Secondary organic aerosol generated from the photo-oxidation of 1,3,5-trimethylbenzene and α-pinene in a smog chamber was investigated. Fourier Transform Infrared Spectroscopy (FT-IR) was used to monitor the time dependent change of five different functional groups in the aerosol (carboxylic acids, alcohols, organonitrates, ketones/aldehydes═carbonyls, and aliphatic carbon) sampled with an impactor on zinc-selenide discs. Based on model compounds for oxidation products of 1,3,5-trimethylbenzene and α-pinene, calibration factors for the different functional groups were calculated, and relative molar fractions of the functional groups were estimated from the analysis of the FT-IR spectra of the smog chamber samples. We show chemical evolutions of secondary organic aerosol on a time scale of up to 20 h. Time series with up to eight measurements per experiment show a strong increase in the relative amounts of carboxylic acid groups and a moderate increase of alcohol and carbonyl groups, whereas the relative amounts of organonitrates and the aliphatic carbon decrease. These findings support the assumption that the chemical composition of the aerosol continues to change for a long time after the particle formation has considerably slowed down. According to these observed changes with time, average, sum formulas of the molecules in the secondary organic aerosol are suggested.
INTRODUCTION
Hydrocarbons, aromatic and nonaromatic, are abundant pollutants in the atmosphere. The atmosphere is a well known system for transport and deposition of organic compounds (CitationBidleman 1988). Atmospheric aerosols contribute to many processes, such as cloud formation and visibility reduction, and have a significant impact on air quality, climate, and human health. To understand the origin of aerosols and the importance of their different sources, it is necessary to understand the chemical composition of the aerosols. Organic compounds consist of a major fraction of the aerosols. Although hundreds of individual organic compounds have been identified (CitationSaxena and Hildemann 1996), only a minor fraction of the total organic content has been identified on a molecular level (CitationPuxbaum et al. 2000; CitationRogge et al. 1993). Up to 90% of the organic aerosol mass in urban areas is of secondary origin (CitationLim and Turpin 2002); i.e., particles that are not directly emitted but formed during oxidation processes of gaseous precursors in the atmosphere. These gaseous precursors can be of anthropogenic or biogenic origin. The total biogenic, organic emissions are estimated to range up to 1150Tg year−1 (CitationGuenther et al. 1995), which exceed by far the anthropogenic emissions an estimated 103Tg year−1 (CitationSingh and Zimmermann 1992). An estimated average of 18.5Tg year−1 of secondary organic aerosol (SOA) is formed from biogenic precursors (CitationGriffin et al. 1999).
Fourier Transform Infrared (FT-IR) spectroscopy has been used by a number of authors to investigate the chemical composition of organic aerosols. FT-IR is used to determine functional groups in the organic mass rather than individual compounds. It provides information on the overall functional-group composition that helps to characterize the degree of oxidation of SOA. Very little sample mass is needed due to the high sensitivity of FT-IR. Measurements can be directly made from deposited particles without extraction or other sample processing. FT-IR has been used to measure ambient aerosol (CitationBlando et al. 1998; CitationMaria et al. 2002), to detect and/or quantify carbonyl-, aliphatic CH-, and organonitrate functional groups (CitationGarnes and Allen 2002; CitationMylonas et al. 1991), and determine the fraction of inorganic mass in an aerosol (CitationAllen et al. 1994; CitationMaria et al. 2002). CitationRussell (2003) used FT-IR spectra from ambient aerosol to calculate organic mass to organic carbon ratios.
FT-IR has been employed previously for the analysis of functional groups of SOA components in smog chamber experiments (CitationJang and Kamens 2001a), in which FT-IR was used in addition to gas chromatography-mass spectrometry (GC-MS) to confirm the presence of organonitrates in SOA. Heterogeneous reactions involved in SOA formation were also investigated with FT-IR by comparing carbonyl absorbances under different conditions (CitationCzoschke et al. 2003; CitationJang et al. 2002; CitationJang and Kamens 2001b). It was shown that the addition of acidic seed aerosol increases the SOA yield (CitationJang et al. 2002), and that chemical changes in the particle occur over prolonged time scales (CitationKalberer et al. 2004).
Various publications (CitationAllen et al. 1994; CitationDekermenjian et al. 1999a, Citation1999b; CitationGarnes and Allen 2002; CitationHoles et al. 1997; CitationLaurent and Allen 2004; CitationPalen et al. 1992) used FT-IR spectroscopy to calculate molar fractions of various functional groups of organic aerosols—generated in a reaction chamber or from ambient samples. Smog chamber samples focused mainly on carbonyls, which showed that the molar loadings of carbonyl groups were significant. The spectra were interpreted using model compounds and were referenced to the aliphatic C─H absorption. This allows us to estimate the molar loading of functional groups in SOA and to calculate the functional group distribution of average product molecules.
In this study we generated SOA particles in smog chamber experiments from an anthropogenic gaseous precursor, 1,3,5-trimethylbenzene (TMB), and a biogenic gaseous precursor—α-pinene (APIN). The gas-phase oxidation of APIN (CitationJang and Kamens 1999; CitationKamens et al. 1981; CitationYu et al. 1999; CitationZhang et al. 1992) and TMB (CitationCocker et al. 2001; CitationHamilton et al. 2003; CitationKleindienst et al. 1999; CitationSmith et al. 1999; CitationYu et al. 1997) has been well studied in the literature. The methods mostly included filter sampling of the particle phase, and extraction and gas chromatography mass-spectrometry analysis of single compounds.
Using FT-IR spectroscopy, the focus of our study was to determine the temporal changes of a variety of functional groups in the SOA as indicators of an ongoing oxidation in the SOA particles. We present, for the first time, a temporal evolution of five different functional groups in SOA generated in a smog chamber over an extended time period. During the SOA formation in TMB and APIN systems, aerosol-mass growth slows considerably after 4 to 5 h in the smog chamber. However, a slow oxidation of the particle bulk continues over a time period of more than 20 h. We observed a strong increase in carboxylic acids, a moderate increase in alcohols and carbonyls, and a decrease in organonitrates.
EXPERIMENTAL METHOD AND DATA ANALYSIS
Sample Preparation and Measurement
SOA was produced in photo-oxidation experiments performed in the new indoor smog chamber of the Paul Scherrer Institute (PSI), Villigen, Switzerland. The smog chamber facility has been described in detail by CitationPaulsen et al. (2005). Briefly, the chamber consists of a 27 m3 Tedlar bag in a wooden housing, which is equipped with air conditioning. Four xenon arc lamps (4 kW each) are used to simulate the solar-light spectrum as closely as possible and mimic natural photochemistry. The primary gas components such as organics, oxides of nitrogen, purified air, and water vapor are flushed into the chamber where they diffuse and mix for 30 to 45 min before the experiment is started by turning on the lights. In our experiments TMB and APIN are injected into a heated glass tube (85°C) to completely evaporate the liquid while purified air carries the vapor into the chamber. The initial hydrocarbon mixing ratios were 656 and 1312 ppb for TMB (three experiments) and 300 ppb for APIN (two experiments). The NO and NO2 mixing ratios were 160 ppb each (for 656 ppb TMB) and 320 ppb (for 1312 ppb TMB) each for TMB experiments, and 120 ppb for APIN (300 ppb). Propene was used as a radical initiator at mixing ratios of 300 ppb. Relative humidity in the chamber was between 57 and 64%.
Particle formation started about 15 min (for TMB) and 30 min (for APIN) after turning on the lights. The size distribution was measured using a scanning mobility particle sizer (SMPS) system. The decrease of the gas-phase concentration of the hydrocarbons was monitored with gas chromatography—flame ionization detection and proton transfer reaction mass-spectrometry, NO and NO2 were measured via chemiluminescence detection, and ozone was monitored by UV absorption.
The main focus of this work was to monitor the temporal change of the functional groups present in the SOA. Seven to eight samples were collected from the beginning of particle formation up to 22 h after the start of the experiment. SOA was sampled with a 12-stage impactor (CitationMaenhaut et al. 1996) at a flow rate of 11/l min−1 for 50 to 120 min. Zinc-selenide plates (25 mm * 2 mm, Fluka, Switzerland) were used as impaction plates in stages 2, 3, and 4, which correspond to particle sizes of 86–153 nm, 154–231 nm, and 232–343 nm, respectively. Depending on the size of the SOA, we used the corresponding impactor stage. At about 12 h after switching on the light, the sampling interval was extended to 4 h because the particle-number concentration decreased because of wall losses. The size distribution of the aerosol generated in a smog chamber is very narrow and no size dependence of the SOA composition could be observed. Thus, results are not reported as a function of size.
Prior to the experiments ZnSe plates were cleaned in a sonication bath with acetonitrile and methanol. They are transparent to IR radiation and allow for direct analysis after sampling without further treatment. Spectra were taken with a Perkin Elmer Spektrum BX II FT-IR instrument (Software Spektrum, version 5.0.1.). Prior to analysis the sample holder was flushed with N2 to decrease the absorption because of CO2, then 10 scans were taken at a resolution of 2 cm−1 from 4000–600 cm−1 using a clean ZnSe disc as background. The flushing of the spectrometer with N2 could potentially lead to off-gassing of high volatility particle components. However, the fast IR measurement (<1 min) minimizes such artifacts, and repetitive measurements of the same sample did not result in measurable changes in the spectra. Thus, such artifacts are thought to be not significant. The instrument software and a scientific data processing package (Igor Pro, Version 4.09A, WaveMetrics) were used for integration of the peaks. Spectra of the calibration compounds were taken with the same instrument. All spectra of liquid calibration compounds were obtained by directly applying a small amount of liquid between two ZnSe discs, whereas the solid calibration compounds were dissolved in a suitable volatile solvent and dropped onto a single disc where the solvent was allowed to evaporate.
Spectral Analysis
In the current section, a thorough description of the spectral analysis is given explaining the method to obtain the integrated peak areas for interpreting the results in terms of possible uncertainties and intercomparison of various experiments.
The following absorbances of the smog chamber spectra were analyzed: aliphatic carbon CH (2800–3000 cm−1), alcohols OH (3200–3600 cm−1), carboxylic acids COOH (3200–2400 cm−1 and 1670–1870 cm−1), ketones and aldehyde (1670–1870 cm−1), and organonitrates RONO2 (1230–1320 cm−1). Aromatic CH absorbance is often weak around 3000–3100 cm−1 and covered by the broad COOH peak. No such peaks were observed in the TMB sample. Thus, we assumed a negligible amount of aromatic components for the TMB-SOA. Compounds with organonitrate groups show three bands at 1630 (asymmetric stretch vibration), 1280, and 850 cm−1 (both symmetric), and we chose the absorbance at 1280 cm−1 for integration because of its strong intensity and fairly good isolation. Organonitrate and carbonyl peaks were integrated using the FT-IR instrument software. (TMB) and (APIN) show the spectra of SOA from TMB and APIN with the integration baselines for the single peaks at 1280 cm−1 for organonitrates and 1720 cm−1 for C═O.
To differentiate between the C═O absorbance from ketones/aldehydes and from carboxylic acids additional spectral features have to be used because all three functional groups contribute to the strong peak at 1720 cm−1. Furthermore, C═O in acids has a much higher molar absorptivity than C═O in ketones/aldehydes. In the region from 2400 cm−1 to 3600 cm−1 overlapping peaks from carboxylic acids and alcohols were separated using Gaussian curve fits because Lorentzian curve fits resulted in much less agreement with the spectra. The O─H stretching vibration at 2400–3200 cm−1 (associated/H-bonded OH), which is due to carboxylic acids, was then used for separation of ketones/aldehydes and carboxylic acids. A distinction between aldehydes and ketones was not possible because the weak C─H absorbance of aldehydes (2720–2820 cm−1) could not be observed in any of the smog chamber spectra. shows the fitted Gaussian peaks underneath the overall fit, resulting in a very good agreement with the measured absorbances. The C─H absorbance between 2800 and 3000 cm−1 was replaced with a linear fit assuming that the C─H band is superimposed on the broad O─H peak and does not contribute to the dominant O─H valence vibration from carboxylic acids in this region. The resulting broad peak originates from alcohols and acids with different hydrogen bonding features. The OH group in alcohols, usually absorbing between 3600 and 3200 cm−1, was assigned to the Gaussian fits 1 and 2 ( and ), and OH in carboxylic acids absorbing mostly below 3200 cm−1 was assigned to fit 3 () and fit 3 and 4 (). For the area from carboxylic acids in APIN, two Gaussian curves were necessary to fit the measured O─H absorption band (3034 cm−1 and 2611 cm−1). The small peak at 2611 cm−1, interpreted as absorption band from associated O─H groups of carboxylic acids, is hardly visible in the TMB spectra and, therefore, the COOH area in TMB spectra could be fitted with one Gaussian curve (3000 cm−1).
FIG. 1 Original FT-IR spectra from 4000–600 cm−1 for SOA generated in the PSI smog chamber from TMB (a) and APIN (b). Lower traces are sections of the spectra from 4000–2000 cm−1 without the aliphatic CH bands. The bold dotted line is the sum of the three (TMB) and four (APIN) Gaussian functions, respectively.
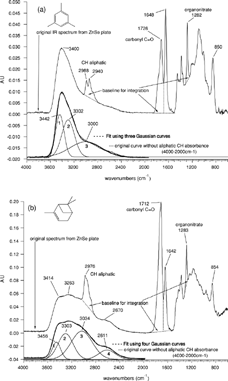
Water present in the particles would also contribute to the OH band leading to an overestimation of acids and alcohols. However, due to the following reasons the water content of the particles during the IR measurements is negligible. Liquid water has an absorption band reaching >3600 cm−1, which is clearly not observed in the SOA spectra ( and ). In addition, particles experience an atmosphere in the spectrometer with a relative humidity close to 0% because of the flushing with N2. Hygroscopic growth measurements for TMB-SOA were performed down to 15% relative humidity for which a hygroscopic growth of only 1.01 was measured (CitationBaltensperger et al. 2005). Extrapolating this value to lower relative humidities—as expected during the measurement—shows that the water content of the SOA particles during the measurement was well below 1%.
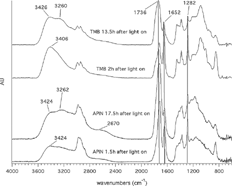
FIG. 2 Comparison of TMB (upper two traces) and APIN-IR spectra at the beginning of the experiment (2 h and 1.5 h, respectively, after turning on the lights) and after 13.5 h and 17.5 h, respectively (the vertical lines at 1726, 1652, and 1282 cm−1 indicate the peaks in all four spectra).
The integrated areas were used to determine the relative mole fractions of five functional groups in the SOA: alcohol group, carboxylic acid group, aliphatic carbon group, organonitrate group, and carbonyl group from ketones and aldehydes. The equations presented below are adapted from CitationPalen et al. (1992) and CitationHoles et al. (1997). Note that the term “mole fraction” here refers to functional groups in a molecule and not entire molecules. The equations relate the integrated areas for each functional group, which is normalized to the CH absorbance area, to the relative molar absorptivities for each functional group derived from calibration compounds.
The relative mole fraction of the organonitrate group is calculated according to Equation (Equation1), (n = number of moles, A = absorbance area of the integrated peak of the SOA spectrum, nc = moles of calibration compound, and Ac = absorbance area from calibration compound).
TABLE 1a Relative molar absorptivities of calibration compounds used for TMB—individual values and average
TABLE 1b Relative molar absorptivities of calibration compounds used for APIN—individual values and standard deviation (SD) for more than two compounds
The relative molar absorptivities in Equation (Equation3a) and (Equation3b) (8.07, 54.01, 7.94, and 13.84) are obtained empirically from the calibration compounds, with an average of aldehydes and ketones because a separation of the two groups was not possible. and present the calibration compounds and the calculated relative molar absorptivities. They compare well with literature values from CitationPalen et al. (1992) and CitationHoles et al. (1997). The calibration compounds were chosen to represent the known oxidation products of TMB and APIN. For example, acetic and formic acid were found to be abundant compounds in TMB-SOA (CitationFisseha et al. 2004). Some known SOA compounds could not be obtained commercially, or did not result in satisfying liquid-phase or solid-phase IR-spectra, therefore surrogates with similar ratios of CH and the functional group of interest were used. and list the individual values for each compound measured. For each functional group with more than two calibration compounds the average values with standard deviations are given. The calibration factors in and were used to calculate the relative mole fractions () of the respective functional groups in the SOA with Equations (Equation1 Equation2 Equation3), assuming that only these five groups are present in the molecule, and the mole fractions add up to one. Note that the term “mole fraction” denotes the ratio of moles of functional groups, not molecules.
TABLE 2 Mole fractions of five functional groups in SOA from TMB and APIN
The relative temporal changes as shown in and are not dependent on the calibration factors because they cancel out (see Equation (Equation1)), except for the CO group. The mole fraction for the carbonyl group depends on the calibration factors of both acids and carbonyls (Equations (Equation2) and (Equation3)). The calibration factors are only relevant for the results shown in and . Thus, these results have to be taken with care because of the partially large variations of the calibration factors in .
FIG. 3 Time-resolved changes of mole fractions of five functional groups present in SOA from TMB (a) and APIN (b).
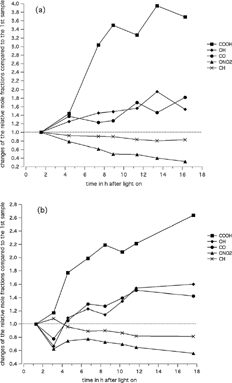
TABLE 3 Functional group distribution in an hypothetical average SOA molecule from TMB and APIN
RESULTS AND DISCUSSION
gives a qualitative overview of the chemical changes of the SOA with time. The ONO2 band at 1282 cm−1 and 1648 cm−1 clearly decreases, whereas the C═O band at 1726 cm−1 increases and broadens. Also the O─H vibration at 2400 to 3600 cm−1 clearly broadens. With increasing time a peak evolves at 2670 cm−1for APIN (Gaussian fit 4 in ), which is not the case for TMB. The APIN spectra also shows an increase of the peak around 3260 cm−1 with time. For TMB-SOA the peak around 3400 cm−1 features a gradual separation into two peaks. The already broad peak around 2900 cm−1 becomes broader with time but no second peak around 2600 cm−1 evolves.
shows the temporal changes of the relative mole fraction of the functional groups C═O (from aldehydes and ketones), OH, COOH, ONO2 and CH, calculated according to Equations (Equation1 Equation2 Equation3). gives the relative mole fractions for 7 and 8 samples as a function of time for the TMB and APIN experiment, respectively, as shown in . Note that the mole fractions relate to the relative abundance of bonds of the respective functional group replacing C─H bonds in a molecule and that the five mole fractions add up to one.
For TMB ( and ) an increase of the abundance of COOH, C═O and OH is measured, which is in agreement with the photo-oxidation products found by GC-MS for TMB in the aerosol phase (CitationYu et al, 1997; CitationCocker et al. 2001). According to , the mole fractions of the carboxylic acid group show a clear increase. The mole fractions of the samples start with less than 1% (see ) at the beginning and reads about 4% in the last sample. The alcohol and carbonyl functional groups also increase with time, yet to a smaller extent. Because the carboxylic acid, alcohol, and carbonyl functional groups increase with time, the CH group decreases accordingly.
The main oxidation pathway of TMB is via the addition of a hydroxyl radical to the aromatic ring to form an alkylated hydroxycyclohexadienyl radical (OH-aromatic adduct). The OH-aromatic adducts react predominantly with O2 leading mainly to ring-opened products (CitationAtkinson 1998). Only about 3% of the initial oxidation starts with an H abstraction from one of the methyl groups maintaining the aromaticity (CitationAtkinson 1994). However, it is likely that part of these first generation aromatic-oxidation products also undergo ring cleavage at a later stage. Therefore, it can be expected that the aromatic content in aged SOA (i.e., after 2 to 3 h) is likely very small, which is supported by the lack of aromatic CH bonds in the IR spectra of TMB-SOA. The trends described here are in agreement with other TMB experiments performed under the same conditions (not shown here).
In APIN-SOA (b and ) the carboxylic acid mole fraction more than doubles over the 22 h of the experiment (from 3% to 8%, ), which is a smaller increase than for TMB. The significance of carboxylic acids in APIN-SOA was also reported by others (CitationJang and Kamens 1999; CitationYu et al. 1999). The ketone/aldehyde and the alcohol fraction increase by a factor of about 1.5, which is similar to the TMB-SOA. Other authors found that gas-phase carbonyls are the major reaction products in APIN oxidation (CitationGlasius et al. 2000; CitationYu et al. 1999; CitationTolocka et al. 2004) with all ring-opening products because ozonolysis of the double bond leads to carbonyls. However, in SOA only low concentrations were found. The ONO2 and aliphatic CH groups decrease in APIN at about the same rate as in TMB-SOA. For both SOA types the increase in COOH is the highest, but major changes are also observed for the carbonyl and OH functional groups.
In the relative mole fractions from are converted into an average amount of each functional group in a hypothetical molecule, and respective sum formula are given. For TMB, an average molecular structure was assumed with 6 aliphatic carbons and a maximum of 14 C─H groups as well as assuming ring-cleavage reaction products. C6 molecules are abundant oxidation products of TMB (see CitationKleindienst et al. 1999). For APIN, hypothetical oxidation molecules with a four-membered ring after ozonolysis of the double bond were assumed, which resulted in a molecule with 10 carbon atoms and at most 15 C─H bonds.
For TMB, a major increase with time is observed for carbonyls, alcohols, and carboxylic acids, with carbonyls and alcohols as dominating groups (about 2 carbonyl and alcohol groups per molecule in a TMB-C6 molecule). In a recent publication by CitationFisseha et al. (2004) analyzing TMB-SOA with ion chromatography combined with mass spectrometry, an acid contribution up to 45% of the total aerosol mass was observed. Fisseha et al's experiments show a relative increase of the total acid fraction of about a factor of 1.5 in the first 7.5 h of the experiment. Our data compare reasonably well, as we observe an increase of the acid mole fraction by a factor of three in the first 7.5 h. This translates for TMB-C6 molecules into an increase in carboxylic acids from one in ten molecules (e.g., V1 in ) to 4 in 10 molecules (e.g., V7 in ) corresponding to an acid concentration of about 40%, which compares well with the results from CitationFisseha et al. (2004).
The SOA from APIN is also dominated by carbonyl and OH groups, although slightly fewer alcohols are present than in TMB. The number of COOH functional groups more than doubled during the experiment. CitationAlfarra et al. (2004) showed, in a recent study with an aerosol mass-spectrometer, that the relative intensity of a mass fragment, assigned to carboxylic acids, increases significantly during the 8 h of the experiment. They also observed a substantially larger increase of acids in TMB-SOA compared to APIN experiments. Spectra from both compounds were dominated by mass fragments assigned to carbonyls and carboxylic acids. Generally, the studies of CitationFisseha et al. (2004) and CitationAlfarra et al. (2004) are in agreement with the main results reported here.
In and no error bars for the mole fractions are indicated. Equations (Equation1 Equation2 Equation3) show that the calculated mole fractions depend directly on the model calibration compounds. CitationPalen et al. (1992) report for a model compound of the same carbon number a 25% difference in molar absorptivity. This generates an uncertainty of 10 to 20% in SOA composition based on FT-IR measurements. Not using the actual aerosol product, but model compounds creates additional uncertainties. According to Equations (Equation1 Equation2 Equation3), and using the calibration factors in , and shows the changes of the relative mole fractions of all functional groups with time. However, because the first value is set to 1 the calibration factors of do not influence the slope of the curves, except for the mole fraction of the C═O group. For the C═O group the molar absorptivity of the COOH group has to be considered because the peak at 1726 cm−1 is due to C═O from aldehydes, ketones, and carboxylic acids. The accuracy of the calibration factors influences only the absolute numbers of the relative mole fractions () and the average sum formula deduced (), but not the relative changes of the mole fractions shown in and , except for the C═O group.
Three experiments were conducted for TMB. By comparing the trends of the functional groups over 16.3 h (i.e., the running time of the shortest experiment) agreement with can be observed for all experiments. At the end of the experiment, i.e., after 16.3 h, the relative standard deviation ranges between 4 and 39% for the different functional groups (19% for OH, 30% for CO, 4% for CH, 23% for ONO2, and 39% for COOH). For APIN only two experiments were performed, both with very good trend agreement with during 15.3 h (i.e., the running time of the shortest experiment). The values at 15.3 h after the lights were switched on differ only by about 3% for OH, 16% for CO, 4% for CH, 13% for ONO2, and 15% for COOH. In both cases, the COOH and CO values show the largest variation.
Despite these uncertainties FT-IR spectroscopy can be used to determine the distribution of functional groups in the aerosol, especially their relative changes with time during the course of an experiment. By intercomparison of different experiments the above-mentioned uncertainties have to be taken into account, but the observed trends are consistent throughout the experiments.
CONCLUSIONS
This work demonstrates the use of FT-IR spectroscopy to monitor the continuous changes of functional groups of SOA from the photo-oxidation of TMB and APIN in a smog chamber. A time resolution of 2–4 h during 22 h was obtained for five different functional groups (alcohol, organonitrate, carboxylic acid, carbonyl, and aliphatic CH) by integrating their absorbances in the spectra. TMB and APIN both show a strong increase in COOH, carbonyls, and OH groups, while carbonyls and alcohols are the dominant functional groups in the SOA. ONO2 plays a minor role for both SOA types because the low content per average molecule decreases with time. The data show an ongoing oxidation in the SOA over the course of an experiment (up to 22 h). This is in contrast to the aerosol mass evolution, which slows down considerably after about 4 to 5 h. This longterm continuous change of the chemical composition of the particle is not inconsistent with recent studies showing oligomerization processes (CitationKalberer et al. 2004), which also proceed over tens of hours. Heterogeneous oxidation reactions of aerosol components or continuous uptake of highly oxidized compounds from the gas phase could contribute to the observed increase in oxidized functional groups. Incorporation of carbonyl and carboxylic acid functionalities into the oligomers could be a possible reason for their strong and continuous increase. The organonitrate functional group, however, might not take part in the oligomerization process, so that possible losses such as further reactions or desorption into the gas phase could occur.
Acknowledgments
This work was supported by the ETH grant TH-10./01-2, the Swiss National Science Foundation, and the EC project EUROCHAMP. We thank the smog chamber crew at the Paul Scherrer Institute for their help during the experiments.
Notes
a average of 8.07 as used in Equation Equation(3a).
a average of 7.94 as used in Equation (Equation3b).
aaverage of typical APIN oxidation products (CitationGlasius et al. 2000; CitationTolocka et al. 2004).
REFERENCES
- Alfarra , M. R. , Paulsen , D. , Gysel , M. , Garforth , A. A. , Dommen , J. , Prevot , A. S. H. , Worsnop , D. R. , Baltensperger , U. and Coe , H. 2004 . A Mass Spectrometric Study of Secondary Organic Aerosols Formed from the Photooxidation of Anthropogenic and Biogenic Precursors in a Reaction Chamber . Submitted to Environ. Sci. Tech. ,
- Allen , D. T. , Palen , E. J. , Haimov , M. I. , Hering , S. V. and Young , J. R. 1994 . Fourier-Transform Infrared-Spectroscopy of Aerosol Collected in a Low-Pressure Impactor—Method Development and Field Calibration . Aerosol Sci. Technol. , 21 : 325 – 342 . [CSA]
- Atkinson , R. 1994 . Gas Phase Tropospheric Chemistry of Organic Compounds . J. Phys. Chem. Ref. Data , : 1 – 216 . Monograph No. 2
- Atkinson , R. 1998 . Gas-phase Degradation of Organic Compounds in the Troposphere . Pure & Appl. Chem. , 70 : 1327 – 1334 .
- Baltensperger , U. , Kalberer , M. , Dommen , J. , Paulsen , D. , Alfarra , R. , Coe , H. , Fisseha , R. , Gascho , A. , Gysel , M. , Nyeki , S. , Sax , M. , Steinbacher , M. , Prevot , A. , Sjgren , S. , Weingartner , E. and Zenobi , R. 2005 . Secondary Organic Aerosols from Anthropogenic and Biogenic Precursors . Faraday Disc. , doi: 10.1039/b417367h, in press
- Bidleman , T. F. 1988 . Atmospheric Processes—Wet and Dry Deposition of Organic Compounds Are Controlled by Their Vapor Particle Partitioning . Environ. Sci. Technol. , 22 : 361 – 367 . [CSA] [CROSSREF]
- Blando , J. D. , Porcja , R. J. , Li , T. H. , Bowman , D. , Paul , J. L. and Turpin , B. J. 1998 . Secondary Formation and the Smoky Mountain Organic Aerosol: An Examination of Aerosol Polarity and Functional Group Composition During SEAVS . Environ. Sci. Technol , 32 : 604 – 613 . [CSA] [CROSSREF]
- Cocker , D. R. , Mader , B. T. , Kalberer , M. , Flagan , R. C. and Seinfeld , J. H. 2001 . The Effect of Water on Gas-Particle Partitioning of Secondary Organic Aerosol. Part 2 m-Xylene and 1,3,5-Trimethylbenzene Photooxidation Systems . Atmos. Environ. , 35 : 6073 – 6085 . [CROSSREF]
- Czoschke , N. M. , Jang , M. and Kamens , R. M. 2003 . Effect of Acidic Seed on Biogenic Secondary Organic Aerosol Growth . Atmos. Environ. , 37 : 4287 – 4299 . [CROSSREF]
- Dekermenjian , M. , Allen , D. T. , Atkinson , R. and Arey , J. 1999a . FT-IR Analysis of Aerosol Formed in the Ozone Oxidation of Sesquiterpenes . Aerosol Sci. Technol. , 30 : 349 – 363 . [CROSSREF]
- Dekermenjian , M. , Allen , D. T. , Atkinson , R. and Arey , J. 1999b . FT-IR Analysis of Aerosol Formed in the Photooxidation of Naphthalene . Aerosol Sci. Technol. , 30 : 273 – 279 . [CROSSREF]
- Fisseha , R. , Dommen , J. , Sax , M. , Paulsen , D. , Kalberer , M. , Maurer , R. , Höfler , F. , Weingartner , E. and Baltensperger , U. 2004 . Identification of Organic Acids in Secondary Organic Aerosol and the Corresponding Gas Phase from Chamber Experiments . Anal. Chem. , 76 : 6535 – 6540 . [PUBMED] [INFOTRIEVE] [CROSSREF]
- Garnes , L. A. and Allen , D. T. 2002 . Size Distributions of Organonitrates in Ambient Aerosol Collected in Houston, Texas . Aerosol Sci. Technol. , 36 : 983 – 992 . [CROSSREF]
- Glasius , M. , Lahaniati , M. , Calogirou , A. , Di Bella , D. , Jensen , N. R. , Hjorth , J. , Kotzias , D. and Larsen , B. R. 2000 . Carboxylic Acids in Secondary Aerosols from Oxidation of Cyclic Monoterpenes by Ozone . Environ. Sci. Technol. , 34 : 1001 – 1010 . [CSA] [CROSSREF]
- Griffin , R. J. , Cocker , D. R. , Seinfeld , J. H. and Dabdub , D. 1999 . Estimate of Global Atmospheric Organic Aerosol from Oxidation of Biogenic Hydrocarbons . Geophys. Res. Lett. , 26 : 2721 – 2724 . [CSA] [CROSSREF]
- Guenther , A. , Hewitt , C. N. , Erickson , D. , Fall , R. , Geron , C. , Graedel , T. , Harley , P. , Klinger , L. , Lerdau , M. , McKay , W. A. , Pierce , T. , Scholes , B. , Steinbrecher , R. , Tallamraju , R. , Taylor , J. and Zimmerman , P. 1995 . A Global-Model of Natural Volatile Organic-Compound Emissions . J. Geophys. Res. , 100 : 8873 – 8892 . [CSA] [CROSSREF]
- Hamilton , J. F. , Lewis , A. C. , Bloss , C. , Wagner , V. , Henderson , A. P. , Golding , B. T. , Wirtz , K. , Martin-Reviejo , M. and Pilling , M. J. 2003 . Measurements of Photo-Oxidation Products from the Reaction of a Series of Alkyl-Benzenes with Hydroxyl Radicals During Exact Using Comprehensive Gas Chromatography . Atmos. Chem. Phys. , 3 : 1999 – 2014 .
- Holes , A. , Eusebi , A. , Grosjean , D. and Allen , D. T. 1997 . FT-IR Analysis of Aerosol Formed in the Photo-oxidation of 1,3,5-Trimethylbenzene . Aerosol Sci. Technol. , 26 : 516 – 526 .
- Jang , M. and Kamens , R. M. 1999 . Newly Characterized Products and Composition of Secondary Aerosols from the Reaction of Alpha-Pinene with Ozone . Atmos. Environ. , 33 : 459 – 474 . [CROSSREF]
- Jang , M. S. and Kamens , R. M. 2001a . Characterization of Secondary Aerosol from the Photooxidation of Toluene in the Presence of NOx and 1-Propene . Environ. Sci. Technol. , 35 : 3626 – 3639 . [PUBMED] [INFOTRIEVE] [CSA] [CROSSREF]
- Jang , M. S. and Kamens , R. M. 2001b . Atmospheric Secondary Aerosol Formation by Heterogeneous Reactions of Aldehydes in the Presence of a Sulfuric Acid Aerosol Catalyst . Environ. Sci. Technol. , 35 : 4758 – 4766 . [PUBMED] [INFOTRIEVE] [CSA] [CROSSREF]
- Jang , M. S. , Czoschke , N. M. , Lee , S. and Kamens , R. M. 2002 . Heterogeneous Atmospheric Aerosol Production by Acid-Catalyzed Particle-Phase Reactions . Science , 298 : 814 – 817 . [PUBMED] [INFOTRIEVE] [CROSSREF]
- Kalberer , M. , Paulsen , D. , Sax , M. , Steinbacher , M. , Dommen , J. , Prevot , A. S. H. , Fisseha , R. , Weingartner , E. , Frankevich , V. , Zenobi , R. and Baltensperger , U. 2004 . Identification of Polymers as Major Components of Atmospheric Organic Aerosols . Science , 303 : 1659 – 1662 . [PUBMED] [INFOTRIEVE] [CROSSREF]
- Kamens , R. M. , Jeffries , H. E. , Gery , M. W. , Wiener , R. W. , Sexton , K. G. and Howe , G. B. 1981 . The Impact of Alpha-Pinene on Urban Smog Formation—an Outdoor Smog Chamber Study . Atmos. Environ. , 15 : 969 – 981 . [CROSSREF]
- Kleindienst , T. E. , Smith , D. F. , Li , W. , Edney , E. O. , Driscoll , D. J. , Speer , R. E. and Weathers , W. S. 1999 . Secondary Organic Aerosol Formation from the Oxidation of Aromatic Hydrocarbons in the Presence of Dry Submicron Ammonium Sulfate Aerosol . Atmos. Environ. , 33 : 3669 – 3681 . [CROSSREF]
- Laurent , J. P. and Allen , D. T. 2004 . Size Distributions of Organic Functional Groups in Ambient Aerosol Collected in Houston, Texas . Aerosol Sci. Technol. , 38 : 82 – 91 . [CROSSREF]
- Lim , H. J. and Turpin , B. J. 2002 . Origins of Primary and Secondary Organic Aerosol in Atlanta: Results of Time-Resolved Measurements During the Atlanta Supersite Experiment . Environ. Sci. Technol. , 36 : 4489 – 4496 . [PUBMED] [INFOTRIEVE] [CSA] [CROSSREF]
- Maenhaut , W. , Hillamo , R. , Makela , T. , Jaffrezo , J. L. , Bergin , M. H. and Davidson , C. I. 1996 . A New Cascade Impactor for Aerosol Sampling with Subsequent Pixe Analysis . Nucl. Instrum. Methods Phys. Res. B , 109 : 482 – 487 .
- Maria , S. F. , Russell , L. M. , Turpin , B. J. and Porcja , R. J. 2002 . FT-IR Measurements of Functional Groups and Organic Mass in Aerosol Samples over the Caribbean . Atmos. Environ. , 36 : 5185 – 5196 . [CROSSREF]
- Mylonas , D. T. , Allen , D. T. , Ehrman , S. H. and Pratsinis , S. E. 1991 . The Sources and Size Distributions of Organonitrates in Los Angeles Aerosol . Atmos. Environ. , 25 : 2855 – 2861 .
- Palen , E. J. , Allen , D. T. , Pandis , S. N. , Paulson , S. E. , Seinfeld , J. H. and Flagan , R. C. 1992 . Fourier-Transform Infrared-Analysis of Aerosol Formed in the Photooxidation of Isoprene and Beta-Pinene . Atmos. Environ. , 26 : 1239 – 1251 .
- Paulsen , D. , Dommen , J. , Kalberer , M. , Prévôt , A. S. H. , Richter , R. , Sax , M. , Steinbacher , M. , Weingartner , E. and Baltensperger , U. 2005 . Secondary Organic Aerosol Formation by Irradiation of 1,3,5-Trimethylbenzene-NOx-H2O in a New Reaction Chamber for Atmospheric Chemistry and Physics . Environ. Sci. Tech. , 39 : 2668 – 2678 . [CSA] [CROSSREF]
- Puxbaum , H. , Rendl , J. , Allabashi , R. , Otter , L. and Scholes , M. C. 2000 . Mass Balance of the Atmospheric Aerosol in a South African Subtropical Savanna (Nylsvley, May 1997) . J. Geophys. Res. , 105 : 20697 – 20706 . [CSA] [CROSSREF]
- Rogge , W. F. , Mazurek , M. A. , Hildemann , L. M. , Cass , G. R. and Simoneit , B. R. T. 1993 . Quantification of Urban Organic Aerosols at a Molecular-Level: Identification, Abundance and Seasonal-Variation . Atmos. Environ. , 27 : 1309 – 1330 .
- Russell , L. M. 2003 . Aerosol Organic-Mass-to-Organic-Carbon Ratio Measurements . Environ. Sci. Technol. , 37 : 2982 – 2987 . [PUBMED] [INFOTRIEVE] [CSA]
- Saxena , P. and Hildemann , L. M. 1996 . Water-Soluble Organics in Atmospheric Particles: A Critical Review of the Literature and Application of Thermodynamics to Identify Candidate Compounds . J. Atmos. Chem. , 24 : 57 – 109 . [CSA] [CROSSREF]
- Singh , H. B. and Zimmermann , P. B. 1992 . “ Atmospheric Distribution and Sources of Nonmethane Hydrocarbons ” . In Gaseous Pollutans, Characterization and Cycling , Edited by: Nriagu , J. O. 177 – 236 . New York : John Wiley & Sons .
- Smith , D. F. , Kleindienst , T. E. and McIver , C. D. 1999 . Primary Product Distributions from the Reaction of OH with m-, p-Xylene, 1,2,4- and 1,3,5-Trimethylbenzene . J. Atmos. Chem. , 34 : 339 – 364 . [CSA] [CROSSREF]
- Tolocka , M. P. , Jang , M. , Ginter , J. M. , Cox , F. J. , Kamens , R. M. and Johnston , M. V. 2004 . Formation of Oligomers in Secondary Organic Aerosol . Environ. Sci. Technol. , 38 : 1428 – 1434 . [PUBMED] [INFOTRIEVE] [CSA] [CROSSREF]
- Yu , J. Z. , Jeffries , H. E. and Sexton , K. G. 1997 . Atmospheric Photooxidation of Alkylbenzenes. Part 1: Carbonyl Product Analyses . Atmos. Environ. , 31 : 2261 – 2280 . [CROSSREF]
- Yu , J. Z. , Cocker , D. R. , Griffin , R. J. , Flagan , R. C. and Seinfeld , J. H. 1999 . Gas-Phase Ozone Oxidation of Monoterpenes: Gaseous and Particulate Products . J. Atmos. Chem. , 34 : 207 – 258 . [CSA] [CROSSREF]
- Zhang , S. H. , Shaw , M. , Seinfeld , J. H. and Flagan , R. C. 1992 . Photochemical Aerosol Formation from Alpha-Pinene and Beta-Pinene . J. Geophys. Res. , 97 : 20717 – 20729 . [CSA]