5 Sulfate Instruments . Atmos. Environ. , 37 : 3335 – 3350 . [CROSSREF]
- Hogrefe , O. , Schwab , J. J. Drewnick , F. 2004 . Semicontinuous PM2.5 Sulfate and Nitrate Measurements at an Urban and a Rural Location in New York: PMTACS-NY Summer 2001 and 2002 Campaigns . J. Air Waste Manag. Assoc. , 54 : 1040 – 1060 . [PUBMED] [INFOTRIEVE] [CSA]
- Jayne , J. T. , Leard , D. C. Zhang , X. F. 2000 . Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles . Aerosol Sci. Technol. , 33 : 49 – 70 . [CROSSREF]
- Jimenez , J. L. , Jayne , J. T. Shi , Q. 2003a . Ambient Aerosol Sampling using the Aerodyne Aerosol Mass Spectrometer . J. Geophys. Res. , 108 : 8425 [CROSSREF]
- Jimenez , J. L. , Bahreini , R. Cocker , D. R. 2003b . New Particle Formation from Photooxidation of Diiodomethane (CH2I2) . J. Geophys. Res. , 108 : 4318 [CROSSREF]
- Liu , P. , Ziemann , P. J. , Kittelson , D. B. and McMurry , P. H. 1995 . Generating Particle Beams of Controlled Dimensions and Divergence: I. Theory of Particle Motion in Aerodynamic Lenses and Nozzle Expansions . Aerosol Sci. Technol. , 22 : 293 – 313 .
- Ma , Y. , Weber , R. J. Meier , K. 2003 . Intercomparisons of Airborne Measurements of Aerosol Ionic Chemical Composition during TRACE-P and ACE-Asia . J. Geophys. Res. , 109 : D15S07
- Ma , Y. 2004 . Developments and Improvements to the Particle-Into-Liquid Sampler (PILS) and Its Application to Asian Outflow Studies , Atlanta, GA : Georgia Institute of Technology . Ph.D. thesis
- McLafferty , F. W. and Turecek , F. 1993 . Interpretation of Mass Spectra , Mill Valley, CA : Univ. Sci. .
- McMurry , P. H. 2000 . A Review of Atmospheric Aerosol Measurements . Atmos. Environ. , 34 : 1959 [CROSSREF]
- Middlebrook , A. M. , Murphy , D. M. Lee , S.-H. 2003 . A Comparison of Particle Mass Spectrometers during the 1999 Atlanta Supersite Project . J. Geophys. Res. , 108 : 8424 [CROSSREF]
- Orsini , D. , Ma , Y. , Sullivan , A. , Sierau , B. , Baumann , K. and Weber , R. 2003 . Refinements to the Particle-Into-Liquid Sampler (PILS) for Ground and Airborne Measurements of Water Soluble Aerosol Composition . Atmos. Environ. , 37 : 1243 – 1259 . [CROSSREF]
- Russell , L. M. 2003 . Aerosol Organic-Mass-to-Organic-Carbon Ratio Measurements . Environ. Sci. Technol. , 37 : 2982 – 2987 . [PUBMED] [INFOTRIEVE] [CSA]
- Schneider , J. , Borrmann , S. Wollny , A. G. 2004 . Online Mass Spectrometric Aerosol Measurements during the MINOS Campaign (Crete, August 2001) . Atmos. Chem. Phys. , 4 : 65 – 80 .
- Slowik , J. G. , Stainken , K. Davidovits , P. 2004 . Particle Morphology and Density Characterization by Combined Mobility and Aerodynamic Diameter Measurements. Part 2: Application to Combustion Generated Soot Particles as a Function of Fuel Equivalence Ratio . Aerosol Sci. Technol. , 38 : 1206 – 1222 . [CROSSREF]
- Topping , D. , Coe , H. McFiggans , G. 2004 . Aerosol Chemical Characteristics from Sampling Conducted on the Island of Jeju, Korea during ACE-Asia . Atmos. Environ. , 38 : 2111 – 2123 . [CROSSREF]
- Turpin , B. J. and Lim , H. J. 2001 . Species Contributions to PM2.5 Mass Concentrations: Revisiting Common Assumptions for Estimating Organic Mass . Aerosol Sci. Technol. , 35 : 602 – 610 .
- Weber , R. J. , Orsini , D. , Daun , Y. , Lee , Y. N. , Klotz , P. J. and Brechtel , F. 2001 . A Particle-Into-Liquid Collector for Rapid Measurements of Aerosol Bulk Chemical Composition . Aerosol Sci. Technol. , 35 : 718 – 727 . [CROSSREF]
- Weber , R. J. , Orsini , D. Duan , Y. 2003 . Intercomparison of Near Real-Time Monitors of PM2.5 Nitrate and Sulfate at the EPA Atlanta Supersite . J. Geophys. Res. , 108 ( D7 ) [CROSSREF] [CSA]
- Zhang , Q. , Canagaratna , M. R. , Jayne , J. T. , Worsnop , D. R. and Jimenez , J. L. 2005a . Time and Size-Resolved Chemical Composition of Submicron Particles in Pittsburgh. Implications for Aerosol Sources and Processes . J. Geophys. Res. , 110 : D07S09 [CROSSREF]
- Zhang , X. F. , Smith , K. A. , Worsnop , D. R. , Jimenez , J. , Jayne , J. T. and Kolb , C. E. 2002 . A Numerical Characterization of Particle Beam Collimation by an Aerodynamic Lens-Nozzle System: Part I. An Individual Lens or Nozzle . Aerosol Sci. Technol. , 36 ( 5 ) : 617 – 631 . [CROSSREF]
- Zhang , X. , Smith , K. A. and Worsnop , D. R. 2004 . Characterization of Particle Beam Collimation: Part II Integrated Aerodynamic Lens-Nozzle System . Aerosol Sci. Technol. , 38 ( 6 ) : 619 – 638 . [CROSSREF]
- Zhang , Q. , Alfarra , M. R. , Worsnop , D. R. , Allan , J. D. , Coe , H. , Canagaratna , M. R. and Jimenez , J. L. 2005b . Deconvolution and Quantification of Primary and Oxygenated Organic Aerosols Based on Aerosol Mass Spectrometry . Environ. Sci. Technol. , 39 : 4938 – 4952 . [PUBMED] [INFOTRIEVE] [CROSSREF]
- Ziemann , P. J. , Liu , P. , Rao , N. P. , Kittleson , D. B. and McMurry , P. H. 1995 . Particle Beam Mass Spectrometry of Submicron Particles Charged to Saturation in an Electron Beam . J. Aerosol Sci. , 26 : 745 – 756 . [CROSSREF]