Sintering rates of pristine and V-doped TiO 2 were obtained using a tandem DMA system. A range of experiments were conducted to first map out the variation of mobility size of a monodisperse (by mobility) agglomerate with time at three fixed temperatures. Using relationships of the surface area to the mobility size, the sintering equation was solved to determine the activation energy and pre-exponential factor. The value of the activation energy was 236 (± 46) kJ/mol for pristine TiO 2 and 363 (± 1) kJ/mol for V-doped TiO 2 . The corresponding pre-exponential factors were 7.22 × 10 19 and 2.22 × 10 12 s/m 4 K, respectively. These values were then used to predict changes in mobility diameter at different temperatures, and good agreement was obtained with measurements. Possible reasons for faster sintering rates of V-TiO 2 relative to pristine TiO 2 were conjectured.
INTRODUCTION
Ceramic powders such as titanium dioxide, zinc oxide, and aluminum oxide are produced via gas phase processes on the order of several million tons per year (CitationLayman 1995). For example, the rutile phase of titanium dioxide is a major ingredient of pigmentary material due to its hiding power. The anatase phase of titanium oxide is a photocatalyst and has applicability for remediation of contaminated air and water streams (CitationFujishima et al. 2000). Nanostructured forms of TiO2 are being touted for use as a self-cleaning coating (CitationWatanabe et al. 1999), as a sorbent for toxic metal emissions control (CitationBiswas and Wu 1998), synthesis of partial oxygenates (CitationSahle-Demessie et al. 1999), and in various doped forms for activation by visible light for hydrogen production by photo-splitting water (CitationFujihara et al. 1998).
In addition to the crystallinity (rutile and anatase), size is also an important parameter for optimal functionality. For example, near monodisperse rutile titanium dioxide of 250 nm is considered to be optimal for pigmentary applications (CitationPratsinis 1998), and particles in the 25 to 40 nm are considered optimal for photooxidation of organic compounds such as phenol (CitationAlmquist and Biswas 2002). Dopants are added to the TiO2 matrix for a variety of reasons such as altering the band gap energies (CitationWang et al. 2001) and for controlling crystallinity (CitationAkhtar et al. 1994). Dopants in trace quantities have been observed to alter the primary particle sizes, for example, addition of 1.3% vanadium to TiO2 during synthesis in a furnace reactor resulted in an increase in the primary particle size (from 10 nm to 20 nm, approximately) of the agglomerates (CitationNamiki et al. 2005). Similar trends have been also observed by CitationPaul (1996) for V-doped TiO2, however in doping with Al, a decreased rate of growth was observed. CitationHelble and Sarofim (1989) reported that the primary particle size increased from 11.4 nm to 26.0 nm on addition of sodium during synthesis of silicon due to faster sintering as a result of reduced viscosity.
The resulting primary particle size and agglomerate size are a function of processing conditions and aerosol dynamic phenomena. Sintering is also an important growth process that affects the size of the primary particles (CitationFriedlander 2000). There have been many studies on the sintering of pristine titanium dioxide. CitationAstier and Vergnon (1976) examined the sintering of 100 nm sized titanium dioxide particles and calculated solid state diffusion coefficients. CitationKobata et al. (1991) defined a sintering time as the time required for the ratio of the neck length to initial particle radius to reach 0.83, based on the model of CitationCoblenz et al. (1980). CitationSeto et al. (1995) used a tandem differential mobility analyzer (TDMA) set up to study the sintering of monodisperse agglomerates based on its mobility. Using the characteristic sintering time expression proposed by CitationKobata et al. (1991), they obtained good agreement with the sintering equation proposed by Koch and Friedlander (1990). CitationYang and Biswas (1997) used light scattering to study alterations of agglomerates in flames and established the change in fractal dimension to that in the normalized surface area. Several other researchers have incorporated sintering rate expression into aerosol growth models and studied evolution of the size distributions (CitationXiong and Pratsinis 1993; CitationSeto et al. 1997).
While there have been many studies on determining the sintering characteristic time for pristine titanium dioxide, there are none for doped titanium dioxide. In this paper, a tandem differential mobility analyzer has been used to determine the characteristic sintering time of V-doped TiO2 nanoparticles. A combination of varying temperature and residence time experiments were conducted to determine the pre-exponential term and activation energy of the sintering rate expression.
EXPERIMENTAL
The schematic diagram of the system to determine the sintering rates of pristine and doped TiO2 is shown in . It consists of a furnace aerosol reactor (FuAR) to synthesize nanostructured agglomerates of titania and V-doped titania. An alumina reactor, 91.4 cm in length and 2.54 cm in diameter, is placed in a temperature controlled furnace (Thermolyne, Type 21100) maintained at 750°C. Nitrogen at a flow rate of 0.1 lpm was bubbled (25 ml, Ace glass impinger) through titanium tetraisopropoxide (TTIP, C12H28O4Ti, Aldrich) maintained at 80°C, and introduced into the furnace to produce pristine TiO2 particles. Particle free air at a flow rate of 4.4 lpm was also introduced to oxidize the precursor to produce the nanostructured titanium dioxide agglomerates. A 1090 cm3 growth chamber was used to provide additional residence time to grow agglomerates to 100 nm and increase the concentration in this size regime to approximately 105 particles/cm3. A lower flow rate of air could have been used to increase the residence time in the furnace reactor, however, this was not done as it resulted in an increase in the primary particle size. V-doped titanium dioxide particles were produced by introducing a vanadium precursor in conjunction with the TTIP vapor. Vanadyl acetylacetonate (C10H14O5V, Aldrich) powder was dissolved in methanol and then diluted in water. Particle free air at a flow rate of 1.4 lpm was used to atomize (TSI, model 3076) this solution, followed by a diffusion drier and then introduced into the furnace.
FIG. 1 Schematic diagram of tandem DMA experimental system for determining the characteristic sintering time.
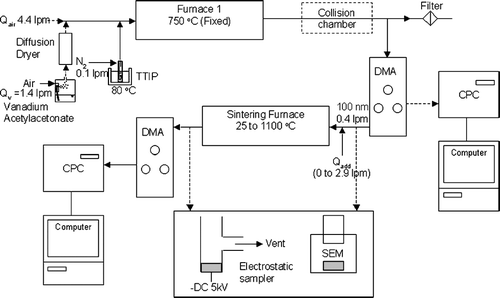
The as produced pristine or V-doped TiO2 was inlet into a differential mobility analyzer (TSI, Model 3071) to classify the aerosol and select a mobility diameter of 100 nm. Particle concentrations at the outlet of the classifier were on the order of 104 cm−3 and they all carried a positive charge; hence, collisional growth did not occur in the second reactor. The second reactor (same dimensions as the first) was heated by a furnace (Lindberg, Model HTF55342C) to temperatures varying from 25 to 1100°C. Additional particle free air was added to vary the residence time from 1.29 to 10.7 s. At the outlet of the second reactor, a scanning mobility particle sizer (SMPS, TSI, Model 3936) was used to measure the mobility equivalent size distribution of particles. Particles were also collected on a TEM grid (Ted Pella) by an electrostatic sampler and analyzed by a scanning electron microscope (Hitachi, S-4500).
The experimental plan consists of a series of tests for the pristine and vanadium doped titanium dioxide (). The initial tests (Tests 1, 2, and 3 for pristine TiO2; and Tests 5, 6, and 7 of V-doped TiO2) were conducted at three different temperatures with varying residence time to determine the activation energy and pre-exponential factor in the sintering rate expression. Experiments were then conducted over a range of temperatures at a fixed residence time (Test 4 for TiO2, Test 8 for V-doped TiO2) to compare the predicted diameter change with the measured values to validate the sintering rate expression.
TABLE 1 Experimental conditions for estimating the characteristic sintering time for pristine and vanadium doped titanium dioxide.
RESULTS AND DISCUSSIONS
Sintering Rates
Sintering resulting in coalescence of particles occurs via three primary routes; evaporation-condensation, viscous flow for liquids and solid-state diffusion. Sintering by evaporation-condensation is a viable pathway for low melting point materials. Ceramic oxides such as titanium dioxide have a high melting point (∼ 1830 °C) and thus sintering proceeds primarily by solid-state phenomena such as volume, grain boundary and surface diffusion (CitationJohnson 1969). Sintering rate expressions have been derived by examining neck radius growth and shrinkage of particles. CitationHiram and Nir (1983) used a viscous flow coalescence model and found that the neck radius, x, approaches its final value, x final (the radius of the resulting sphere) exponentially:
where τ is the characteristic sintering time. This equation is valid in the latter stages of sintering, after x has increased by 10–15% from its initial value of zero. By assuming that the remaining parts of the coalescing particles retain their spherical shape, an expression for the change of the surface area, a was proposed by Koch and Friedlander (1990). For long times (t > > τ),
Initial Sintering Rates
Researchers (CitationGerman and Munir 1976; CitationYadha and Helble 2004) have indicated that initial sintering rates may be faster than that at latter stages. To examine whether this would be significant, an approach has been developed to study this. First, characteristic time expressions have to be determined for the different stages of sintering. CitationCoblenz et al. (1980) developed an expression for the neck radius variation with time for various models such as surface, grain boundary and volume diffusion. As surface and volume diffusion are important in the initial and final stages, the expression assuming grain boundary diffusion is applicable for a broader range.
where x is the neck radius (m), γ is the surface tension (N m−1), Ω is the atomic volume (m3), r is the particle radius (m), k is the Boltzmann constant (J K−1), T is the absolute temperature (K), t is time (s), b is the grain boundary width (m) and D b is the grain boundary diffusion coefficient (m2 s− 1). The two stages of sintering can be derived based on the value of x/r initial sintering is defined for x/r from 0 to 0.3; and latter stages for x/r to 0.83. Substituting these values in Equation (Equation3),
where τ i and τ f are the characteristic times (s) for initial and final sintering, respectively.
To examine the effect of the two stages of sintering on the surface area change, the following equations are used
where a1 is the surface area that decreases in the initial sintering stage; and
where a2 is the surface area that decreases in the final sintering stage. Thus, it is assumed that there are two fractions of the surface area that follow sintering expressions (Equation5) and (Equation6). Based on an approach similar to CitationGerman (1996), the initial values of a1 and a2 are:
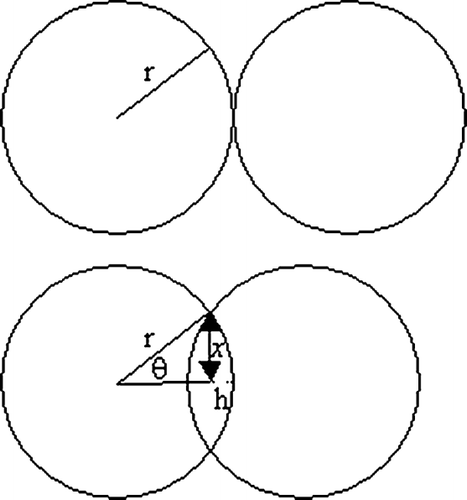
where f is the fraction of surface area for final sintering. One way of determining f is based on the number of contacts (Nc) of the primary particles (), similar to the concept of coordination number (CitationFriedlander 2000). Using geometrical considerations, when x/r = 0.3, θ i is equal to 17.5°, and h is 0.046r (). The area of the spherical cap (= 2π rh) is the area that has undergone initial sintering. Hence 1-f is the ratio of the spherical cap area to the total surface area; or f = 0.977. Using similar geometric considerations, f is calculated to be 0.956, 0.934, and 0.913 for Nc = 2, 3, and 4, respectively; and these values are similar to those reported by CitationGerman (1996).
FIG. 2a Diagram of two equal sized spheres (1) before sintering and (2) during initial sintering.
Equations (Equation5) and (Equation6) are solved with initial conditions (Equation [Equation7]) to determine the area of two sintering titanium dioxide particles, a (= a1+ a2) as a function of time. Values of τ i and τ f (Equation [Equation4]) are determined using property values for titanium dioxide and bD b from CitationAstier and Vergnon (1976). The characteristic times are a function of the particle radius which varies with time and is estimated by r = 3 V /a. illustrates the results for two 10 nm particles undergoing sintering with f = 0.977. There is no discernible difference in the surface area predicted by the two-stage model (Equations [Equation5] and [Equation6]) compared to a single stage model (Equation [Equation2]). The maximum difference in the surface area is 2.12% at a very short time (t = 0.5 ms).
FIG. 2b Surface area decrease for agglomerate consisting of two primary particles (d p = 10 nm) at 1273 K with time for single and two-stage sintering models.
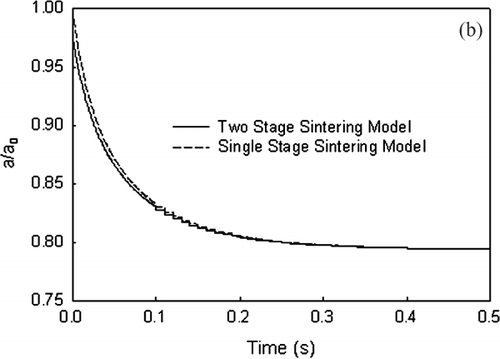
These calculations were carried out without collisional growth. If collisional growth occurs, it maybe conjectured that “fresh” contacts are established and the faster initial rates may be important and result in larger differences in predicted surface areas. Calculations using a monodisperse model (CitationCho 2005) for sintering silica particles with system conditions similar to Kruis et al. (1993) have demonstrated no impact in the total surface area variation as a function of time; with a maximum difference of 1.03% difference at 1 ms and 1273 K. Therefore, in this work, Equation (Equation2) is used for the analysis of the data sets.
Estimation of Surface Area From Mobility Diameter
The objective is to use Equation (Equation2) with experimental data to obtain values of the characteristic sintering time for pristine and V-doped TiO2. Real-time instruments such as differential mobility analyzers provide mobility equivalent diameters. Therefore, a relationship of the surface area of the agglomerate to the mobility equivalent diameter has to be used. Furthermore, a procedure to solve the sintering equation has to be developed.
In the continuum regime, the mobility diameter scales with the radius of gyration if the number of primary particles is sufficiently large and the fractal dimension is larger than 1.3 (CitationKasper et al. 1985; CitationWiltzius 1987; CitationRogak and Flagan 1990). In the free molecular regime, the ratio of mobility diameter of a chain aggregate to the diameter (dA) of the sphere with the same projected area is 1.03 for N > > 1 (CitationChan and Dahneke 1981). CitationRogak et al. (1993) found that the mobility diameter of titanium dioxide was almost the same as the diameter of the sphere with the same projection area for agglomerates with fractal dimension of 2, and particle size less than 400 nm. The projected area equivalent diameter was found to be within ten percent of the mobility diameter for various shapes of titanium dioxide agglomerates produced in furnace reactors at temperatures ranging from 800 to 1300°C (CitationOh et al. 2004). Therefore, the mobility diameter can be assumed to be almost the same as the diameter of the sphere with the same projected area in the free molecular regime regardless of the number of primary particles and fractal dimension of the agglomerate. In this work, the relationship proposed by CitationShimada et al. (1994) is used to estimate the surface area from the measured mobility diameter.
where the subscript 0 represents the initial value, a p is projection area and d m is mobility diameter.
Procedure to Solve the Sintering Equation: Determination of E and A1
Assuming that the grain boundary diffusion coefficient has an exponential dependence on temperature (D b = D0exp(-E/RT), where D0 is a pre-factor (m2 s− 1)), the characteristic sintering time (Equation [Equation4b]) can be expressed as
where B(T) = A
1
Texp(E/RT), A
1 is the pre-exponent term (= 1.06 × 10− 4
k /(bD
0γ Ω)), and E is the activation energy. The initial experiments are carried out at fixed T, and varying time. On differentiation of Equation (Equation8), d(a) = 2a
0
![]() d(d
m
). Substituting this expression in equation (Equation2), τ (from Equation Equation9), a (from Equation [Equation8]), d
p
=
d(d
m
). Substituting this expression in equation (Equation2), τ (from Equation Equation9), a (from Equation [Equation8]), d
p
= ![]() , setting x = d
m
/d
f
, and integrating from time 0 to t,
, setting x = d
m
/d
f
, and integrating from time 0 to t,
FIG. 3 SEM pictures of pristine titanium dioxide at various sintering furnace temperatures: (1) room temperature, (2) 1000°C, and (3) 1100°C and vanadium doped titania at various sintering furnace temperatures: (4) room temperature, (5) 900°C, and (6) 1100°C.

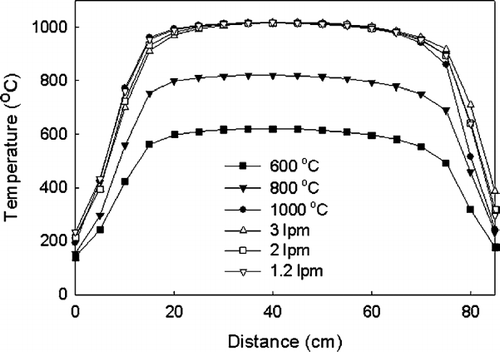
An accurate estimate of the residence time is required to calculate the RHS of Equation (Equation11). Measured temperature profiles shown in (at a 1000°C set point for four different flow rates) are used to determine the density of the gases for calculation of the residence time. A radiation correction term was used for the temperature measurement, however, it is negligible for in-furnace measurements. The fluid (gas) temperature is given by T
f
= T
t
+ ![]() (T
t
4 − T
∞
4) where T
t
is the measured thermocouple temperature, σ is Stefan-Boltzmann constant (= 5.67 × 10− 8 W m− 2 K− 4), ϵ is emissivity of thermocouple (∼ 0.2), d is the diameter of thermocouple bead (= 1 mm), k
f
is the thermal conductivity of air (= 8.19 × 10− 2 W m− 1 K− 1 at 1273 K), and T
∞ is the wall temperature (measured in the furnace control system). For T
t
equal to 1278 K and T
∞ is 1273 K, the fluid temperature is only 2.87 K higher than the measured thermocouple temperature. Based on the temperature profile, a length of 60 cm was used for calculation of the residence time. The errors due to the end effects are expected to be negligible as the sintering rate has an exponential dependence on temperature.
(T
t
4 − T
∞
4) where T
t
is the measured thermocouple temperature, σ is Stefan-Boltzmann constant (= 5.67 × 10− 8 W m− 2 K− 4), ϵ is emissivity of thermocouple (∼ 0.2), d is the diameter of thermocouple bead (= 1 mm), k
f
is the thermal conductivity of air (= 8.19 × 10− 2 W m− 1 K− 1 at 1273 K), and T
∞ is the wall temperature (measured in the furnace control system). For T
t
equal to 1278 K and T
∞ is 1273 K, the fluid temperature is only 2.87 K higher than the measured thermocouple temperature. Based on the temperature profile, a length of 60 cm was used for calculation of the residence time. The errors due to the end effects are expected to be negligible as the sintering rate has an exponential dependence on temperature.
FIG. 4 Temperature profile in the sintering furnace at various set point temperatures (600, 800, 1000 C, 1.2 lpm) and flowrates (for a set point of 1000°C).
The overall approach is thus summarized as follows: a plot of the left-hand side (LHS) of Equation (Equation11) as a function of the residence time yields a slope that is equal to 1/2B(T)d f 4. The slope, –1/2B(T)d f 4 is thus determined for the experiments at different temperatures. A plot of ln(2B(T)d f 4/T) versus 1/RT then yields E as the slope and ln(2d f 4 A 1) as the intercept.
is a plot of the measured mobility diameter variation with time at three different temperatures for pristine TiO2 and V-doped TiO2. As discussed earlier, this data is used to calculate the LHS of (11) and is plotted as a function of the residence time for the three different temperatures in . The slope of the best fit line is equal to –1/2Bd f 4, and values are obtained for three temperatures for both pristine and vanadium doped titanium dioxide. The slope at time close to 0 appears to be greater, probably due to the faster initial sintering rates which are not accurately represented by the Koch and Friedlander (1990) expression (CitationYadha and Helble 2004). However, in this work, the slopes are inferred from the later stages of sintering and the use of the Koch and Friedlander expression (1990) is justified.
FIG. 5a Mobility particle size change of pristine titanium dioxide with time at three different temperatures: (a) 1000°C, (b) 1025°C, and (c) 1050°C.
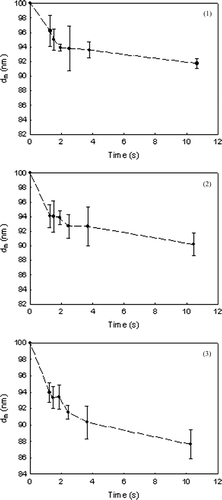
FIG. 5b Mobility particle size change of vanadium doped titanium dioxide with time at three different temperatures: (a) 900°C, (b) 950°C, and (c) 1000°C.
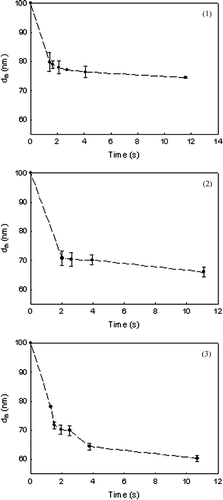
FIG. 6a Plot of LHS on time of equation (Equation11) for pristine titanium dioxide at three temperatures of (1) 1000°C, (2) 1025°C, and (3) 1050°C. The slope is equal to −1/2Bd f 4.
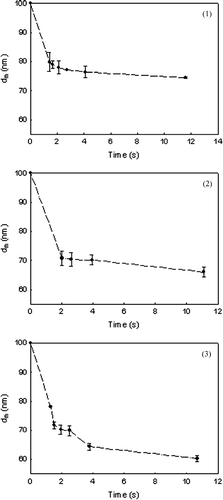
FIG. 6b Plot of LHS on time of equation (Equation11) for V doped titanium dioxide at three temperatures of (1) 900°C, (2) 950°C, and (3) 1000°C. The slope is equal to −1/2Bd f 4.
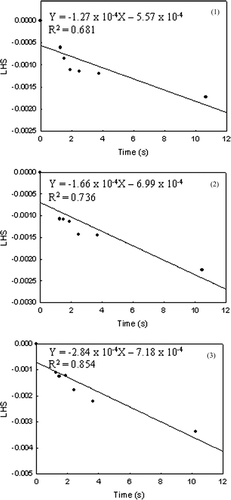
Using the value of B and d f , ln (2Bd f 4/T) is plotted versus 1/RT in . The activation energy for pristine TiO2 is estimated to be 236 kJ/mol (). This value is in a similar range of values reported by earlier researchers (CitationAstier and Vergnon 1976; CitationYang and Biswas 1997). To examine the impact of different d f (assumed to be 55.6 nm for pristine TiO2 same as that of V-TiO2), this was varied and estimates of E determined. Though the primary particle size is not clearly evident from , synthesis reactor conditions were selected that yield a smaller primary particle size for pristine TiO2 (CitationNamiki et al. 2005). Thus, for the same mobility diameter, the final size, d f , is expected to be smaller. Hence, the calculations were repeated for d f = 50 and 40 nm for the pristine TiO2 case. The activation energies (E) estimated were 234 and 231 kJ/mol for d f = 50 and 40 nm, respectively, and this is not very sensitive to the value of d f .
FIG. 7 Plot of ln(2Bd f 4/T) (= y) with 1/RT (= x) to obtain the activation energy (slope, J mol− 1) and the pre-exponential factor (exp(intercept)/2d f 4, s nm− 4 K− 1) for (a) pristine TiO2 and (b) V-TiO2.
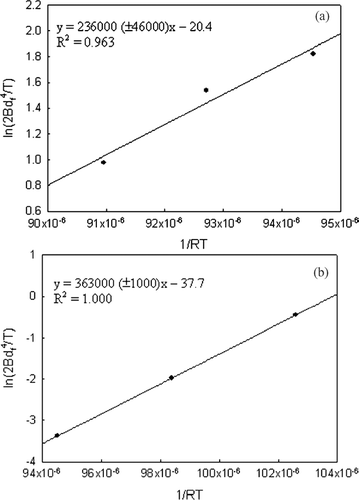
TABLE 2 Comparison of values of the sintering activation energy and pre-exponential factor for pristine (this work and literature reported) and vanadium doped (this work only) titanium dioxide
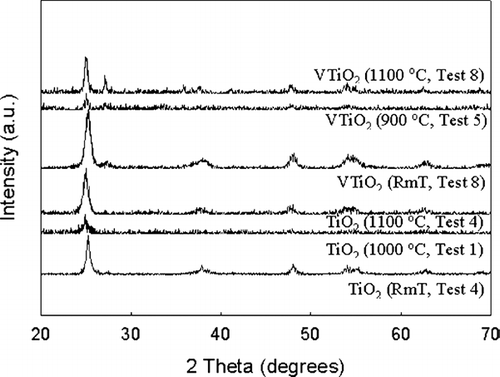
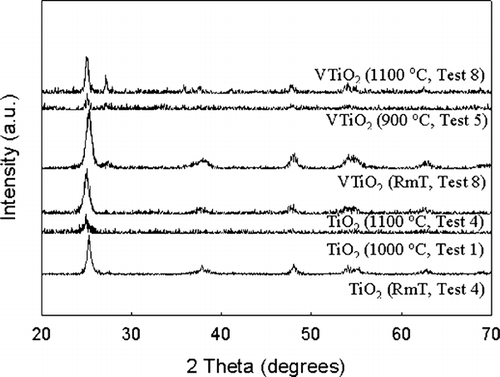
The activation energy for the V-doped TiO2 particles was 363 kJ/mol. The increase of activation energy can be attributed to the increase of enthalpies of jump frequency, vacancy formation, and vanadium—vacancy binding in vanadium doped titania (CitationBorg and Dienes 1988). The characteristic sintering time for vanadium doped TiO2 was lower than that of pristine TiO2. Addition of the dopant could be altering the solid state diffusion coefficient. High resolution transmission electron microscopy revealed lattice spacing gradients in the vicinity of the dopant, and differences in the lattice spacing from pristine TiO2 (). Another plausible reason is the alteration of the crystal structure. X-ray diffraction patterns (the peak heights are not important since the sample amounts are different) of the samples are illustrated in , and show a presence of the rutile phase in the V-doped samples. The ionic radius of V5 + ion is 0.68 Å compared to 0.75 Å for Ti4 +. Thus V5 + can readily substitute some Ti4 + and promote a tighter packing and local transformation to the rutile phase. Similar trends were observed on iron doped TiO2 samples synthesized in flame reactors (CitationWang et al. 2001). It should be noted that the pristine TiO2 did not convert to the rutile phase even at 1100°C due to the relatively short residence times. The presence of the rutile phase in the V-doped samples may also result in enhanced solid state diffusion rates readily to faster sintering. While these observations provide qualitative explanations for the differences in the solid state diffusion coefficients in pristine and V-doped TiO2, detailed characterization studies on the distribution of the dopant are required to quantitatively explain the observed differences in sintering rates.
FIG. 8a High-resolution TEM image of pristine titanium dioxide (above) and vanadium doped titanium dioxide (below). There are regions in the doped sample where the lattice spacing is lower than that of the pristine sample.
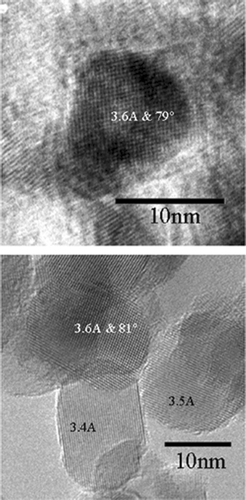
FIG. 8b X-ray diffraction patterns of the resultant particles at the exit of the sintering furnace for the various tests conducted at different temperatures. The predominant phase is anatase for all the samples, with trace quantities of rutile in the V-doped samples.
Prediction of Mobility Diameter at Different Temperatures
Knowing E and A1, Equation (Equation11) can be used to determine the mobility diameter at a given temperature. Substituting x = d m / d f and B(T) = A 1 Texp(E/RT) and using the estimated activation energy and pre-exponential factor () in Equation (Equation11), d m was calculated at various temperatures for the used flow rate (0.4 lpm). The results are plotted in and compared to the measured mobility sizes at the exit of the reactor.
FIG. 9 A comparison of predicted (— for TiO2 and - - - for V-TiO2) and measured mobility diameter at various temperatures for a fixed flowrate (0.4 lpm) for pristine (▪ and vanadium (•) doped titanium dioxide.
CONCLUSIONS
Using expressions for characteristic times for initial sintering and final sintering, it was shown that there is not much difference in the predicted surface area. Hence, the single stage sintering equation was fit to the measured data from a tandem DMA system to determine the activation energy and pre-exponential factors for pristine and V-doped titanium dioxide. Measurements indicate that V-doped TiO2 sinters at a faster rate than pristine TiO2. The activation energy for V-doped TiO2 (363 ± 1 kJ mol−1) was higher than that for pristine TiO2 (236 ± 46 kJ mol−1). The sintering times for pristine and V-doped titanium dioxide were 4.5 and 0.023 s, respectively, for 10 nm particles at 1273 K. The faster sintering rate for the V-doped sample was attributed to a higher solid state diffusion coefficient. The reasons for this were conjectured to be variations in the lattice spacings (reduction in the doped sample) and due to the presence of lattice spacing gradients (in the V-doped sample), or possibly due to the presence of trace amounts of the rutile phase.
Acknowledgments
This work was partially supported by grants from the Center of Materials Innovation (CMI), NSF NIRT 0304649 and DOD MURI UR 523873. Help provided by Dr. P. Fraundorf, University of Missouri, St. Louis for the HRTEM is gratefully acknowledged. Kuk Cho acknowledges the award of a McGrath Fellowship for Doctoral Studies at Washington University in St. Louis.
REFERENCES
- Akhtar , M. K. , Pratsinis , S. E. and Mastrangelo , S. V. R. 1994 . Vapor-Phase Synthesis of Al-Doped Titania Powders . J. Materials Res. , 9 : 1241 – 1249 . [CSA]
- Almquist , C. B. and Biswas , P. 2002 . Role of Synthesis Method and Particle Size of Nanostructured Tio2 on Its Photoactivity . J. Catalysis , 212 : 145 – 156 . [CSA] [CROSSREF]
- Anderson , H. U. 1967 . Initial Sintering of Rutile . J. Amer. Ceram. Soc. , 50 : 235 – 238 . [CSA]
- Astier , M. and Vergnon , P. 1976 . Determination of the Diffusion Coefficients from Sintering Data of Ultrafine Oxide Particles . J. Solid State Chem. , 19 : 67 – 73 . [CSA] [CROSSREF]
- Biswas , P. and Wu , C. Y. 1998 . Control of Toxic Metal Emissions from Combustors Using Sorbents: A Review . J. Air & Waste Manag. Assoc. , 48 : 113 – 127 . [CSA]
- Borg , R. J. and Dienes , G. J. 1988 . An Introduction to Solid State Diffusion. , San Diego : Academic Press .
- Chan , P. and Dahneke , B. 1981 . Free-Molecular Drag on Straight Chains of Uniform Spheres . J. Appl. Phys. , 52 : 3106 – 3110 . [CSA] [CROSSREF]
- Coblenz , W. S. , Dynys , J. M. , Cannon , R. M. and Coble , R. L. 1980 . “ Initial State Solid State Sintering Models. A Critical Analysis and Assessment ” . In Sintering Processes. , Edited by: Kuczynski , G. C. 141 – 157 . New York : Plenum Press .
- Friedlander , S. K. 2000 . Smoke, Dust, and Haze. , New York : Oxford University Press .
- Fujihara , B. , Ohno , T. and Matsumura , M. 1998 . Splitting of Water by Electrochemical Combination of Two Photocatalytic Reactions on Tio2 Particles . J. Chemical Society-Faraday Transactions , 94 : 3705 – 3709 . [CSA] [CROSSREF]
- Fujishima , A. , Rao , T. N. and Tryk , D. A. 2000 . Titanium Dioxide Photocatalysis . J. Photochem. Photobiol. C: Photochem. Rev. , 1 : 1 – 21 . [CSA] [CROSSREF]
- German , R. M. 1996 . Sintering Theory and Practice. , New York : Wiley .
- German , R. M. and Munir , Z. A. 1976 . Surface Area Reduction During Isothermal Sintering . J. Amer. Ceramic. Soc. , 59 : 379 – 383 . [CSA] [CROSSREF]
- Haul , R. and Dumbgen , G. 1965 . Sauerstoff-Selbstdiffusion in Rutilkristallen . J. Phys. Chem. Solids , 26 : 1 – 10 . [CSA] [CROSSREF]
- Helble , J. J. and Sarofim , A. F. 1989 . Factors Determining the Primary Particle-Size of Flame-Generated Inorganic Aerosols . J. Coll. Interface Sci. , 128 : 348 – 362 . [CSA] [CROSSREF]
- Hiram , Y. and Nir , A. 1983 . A Simulation of Surface Tension Driven Coalescence . J. Coll. Interface Sci. , 95 : 462 – 470 . [CSA] [CROSSREF]
- Johnson , D. L. 1969 . New Method of Obtaining Volume, Grain-Boundary, and Surface Diffusion Coefficients from Sintering Data . J. Applied Physics , 40 : 192 – 200 . [CSA] [CROSSREF]
- Kasper , G. , Niida , T. and Yang , M. 1985 . Measurements of Viscous Drag on Cylinders and Chains of Spheres with Aspect Ratios between 2 and 50 . J. Aerosol Sci. , 16 : 535 – 556 . [CSA] [CROSSREF]
- Kobata , A. , Kusakabe , K. and Morooka , S. 1991 . Growth and Transformation of Tio2 Crystallites in Aerosol Reactor . AIChE Journal , 37 : 347 – 359 . [CSA] [CROSSREF]
- Layman , P. L. 1995 . Titanium-Dioxide Makes Fast Turnaround, Heads for Supply Crunch . Chem. Engineer. News , 73 : 12 – 15 . [CSA]
- Lehtinen , K. E. J. , Windeler , R. S. and Friedlander , S. K. 1996 . A Note on the Growth of Primary Particles in Agglomerate Structures by Coalescence . J. Coll. Interface Sci. , 182 : 606 – 608 . [CSA] [CROSSREF]
- Namiki , N. , Cho , K. , Fraundorf , P. and Biswas , P. 2005 . Tubular Reactor Synthesis of Doped Nanostructured Titanium Dioxide and Its Enhanced Activation by Coronas and Soft X-Ray . Industrial & Engineering Chemistry Research , 44 : 5213 – 5220 . [CSA] [CROSSREF]
- O'Bryan , H. M. Jr. and Parravano , G. 1966 . On the Sintering of Single Crystal Rutile . Mater. Sci. Eng. , 1 : 177 – 182 . [CSA] [CROSSREF]
- Oh , H. , Park , H. and Kim , S. 2004 . Effects of Particle Shape on the Unipolar Diffusion Charging of Nonspherical Particles . Aerosol Sci. Techno. , 38 : 1045 – 1053 . [CSA]
- Paul , S. 1996 . Surface Coatings: Science and Technology. , Chichester : Wiley .
- Pratsinis , S. E. 1998 . Flame Aerosol Synthesis of Ceramic Powders . Progress in Energy Combust. Sci. , 24 : 197 – 219 . [CSA] [CROSSREF]
- Rogak , S. N. and Flagan , R. C. 1990 . Stokes Drag on Self-Similar Clusters of Spheres . J. Coll. Interface Sci. , 134 : 206 – 218 . [CSA] [CROSSREF]
- Rogak , S. N. , Flagan , R. C. and Nguyen , H. V. 1993 . The Mobility and Structure of Aerosol Agglomerates . Aerosol Sci. Techno. , 18 : 25 – 48 . [CSA]
- Sahle-Demessie , E. , Gonzalez , M. , Wang , Z. M. and Biswas , P. 1999 . Synthesizing Alcohols and Ketones by Photoinduced Catalytic Partial Oxidation of Hydrocarbons in Tio2 Film Reactors Prepared by Three Different Method . Industrial & Engineering Chemistry Research , 38 : 3276 – 3284 . [CSA] [CROSSREF]
- Schmid , H. J. , Al-Zaitone , B. , Artelt , C. and Peukert , W. 2005 . Evolution of the Fractal Dimension for Simultaneous Coagulation and Sintering . Chemical Engineering Science , 61 : 293 – 305 . [CSA] [CROSSREF]
- Seto , T. , Hirota , A. , Fujimoto , T. , Shimada , M. and Okuyama , K. 1997 . Sintering of Polydisperse Nanometer-Sized Agglomerates . Aerosol Sci. Technol. , 27 : 422 – 438 . [CSA]
- Seto , T. , Shimada , M. and Okuyama , K. 1995 . Evaluation of Sintering of Nanometer-Sized Titania Using Aerosol Method . Aerosol Sci. Technol. , 23 : 183 – 200 . [CSA]
- Shimada , M. , Seto , T. and Okuyama , K. 1994 . Size Change of Very Fine Silver Agglomerates by Sintering in a Heated Flow . J. Chem. Engineer. Japan , 27 : 795 – 802 . [CSA] [CROSSREF]
- Spiegel , M. R. and Liu , J. L. 1999 . Mathematical Handbook of Formulas and Tables. , New York : McGraw-Hill .
- Venkatu , D. A. and Poteat , L. E. 1969 . Diffusion of Titanium in Single Crystal Rutile . Mater. Sci. Eng. , 5 : 258 – 262 . [CSA]
- Wang , Z. M. , Yang , G. , Biswas , P. , Bresser , W. and Boolchand , P. 2001 . Processing of Iron-Doped Titania Powders in Flame Aerosol Reactors . Powder Technology , 114 : 197 – 204 . [CSA] [CROSSREF]
- Watanabe , T. , Nakajima , A. , Wang , R. , Minabe , M. , Koizumi , S. , Fujishima , A. and Hashimoto , K. 1999 . Photocatalytic Activity and Photoinduced Hydrophilicity of Titanium Dioxide Coated Glass . Thin Solid Films , 351 : 260 – 263 . [CSA] [CROSSREF]
- Whitemore , D. H. and Kawai , T. 1962 . J. Am. Ceram. Soc. , 45 : 375 – 379 . [CSA]
- Wiltzius , P. 1987 . Hydrodynamic Behavior of Fractal Aggregates . Phys. Rev. Lett. , 58 : 710 [CSA] [CROSSREF]
- Xiong , Y. , Akhtar , M. K. and Pratsinis , S. E. 1993 . Formation of Agglomerate Particles by Coagulation and Sintering. 2. The Evolution of the Morphology of Aerosol-Made Titania, Silica and Silica-Doped Titania Powders . J. Aerosol Sci. , 24 : 301 – 313 . [CSA] [CROSSREF]
- Xiong , Y. and Pratsinis , S. E. 1993 . Formation of Agglomerate Particles by Coagulation and Sintering. 1. A 2-Dimensional Solution of the Population Balance Equation . J. Aerosol Sci. , 24 : 283 – 300 . [CSA] [CROSSREF]
- Yadha , V. and Helble , J. J. 2004 . Modeling the Coalescence of Heterogenous Amorphous Particles . J. Aerosol Sci. , 35 : 665 – 681 . [CSA] [CROSSREF]
- Yang , G. X. and Biswas , P. 1997 . Study of the Sintering of Nanosized Titania Agglomerates in Flames Using In Situ Light Scattering Measurements . Aerosol Sci. Technol. , 27 : 507 – 521 . [CSA]