We introduce a new in-situ instrument, Thermal desorption Aerosol GC/MS-FID (TAG), capable of hourly measurements of speciated organic compounds in atmospheric aerosols. Aerosol samples are collected into a thermal desorption cell by means of humidification and inertial impaction. The sample is thermally desorbed and transferred with helium carrier gas into a gas chromatography (GC) column, with subsequent detection by both quadrupole mass spectrometer (MS) and a flame ionization detector (FID). The collection and analysis steps are automated, yielding around the clock speciation. This approach builds on the extensive body of knowledge available for quantification and source apportionment of organic aerosols from past research using filter-based GC/MS analyses, but it is the first instrument to achieve in-situ time resolved measurements for an essentially unlimited number of samples, making it possible to analyze changes in organic aerosol speciation over timescales ranging from hours to seasons.
INTRODUCTION
Atmospheric aerosols impact atmospheric visibility, directly and indirectly affect the global radiation balance, and are detrimental to human health (CitationCharlson et al. 1992; CitationRamanathan et al. 2001; CitationSchwartz et al. 1996). Over the last decade atmospheric aerosol studies have increasingly pointed to the importance of the carbonaceous component.
Organic matter typically accounts for 20–50% of the mass of airborne particles below 2.5 μ m in diameter (CitationChow et al. 1993; CitationSchauer and Cass 2000; CitationKim et al. 2000; CitationChristoforou 2000; NARSTO, Ch. 6, 2003). The chemical composition of organic matter in aerosols is complex and largely not understood. Many hundreds of organic compounds have been identified through chromatography and mass spectrometry techniques (CitationRogge et al. 1993a, Citation1993b, Citation1993c, Citation1997a, Citation1997b, Citation1998; CitationSchauer et al. 1999; CitationNolte et al. 1999; CitationFine et al. 2001). These include alkanes, alkanals, ketones, alcohols, amides, amines, polycyclic aromatic hydrocarbons, hopanes, substituted phenols, alkanoic acids, alkenoic acids, resin acids, fatty acid methyl esters, mono- and di-carboxylic acids, sugar derivatives, and more.
These identified compounds typically comprise less than 20% of the total organics in aerosols, yet they serve as valuable tracers for particle sources. For example, hopanes, which are remnants of the biological material from which petroleum originated, serve as a unique tracer for fossil fuel in ambient organic aerosols from such sources as lubricating oils, vehicle exhaust, tire wear, road dust, fuel oil combustion, and asphalt roofing tar pot fumes (CitationRogge et al. 1993a, Citation1993b, Citation1993c, Citation1997a, Citation1997b; CitationSimoneit 1984, Citation1985). Levoglucosan is a product of the breakdown of cellulose, and is a unique tracer for wood combustion (CitationSimoneit et al. 1999). Biogenic alkanes are distinguished from fossil-derived alkanes through a carbon preference number which reflects the predominance of odd-carbon number alkanes in plant waxes (CitationSimoneit 1984). These types of differences in organic compound composition have been used to determine the relative contribution of various source types to primary ambient organic matter (CitationSchauer and Cass 2000; CitationFraser et al. 2000; CitationYue and Fraser 2004a, Citation2004b).
The identification and quantification of organic matter at the compound level has historically involved integrated sample collection by filtration or impaction with subsequent extraction and analysis by liquid or gas chromatography. Generally large samples are required, and analyses are time consuming. These methods have provided valuable insight and guidance in our understanding of airborne organic matter, but are limited by their poor time resolution, intensity of manual efforts, and cost.
Temperature programmed thermal desorption GC/MS analysis has successfully been applied to time-integrated, filter and impaction collected aerosol samples (CitationGreaves et al. 1985; CitationWaterman et al. 2000; CitationNeusüss et al. 2000; CitationFalkovich and Rudich 2001). These instruments directly desorb samples into the GC without solvent extraction, thus reducing substantially the total analysis time and complexity, but are still physically and temporally decoupled from the collection process. Quantification of targeted organic marker compounds is straightforward since thermal desorption efficiency is close to 100% (CitationFalkovich and Rudich 2001).
Over the last decade, several types of particle beam mass spectrometry methods have emerged for the study of ambient particles, including two methods that are now available commercially (i.e., Aerodyne AMS, TSI ATOFMS). The Aerodyne aerosol mass spectrometer (AMS) offers high time resolution particle sizing and employs either quadrupole or time of flight mass spectrometry to separate aerosol composition into total organics, nitrate, sulfate, potassium, chloride, and ammonium (CitationAllan et al. 2003, Citation2004; CitationJimenez et al. 2003). Continued analysis of AMS mass spectra is leading to further particle characterization including a separation of total organics into hydrocarbon-like organic aerosol (HOA) and oxygenated organic aerosol (OOA) (CitationZhang et al. 2005). The TSI aerosol time-of-flight mass spectrometer (ATOFMS) produces detailed mass spectra of individual particles, detecting organic carbon functional groups, elemental carbon, sulfate, nitrate, metals, chlorine, ammonium, and more (CitationNoble and Prather 1996). These instruments provide a wealth of real-time data on aerosols and on single particle composition. However, the deconvolution of single mass spectra from multiple organic compounds remains a daunting challenge. These techniques are not designed to identify or quantify individual source marking organic compounds in ambient particles.
Ziemann has accomplished compound separation and identification with his particle beam mass spectrometer through integrated, sample preconcentration followed by programmed thermal desorption (CitationTobias and Ziemann 1999; CitationTobias et al. 2000). This provides separation of compounds by volatility. They report that ramping the temperature of the desorption surface at a rate of ∼ 1°C/minute sufficiently separates compounds having a factor of 5 or more difference in vapor pressure and different mass-to-charge (m/z) values.
Instruments such as the In-Situ Carbon Analyzer provide organic carbon and elemental carbon mass with one- to two- hour time resolution (CitationTurpin et al. 1990). The particle into liquid sampler coupled to a total organic carbon analyzer (PILS-WSOC) provides six-minute integral water soluble organic mass measurements (CitationSullivan et al. 2004). As with the particle beam mass spectrometers, these automated, in-situ instruments are not designed to provide data at the compound level.
Previous analysis of organic compounds in atmospheric aerosols has typically relied on samples collected by filtration of air. An alternate approach to sampling aerosols is impact collection (CitationHering et al. 1990). Impactors use one or more orifices to impact aerosol particles into concentrated spots on a collection substrate, which is later thermally desorbed into a gas analyzer. CitationStolzenburg and Hering (2000) first combined the collection and desorption steps for particle nitrate sampling using a resistively heated metal strip for the collection substrate and placing it within a glass cell. A nitrogen carrier gas is sent through the cell during desorption to transfer the sample into a gas analyzer. This integrated collection and thermal desorption cell (CTD) provides 10-minute resolution for continuous, automated nitrate monitoring. In this paper, we describe an adaption of this integrated CTD design in combination with a GC/MS-FID that allows for hourly in-situ collection and analysis of ambient aerosols for the semi-continuous measurement of speciated organics.
INSTRUMENT DESCRIPTION
Our approach combines inertial impaction, thermal desorption, and GC/MS techniques in an in-situ, automated instrument. Atmospheric aerosol samples are collected into a small thermal desorption cell by means of humidification and inertial impaction. The sample is transferred onto a GC column by thermal desorption, with subsequent GC/MS-FID analysis. The collection and analysis steps are automated, yielding around the clock organic aerosol speciation with hourly time resolution. Here we describe the instrument and each of its major components in order from the sample inlet to the final detection of individual organics.
A schematic of the Thermal desorption Aerosol GC/MS-FID (TAG) instrument is shown in. TAG has two basic modes of operation: (1) ambient sampling with concurrent GC/MS-FID analysis of the previously collected sample (), and () thermal desorption with sample injection (). The Thermal Desorption Mode transfers the previously collected sample onto the GC column. During the Sampling/Analysis Mode, the GC analysis is completed while the CTD cell is cooled to room temperature and the next sample is collected.
FIG. 1 Schematic of the TAG system, showing flow configuration for two modes of operation: (a) concurrent sampling and analysis, and (b) thermal desorption. The thermal desorption mode is used for transfer of collected sample onto the chromatography column.
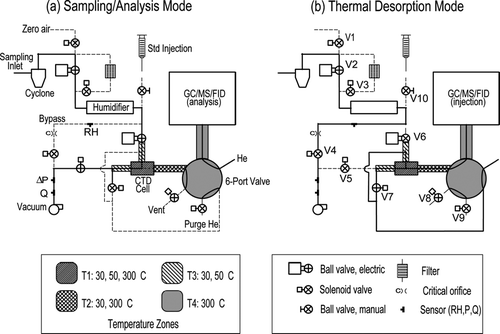
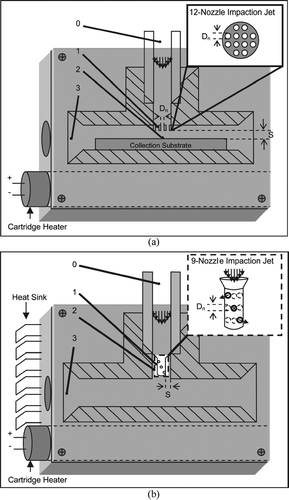
FIG. 2 Schematic diagrams for particle collection-thermal desorption (CTD) cells. Numbers 0–3 correspond to different pressure regions during operation, with region 0 approximately 1 atm and region 3 approximately 0.5 atm. Diagram (a) is a 12-nozzle jet with Dn = 0.28 mm, and S = 3Dn. Diagram (b) is a 9-nozzle jet with Dn = 0.34 mm, and S = 3Dn.
Inlet Precut and Humidity Conditioning
Ambient air is pulled through a BGI sharp cut PM2.5cyclone (SCC BGI Inc., Waltham, MA) (CitationKenny et al. 2000), and 3/8″ tubing by a vacuum pump to achieve a flow rate of 9 L/min, restricted either by the impaction jet on the sample line or the critical orifice on the bypass line depending on the valve positions. At this flow rate, the cyclone excludes particles larger than 2.0 μ m in aerodynamic diameter. All components exposed to the sample (i.e. tubing, fittings, impaction jet) are 316 stainless steel chemically passivated with an Inertium® coating (Advanced Materials Components Express, Lemont, PA). To minimize particle losses, ball valves are used in aerosol sampling lines upstream of the collection cell.
Particles in the sample stream are humidified to increase adhesion and eliminate particle bounce during the following impaction stage (CitationStein et al. 1994). Particle aerodynamic diameter is essentially unchanged after humidification due to a compensating decrease in particle density with water uptake. The humidifier consists of 10 parallel water semi-permeable Nafion tubes, each with 2.2 mm inner diameter and 30 cm length enclosed in a water jacket. The humidifier (MH-110, PermaPure, Toms River, NJ) with a 9 L/min air stream and an input water vapor content of 20–95% relative humidity (RH) consistently produces an output value of approximately 65–95% RH, high enough to minimize particle bounce. Similar humidifiers have been successfully employed in other inertial impaction collection systems (CitationStolzenburg et al. 2003).
Following humidification, the air stream passes through ball value V6 and is collected, or is diverted to the bypass line through V4. RH and temperature are monitored using an in-line RH&T probe (50y, Vaisala Inc., Woburn, MA) on the bypass line. The critical orifice on the bypass line is equivalent to the critical orifice on the sampling line, which creates a constant flow rate of 9 L/min and ensures a constant RH.
Ambient Sampling/Field Blank/System Blank
Following the cyclone, the sample stream can be sent directly through valve V2 () to collect ambient PM2.5, or through V3 and a Teflon membrane filter (Zefluor 2.0 μ m, Pall Corp.) before collection in order to achieve a field blank. These field blanks are used to determine the extent of gas-phase adsorption to the surfaces heated during thermal desorption, including the multijet impactor, impaction substrate, walls of the CTD cell, and the tubing between valves V5 and V6.
Periodically, particle and hydrocarbon free air can be analyzed instead of ambient or filtered ambient air in order to test for contamination coming from within the system, such as desorption of chemical build up on the filter and impaction surface. Clean air is delivered by a pure air generator (737, AADCO, Cleves, OH), and is introduced by opening V1.
Collection-Thermal Desorption (CTD) Cell
The CTD cell collects particles and then thermally desorbs them into helium (He) carrier gas that flows into the GC column. The CTD consists of an inertial impaction collector mounted inside an aluminum block with a cartridge heater controlled through a proportional integral differential (PID) controller (Omega Engineering Inc.). The entire CTD assembly is housed within an insulated box with a time programmed fan that permits cooling of the cell at the end of the desorption step.
Details of the impactor collector are shown in. Two geometries have been tested. The first configuration () is a multijet impactor with 12 nozzles each with a diameter Dn = 0.28 mm impinging upon a glass surface spaced S = 3Dn = 0.84 mm away from the impaction jet. A second geometry, shown in () utilizes 9 more widely spaced nozzles with a diameter Dn = 0.34 mm impinging onto the sidewalls of the CTD, spaced S = 3Dn = 1.02 mm away from the impaction jet. Both have a sample rate of 9 L/min under critical flow conditions. Corresponding Stokes numbers, based on the upstream conditions, are 0.13 and 0.11 for 0.07 μ m particles. The 9-jet geometry, with its more widely spaced orifices, is less susceptible to cross-flow interferences, as described by CitationRouf (1975). The 12-jet geometry has the advantage of more easily changing the collection surface. For the work reported here the impactor jet is mounted in the cross arm of a Swagelok tee (0.25 inch diameter). Subsequent efforts have led to the construction of a collection cell of similar dimensions, but with the added provision for the easy introduction of standards. All parts are fabricated from 316 stainless steel and passivated using the previously mentioned Inertium® treatment. The CTD cell in is an early cell design that does not include a heat sink.
During in-situ automated sampling, aerosols are typically impacted onto the collection substrate at a temperature of 30°C for 30 minutes. After collection, the upstream valve V6 closes off the input air flow and solenoid valve V5 closes off the output end of the CTD cell. As the CTD cell ramps from 30°C to 50°C, solenoid valves V7, V8, and V9 open to purge with He any water vapor adsorbed to surfaces. If a large amount of H2O entered the GC, then it would interfere with the chromatography. Some of the most volatile compounds (30°C < boiling point < 100°C) will be purged along with H2O. This sets the upper vapor pressure limit for organic compounds analyzed by this technique. However, it is our goal to collect as many marker compounds as possible while maintaining consistent chromatography. To reach this goal, compounds with very low boiling points (i.e., < 100°C) and extremely high boiling points (i.e., > 500°C) will not be analyzed.
After approximately 5 min of purging at 50°C, the 6-port valve (Valco Instruments Co. Inc.) is switched to transfer He through the CTD cell and into the GC column. The organic compounds in the collected sample are desorbed by heating the CTD to 300°C at a rate of approximately 30–40°C/min. The sample transfer lines and the 6-port valve are also heated to 300°C. During desorption, the GC oven is held at 45°C to refocus the sample onto the head of the column. Following desorption, the system is switched back to the sampling and analysis configuration, simultaneously allowing GC/MS-FID analysis of the current sample, and collection of the subsequent sample to proceed. More information regarding the desorption process is provided in the following Thermal Desorption and Transfer Efficiency section.
Liquid standards are manually injected with a microliter syringe directly into the CTD cell. While the sample line is in bypass mode, valves V10 and V6 are opened just long enough to apply the standard as illustrated in. After injection, the standard is thermally desorbed and analyzed in an identical manner to an ambient sample.
Gas Chromatographic Separation
Chromatographic separation of the analytes is achieved using a Hewlett-Packard (HP) 6890 GC equipped with a Rtx-5MS column (30 m, 0.25 mm i.d., 0.25 μ m film thickness; Restek Corp.). The temperature program for the GC oven starts at 80°C with an immediate drop to 45°C and held for 12 minutes during sample injection, increases 8.5°C/min to 300°C, and holds at 300°C for 7 minutes. This GC method is typical of published analytical protocols for speciated organic aerosol analyses (CitationYue and Fraser 2004a; CitationFine et al. 2001; CitationNolte et al. 1999). The oven then returns to 80°C in preparation for the next run. The GC oven temperature cycle is temporally minimized by starting and ending each run at 80°C.
Compounds eluting from the column are split between the flame ionization detector (FID) and mass selective detector (MSD) to provide simultaneous mass spectra identification and FID quantification. The carrier He (UHP further purified of oxygen, moisture, and hydrocarbons using traps from Restek Corp.) is kept at a constant head pressure of 25 psi, with a constant 75% split to the MSD and 25% to the FID.
Mass Selective Detection (MSD) and Flame Ionization Detection (FID)
A 70 eV electron impact (EI) ionization, quadrupole mass selective detector (5973 MSD, HP) is operated in total ion scan mode (29–550 m/z) to collect full mass spectral signatures for compound identification. Compounds are identified by matching MSD ion fragment patterns for each resolved peak to compounds found in mass spectral databases or to authentic standards when available. A flame ionization detector (FID), widely utilized for its linear detection of organic compounds (e.g., CitationGoldstein et al. 1995), is also used to provide additional quantification capability. Calibration curves for several compound classes (polar and nonpolar) were obtained by manual injection of multi-component standards prepared by the Wisconsin State Laboratory of Hygiene. Examples are presented in the Instrument Evaluation section.
Instrument Automation and Data Acquisition
TAG is fully automated for unattended field operation. Valve array (V6, V7, V8, V9), the 6-port valve, the CTD cell T1 cartridge heater, and T2 heating tape are controlled through the GC via auxiliary output circuitry. The PC controlling the GC is interfaced with a CR10X datalogger (Campbell Scientific Inc.), which is also triggered via GC auxiliary output circuitry at the start of each analysis. Valve array (V1, V2, V3, V4, V5), the CTD cell box cooling fan, and T3 heating tape are switched at the appropriate times during the sampling cycle by a relay module (SDM-CD16AC, Campbell Scientific Inc.) controlled by the datalogger.
All temperature zones are controlled by PID controllers (CN1166 Series, Omega Engineering Inc.) capable of 16 segments of preprogrammed temperatures triggered by the previously mentioned methods. Actual temperatures are measured using K-type thermocouples (Omega Engineering Inc.). Sample flow is monitored using a Mass-Flo Meter (MKS Instruments Inc.), and pressure is monitored downstream of the CTD cell using a pressure transducer (Honeywell Data Instruments). Relevant engineering data (i.e., time, temperatures, flow rates, pressures, etc.) for each sampling interval are recorded by the CR10X datalogger with an AM416 multiplexer (Campbell Scientific Inc.), then uploaded to the PC and stored with the associated chromatographic data.
Data Reduction and Analysis
Chromatogram integrations are done using HP ChemStation (G1701AA Version A.03.00) software. Mass spectra are identified using the Palisade Complete Mass Spectral Library (600K edition, Palisade Mass Spectrometry, Ithaca, NY) for EI quadrupole mass spectral matching of 495,000 unique compounds using 606,000 available mass spectra. All subsequent data processing and QA/QC is performed using routines created in S-Plus 6.2 (Insightful Corp.).
INSTRUMENT EVALUATION: METHODS
Critical aspects of TAG instrument performance have been evaluated in the laboratory. Here we describe testing methods used for (1) the particle collection efficiency within the CTD cell, (2) the efficiency of sample transfer to the chromatographic column via thermal desorption, (3) the system detection limits of known standards, and (4) the system reproducibility for collocated sampling.
Particle Collection Efficiency
Particle collection efficiencies were measured using monodisperse particles generated by nebulization and mobility selected by means of a high-flow differential mobility analyzer. Upstream particle number concentrations were measured using a TSI 3760 condensation particle counter (CPC). Downstream concentrations were measured using an aerosol electrometer adapted from a TSI Model 3020 EAA, through which all of the flow from the cell was directed. This approach had the advantage of providing a direct measure of the total charge flux associated with aerosol escaping collection, but requires high concentrations and correction for doubly charged aerosol. Efficiency measurements were also made using a pair of Model 3760 CPCs, one upstream and one downstream. The downstream CPC was operated at low pressure, with a bypass flow to account for the difference in impactor and CPC flow rates, and with an auxiliary magnehelic installed in the CPC pressure balance line to ensure that the instrument was not flooded during pump down. For both configurations, the downstream concentration values were compared to that obtained by passing the aerosol through a “bypass” orifice with the same flow rate and a straight path to the downstream particle counter. Simple ball valves directed the flow either through the CTD cell or through the bypass orifice.
Thermal Desorption and Transfer Efficiency
CitationGreaves et al. (1985) studied the heating profile most effective for direct thermal desorption analysis of ambient aerosol samples collected on filters. They reported the most effective response was to heat to the desired maximum temperature as rapidly as possible while passing He over the desorption surface to remove the vaporized compounds. We use a similar heating profile of approximately 30–40°C/min for the CTD cell.
The efficiency of thermal desorption and transfer from the CTD cell to the head of the chromatography column was evaluated using a standard solution (Wisconsin State Laboratory of Hygiene standard) comprised of a wide range of compounds including hexadecane, eicosane, octacosane, decanoic acid, 4,4′-dimethoxybenzophenone, acenaphthene, chrysene, levoglucosan, and cholestane. We compared the results obtained by introducing the standard with a microliter syringe through the splitless GC injection port (traditional approach for filter extracts), to that obtained by thermal desorption of the same size standard aliquot from the CTD cell depicted in. For the CTD analysis, the standard aliquot was placed on a glass substrate within the CTD cell, the solvent was allowed to evaporate, and then the standard was thermally desorbed and transferred through the sampling valve directly onto the GC column. Both injection methods allowed equivalent sample desorption at 300°C for several minutes.
Detection Limits
Detection limits were evaluated on the basis of the same standards used to determine transfer efficiency. Chromatography detection limits for organic compounds are typically defined as the quantity of standard required for the area of the chromatographic peak to equal approximately 3 times the baseline noise level (CitationLamanna and Goldstein 1999; CitationDocherty and Ziemann 2001). Using this definition and our standard calibration curves from CTD cell transfer produces very low limits of detection. For compounds with poor transfer efficiency onto the column, the calibration curves do not respond linearly near the detection limit and therefore we have estimated limits of detection based on our smallest standard quantity thermally desorbed and then detected by the system. Similar observations and analyses have been performed by previous GC/MS users attempting to quantify underivatized oxygenated semivolatile compounds (CitationFalkovich and Rudich 2001).
Reproducibility for Ambient Sampling
Reproducibility for identifying and quantifying individual compounds in ambient aerosol samples was tested using simultaneous off-line collection of multiple ambient aerosol samples in parallel onto glass collection boats. Collection was done in Berkeley, CA in the vicinity of an interstate highway. Following collection, sample boats were sequentially inserted into the CTD cell mounted on the GC/MS-FID for thermal desorption and analysis.
Our sampling configuration consisted of 3 CTD cells operated in parallel with a fourth CTD cell used as a vapor adsorption blank. The fourth cell was immediately preceded by a Teflon filter, but otherwise operated for the entire sample time identical to the other CTD cells. All four cells (of the design shown in) were mounted downstream of a PM2.5 cyclone, and their sampling rates were determined to be within 10% of each other. Glass sample substrates were prepared by solvent washing and baking in an oven at 400°C for two hours, then stored and refrigerated in aluminum lined Petri dishes until used for sample collection. Samples of 1 m3 ambient air were collected. The reproducibility for quantifying individual compounds in ambient air samples can be assessed from these triplicate samples.
INSTRUMENT EVALUATION: RESULTS
Particle Collection Efficiency
Particle collection efficiencies were measured using oleic acid, a non-bouncy liquid particle, and potassium chloride, a solid particle below its deliquescence point of 84% RH at 25°C. For the potassium chloride tests the air stream was humidified to 60–70% RH, as is done in the TAG system under normal operation. Results obtained using a CPC to count particles penetrating the cell are shown in. The 12-jet impactor displays a particle size cutpoint (particle size at which 50% collection efficiency is achieved) of D50 = 0.085 μ m for oleic particles. This is considerably higher than that obtained with a single-jet impactor of the same diameter, indicating cross-flow interference among the jets. These results led to the design of the 9-jet impactor, with more widely spaced jets. The 9-jet impactor displays a particle size cutpoint of D50 = 0.065 μ m for oleic particles and D50 = 0.085 μ m for the more bouncy potassium chloride particles. Particle collection efficiency exceeds 90% on both impactors for all particles larger than 0.17 μ m.
FIG. 3 Collection efficiency curves for 9- and 12-jet impactors. The oleic aerosol is a nonhygroscopic oil, while potassium chloride aerosol is a solid particle (84% deliquescence), which was introduced to the cell at 60–70% RH. Data were obtained using the CPC counting method (see text).
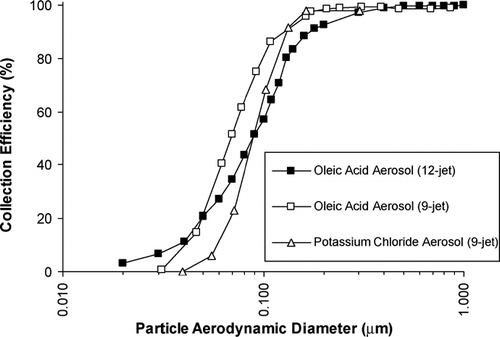
Oleic acid aerosol calibrations were also done using an electrometer for downstream counting, This approach has the advantage of not requiring the flow split below the cell needed for the CPC counting method, but is not sensitive enough for measurements at larger particle sizes where concentrations are small. Calibrations done with the electrometer method yielded similar cutpoints, within 0.05 μ m, to those obtained by the CPC counting method.
Thermal Desorption and Transfer Efficiency
Multipoint calibration curves were generated separately using direct injections and using CTD cell thermal desorptions. The multipoint calibration curves for several representative compounds are shown in. Chrysene is a polycyclic aromatic hydrocarbon formed through combustion. Cholestane is one of the hopanes which serves as a biomarker for petroleum. Levoglucosan is a product of the combustion of cellulose, and is a powerful tracer for wood combustion. Eicosane is one of the many alkanes found in ambient particles. We obtained excellent linear responses (R2 ≥ 0.95) and near zero intercepts for both injection modes for almost all compounds in the standard mixture, as listed in .
FIG. 4 Calibration curves for the CTD cell for four representative marker compounds: eicosane, cholestane, chrysene, and levoglucosan, with comparison to the MSD and FID response for direct injection through the HP injection port.
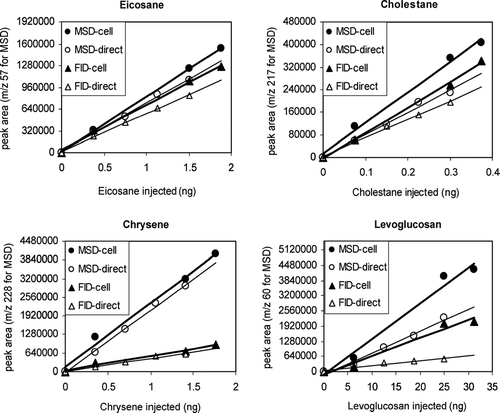
TABLE 1 Thermal desorption response to laboratory standards
Desorption and transfer efficiencies from the CTD relative to that for direct injection are displayed in the fourth column of . Results are calculated as the ratio of the CTD response to that of the direct injection for 10 ng of analyte, where individual response is calculated by the corresponding regression line from the multipoint calibration using data from the FID. The FID was used for this analysis rather than the MSD because it provides a more stable response over time. The relative transfer efficiency among compounds is roughly indicated by the FID response per picomole of carbon (pMC), as listed in columns 5 and 6 of . The transfer efficiency from the CTD cell is equivalent to that for direct injection for most compounds, and even better for levoglucosan, hexadecane, eicosane, octacosane, chrysene, and cholestane. Neither approach shows good efficiency for levoglucosan or decanoic acid. For these compounds, the FID signal relative to the number of carbon atoms is much lower than that for all other compounds, indicating incomplete transfer of these compounds through the analytical system, regardless of the introduction technique. These compounds are known to be very difficult to transfer through gas chromatography systems, so they are generally derivatized prior to analysis. However, they do show a linear response for both detectors and can therefore be analyzed during field measurement if present in ambient air.
Detection Limits
For compounds that transfer efficiently through the CTD cell, detection limits range between 0.027 ng and 0.153 ng. To collect 0.153 ng at 9 L/min for 30 minutes the corresponding ambient concentration must be 0.57 ng/m3. Reported concentrations for Fresno and Bakersfield during the IMS95 experiment are 2 to 200 ng/m3, depending on day and compound (CitationSchauer and Cass 2000). The detection limits determined from injections of our standards, and our initial measurements of ambient air, suggests that TAG is capable of routine analysis of samples significantly smaller than 1 m3 in urban environments, and will therefore be able to collect adequate sample sizes within 30 minutes.
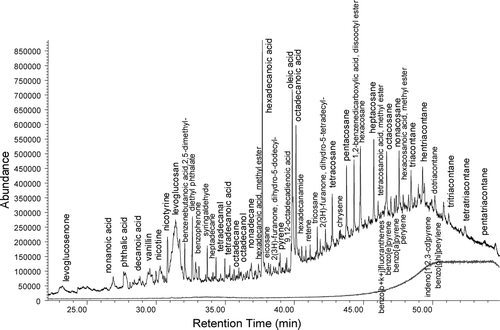
Detection limits are higher for compounds that are not transferred through the instrument with 100% efficiency, as shown in column 8 of . Levoglucosan is not transferred efficiently through the system, and its detection limit is correspondingly larger. However, concentrations of levoglucosan are also high when wood burning occurs (CitationSchauer and Cass 2000), and is clearly seen, as shown in our ambient Berkeley sample presented in (see the Fully Automated Testing section).
FIG. 6 TAG Chromatogram from Berkeley California, with many of the identified peaks labeled. Volume of air sampled is approximately 0.4 m3. Lower gray line shows signal from blank. The rise in background signal after 40 minutes is a result of typical column bleed.
Reproducibility for Ambient Sampling
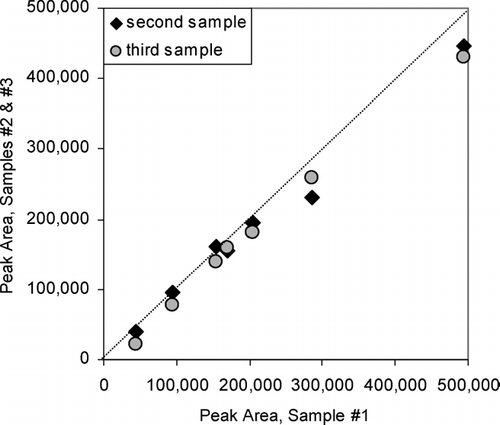
shows a scatter plot of peak areas for a selection of compounds measured between the first of these triplicate samples on the x-axis and the other two on the y-axis. Some variability between the peak areas scales linearly indicating slight differences in sample size, but nearly identical relative responses. Relative standard deviations for measurements of individual compounds in the triplicate samples ranged from 0.04 to 0.33 for 11 selected representative compounds (not present in the vapor adsorption blank) with a pooled standard deviation of 0.12, and reproducibility for the majority of these compounds was better than 10%.
FIG. 5 Comparison of peak areas for independent, collocated samples of ambient air in Berkeley, California. Line shows 1:1 correspondence.
FULLY AUTOMATED TESTING
The fully automated TAG was initially tested at UC Berkeley, an urban environment, by running continuous automated analyses of ambient air and filtered ambient air for approximately 1 full day on 10–11 January, 2004. An example chromatogram from one of the air samples is shown in. We have identified over 100 of the major peaks according to the best matches available in the Palisade Complete 600K mass spectral database, many of which are labeled in.
Notable in the chromatogram is the presence of levoglucosan (31.4 min) which is strongly associated with wood burning. Several alkanoic acids are seen, including tetradecanoic acid, hexadecanoic acid, and octadecanoic acid. All of the alkanes from C17 through C35 are present. Also, note the presence of many polycyclic aromatic hydrocarbons, especially later in the chromatogram. These include phenanthrene, anthracene, fluoranthene, pyrene, retene, benzo[a]anthracene, chrysene, benzo[b+k+j]fluoranthenes, benzo[e]pyrene, benzo[a]pyrene, perylene, indeno[1,2,3-cd]pyrene, and benzo[ghi]perylene. Also present in the chromatogram are alkenes, alkenoic acids, aldehydes, ketones, alcohols, fatty acid methyl esters, substituted phenols, amines, and amides.
The abundance of the measured compounds varies significantly over the one-day measurement period, and several groups of compounds have similar temporal variations. This variability results from multiple sources with differing temporal attributes (e.g., woodsmoke, mobile sources) along with meteorological variability. The covariance of groups of measured compounds provides information about aerosol composition associated with specific source categories, and we can separate marker compounds for specific sources using statistical approaches such as factor analysis. With factor analysis the variation in marker compound profiles measured at the receptor site can be described through the superposition of several independent “factors.” Each factor is associated with a characteristic ratio of compounds, much like a source signature (CitationLamanna and Goldstein 1999). The independent factors can then be compared with source profiles published in the literature (CitationRogge et al. 1993a, Citation1993b, Citation1993c, Citation1997a, Citation1997b, Citation1998; CitationSchauer et al. 1999; CitationFine et al. 2001).
This 1 day data set is too short (i.e., only contains 15 non-filtered ambient air measurement points) to do a complete factor analysis of all 100 compounds measured, but a preliminary analysis of 14 compounds is shown in . These 14 compounds are primarily associated with three different factors, or three different underlying processes or sources. Dominant compounds associated with Factor 1 include oleic acid, octadecanoic acid, and levoglucosan, all markers of wood burning (CitationRogge et al. 1998; CitationSimoneit 1984; CitationSimoneit et al. 1999). The dominant compounds associated with Factor 2 are all alkanes. Alkanes in aerosols potentially come from anthropogenic combustion such as vehicle exhaust, but can also have biogenic sources which favor odd-carbon alkanes. The odd-carbon preference is observed for this data set through the C27–C32 alkane window, indicating a dominant biogenic contribution over vehicle exhaust. Due to a high correlation between only oxygenated compounds such as ketones, aldehydes, and alcohols, Factor 3 is potentially a result of photochemical processing in the atmosphere. With longer data sets which include TAG compounds and meteorological parameters, this type of factor analysis should be able to separate multiple source categories as the basis for receptor based source apportionment studies.
TABLE 2 Factor analysis results for air sampled in Berkeley, California, 10–11 January, 2004
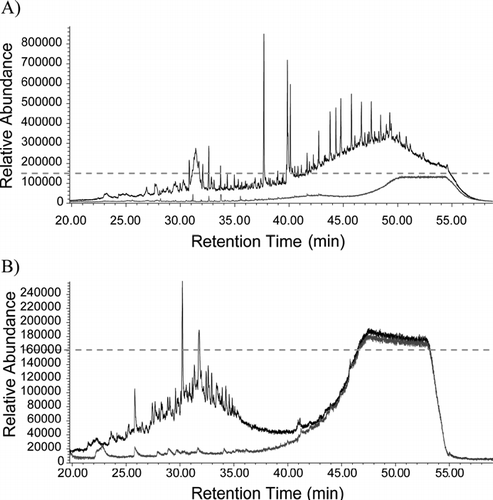
The first field deployment with the TAG system was conducted as part of the ICARTT 2004 campaign. Around-the-clock, automated, in-situ measurements were made on the southwest coast of Nova Scotia (Chebogue Point) during July and August, 2004. In total, 750 atmospheric aerosol samples were collected with hourly time resolution (750 chromatograms × 2 detectors = 1500 total chromatograms). The difference between a typical chromatogram collected during summer at remote Nova Scotia compared to a typical wintertime chromatogram collected in Berkeley, CA, an urban environment, is of interest. displays the same chromatogram from Berkeley, CA which contains many freshly emitted hydrocarbons. Most of these hydrocarbons elute from the gas chromatography column with retention times later than 40 minutes. The earlier retention time compounds are mostly smaller oxygenated compounds (e.g., aldehydes, ketones, alkanoic acids). In, it is shown that the Nova Scotia organic aerosol almost entirely elutes from the GC column before 40 minutes. According to Palisade Complete 600K spectral database matches, nearly all of the resolved compounds collected in Nova Scotia are oxygenated, and a few of these were also measured in the oxygenated section of the Berkeley chromatogram (e.g., phthalic acid, γ -nonalactone, γ -dodecalactone, and 3,5-di-tert-butyl-4-hydroxybenzaldehyde). This frequently occurring high level of oxidation indicates ambient aerosols observed in Nova Scotia were transported over significant distances during which they were oxidized or else must be dominated by secondary aerosol formation, or are impacted by a combination of these processes. Detailed results are presented by Williams et al. (in preparation).
FIG. 7 A comparison between (a) an urban organic aerosol sample, 0.4 m3of air collected in Berkeley, CA, and (b) a remote organic aerosol sample, 0.25 m3of air collected at Chebogue Point, Nova Scotia. The overlapping chromatograms, present in lower abundance and displayed in light gray, are field blanks through a Teflon filter. The dotted gray line represents equivalent detector signal. The rise in background signal after 40 minutes on all chromatograms is a result of typical column bleed.
SUMMARY
TAG is the first continuously operating automated gas chromatography instrument to be successfully used for measuring hourly in-situ organic aerosol chemical speciation. The sampling and data acquisition process are fully automated, allowing around-the-clock operation, free of artifacts associated with filter collection and handling. The system efficiently collects particles in the 0.06–2.5 μ m size range, and efficiently transfers organic compounds via thermal desorption into the GC with quantitatively reproducible results.
While our focus is to use individual organic marker compounds to determine aerosol sources and transformations, and not to characterize the entire chemical makeup of the aerosol, it is still of interest to increase the number of marker compounds measured by TAG. Many of the organic compounds of interest in the ambient aerosol are oxygenated and polar, and are not completely eluted through GC columns. For example, dicarboxylic acids are typically not completely eluted. Often polar compounds are “derivatized” prior to GC analysis to increase their transmission efficiency through the analytical instruments. Organic acids can be converted to their methyl ester analogues by reaction with diazomethane (CitationSchauer and Cass 2000). They may also be reacted with trimethylsilyl reagents (CitationEvershid 1993). Levoglucosan has also been measured by reaction with a silylating agent, though underivatized levoglucosan does pass through the GC column, albeit with a lower response factor (CitationSimoneit et al. 1999). CitationDocherty and Ziemann (2001) report an on-line, inlet-based silylation method via coinjection of the reagent, with derivatization occurring via gas-phase reactions. Their results show quantitative analysis of mono and dicarboxylic acids, and should be adaptable to our thermal desorption analysis. However, our preliminary results do show reasonable recovery of underivatized monocarboxylic acids in thermally desorbed standards.
The 24-hour period from Berkeley, CA is representative of a time period where traditionally only 1 or perhaps 2 sequential filter analyses of organic aerosol composition would typically have been measured. Our TAG measurements clearly show that having hourly time resolution dramatically enhances the amount of information that can be learned about the sources contributing to organic aerosol loading in the atmosphere and aerosol transformation processes, compared to traditional 12- or 24-hour filter sampling.
Field measurements with TAG in a variety of environments will provide higher time resolution data than has previously been available for speciated organics in aerosols, opening new windows into the study of their origins, chemistry, fate, and impacts on the environment and human health.
TAG was developed using a U.S. Department of Energy SBIR Phase I & II grant (DE-FG02-02ER83825), and a graduate research environmental fellowship was provided by DOE's Global Change Education Program. Additional support during ICARTT 2004 was provided by the National Oceanic and Atmospheric Administration. We would also like to thank Megan McKay of the University of California at Berkeley for technical and logistical support, Dr. Dylan Millet (previously of the University of California at Berkeley, currently at Harvard University) for technical assistance and extensive assistance regarding GC/MS operation, and Dr. James Schauer of the University of Wisconsin at Madison for assistance regarding GC/MS analysis of semivolatile compounds.
Related Research Data
REFERENCES
- Allan , J. D. , Jimenez , J. L. , Williams , P. I. , Alfarra , M. R. , Bower , K. N. , Jayne , J. T. , Coe , H. and Worsnop , D. R. 2003 . Quantitative Sampling using an Aerodyne Aerosol Mass Spectrometer 1. Techniques of Data Interpretation and Error Analysis . J. Geophys. Res. , 108 ( D3 ) : 4090 doi:10.1029/ 2002JD002358.[CSA]
- Allan , J. D. , Delia , A. E. , Coe , H. , Bower , K. N. , Alfarra , M. R. , Jimenez , J. L. , Middlebrook , A. M. , Drewnick , F. , Onash , T. B. , Canagaratna , M. R. , Jayne , J. T. and Worsnop , D. R. 2004 . A Generalized Method for the Extraction of Chemically Resolved Mass Spectra from Aerodyne Aerosol Mass Spectrometer Data . J. Aerosol Sci. , 35 ( 7 ) : 909 – 922 . [CSA] [CROSSREF]
- Charlson , R. J. , Schwartz , S. E. , Hales , J. M. , Cess , R. D. , Coakley , J. A. , Hansen , J. E. and Hofmann , D. J. 1992 . Climate Forcing by Anthropogenic Aerosols . Science , 255 ( 5043 ) : 423 – 430 . [CSA]
- Christoforou , C. S. , Salmon , L. G. , Hannigan , M. P. , Solomon , P. A. and Cass , G. R. 2000 . Trends in Fine Particle Concentration and Chemical Composition in Southern California . J. Air and Waste Management Assoc. , 50 : 43 – 53 . [CSA]
- Chow , J. C. , Watson , J. G. , Lowenthal , D. H. , Solomon , P. A. , Magliano , K. L. , Ziman , S. D. and Richards , L. W. 1993 . PM10 and PM2.5 Compositions in California's San Joaquin Valley . Aerosol Sci. Technol. , 18 : 105 – 128 . [CSA]
- Docherty , K. S. and Ziemann , P. J. 2001 . On-line, Inlet-Based Trimethylsilyl Derivatization for Gas Chromatography of Mono- and Dicarboxylic Acids . J. Chromat. A. , 921 : 265 – 275 . [CSA] [CROSSREF]
- Evershid , R. 1993 . Handbook of Derivatives for Chromatography , Edited by: Blau , K. and Halket , J. p. 53 New York : Wiley .
- Falkovich , A. H. and Rudich , Y . 2001 . Analysis of Semivolatile Organic Compounds in Atmospheric Aerosols by Direct Sample Introduction Thermal Desorption GC/MS . Environ. Sci. Technol. , 35 : 2326 – 2333 . [INFOTRIEVE] [CSA] [CROSSREF]
- Fine , P. M. , Cass , G. R. and Simoneit , B. R. T. 2001 . Chemical Characterization of Fine Particle Emissions from Fireplace Combustion of Woods Grown in the Northeastern United States . Environ. Sci. Technol. , 35 : 2665 – 2675 . [INFOTRIEVE] [CSA] [CROSSREF]
- Fraser , M. P. , Kleeman , M. J. , Schauer , J. J. and Cass , G. R. 2000 . Modeling the Atmospheric Concentrations of Individual Gas-Phase and Particle-Phase Organic Compounds . Environ. Sci. Technol. , 34 : 1302 – 1312 . [CSA] [CROSSREF]
- Goldstein , A. H. , Daube , B. C. , Munger , J. W. and Wofsy , S. C. 1995 . Automated In-Situ Monitoring of Atmospheric Nonmethane Hydrocarbon Concentrations and Gradients . J. Atmos. Chem. , 21 : 43 – 59 . [CSA] [CROSSREF]
- Greaves , R. C. , Bakley , R. M. and Sievers , R. E. 1985 . Rapid Sampling and Analysis of Volatile Constituents of Airborne Particulate Matter . Anal. Chem. , 57 : 2807 – 2815 . [CSA] [CROSSREF]
- Hering , S. V. , Appel , B. R. , Cadle , S. H. , Cahill , T. A. , Fitz , D. , Howes , J. E. , Knapp , K. T. , Turpin , B. J. and McMurry , P. H. 1990 . Comparisons of Sampling Methods for Carbonaceous Aerosols in Ambient Air . Aerosol Sci. Technol. , 12 : 200 – 213 . [CSA]
- Jayne , J. T. , Leard , D. C. , Zhang , X.-F. , Davidovits , P. , Smith , K. A. , Kolb , C. E. and Worsnop , D. R. 2000 . Development of an Aerosol Mass Spectrometer fro Size and Composition Analysis of Submicron Particles . Aerosol Sci. Technol. , 33 : 49 – 70 . [CSA] [CROSSREF]
- Jimenez , J. L. , Jayne , J. T. , Shi , Q. , Kolb , C. E. , Worsnop , D. R. , Yourshaw , I. , Seinfeld , J. H. , Flagan , R. C. , Zhang , X. , Smith , K. A. , Morris , J. and Davidovits , P. 2003 . Ambient Aerosol Sampling with an Aerosol Mass Spectrometer . J. Geophys. Res. Atmospheres , 108 ( D7 ) : 8425 [CSA] [CROSSREF]
- Kenny , L. C. , Gussman , R. A. and Meyer , M. 2000 . Development of a Sharp-Cut Cyclone for Ambient Aerosol Monitoring Applications . Aerosol Sci. Technol. , 32 ( 4 ) : 338 – 358 . [CSA] [CROSSREF]
- Kim , B. M. , Teffera , S. and Zeldin , M. D. 2000 . Characterization of PM2.5 and PM10 in the South Coast Air Basin of Southern California: Part 1n–Spatial Variation . J. Air and Waste Management Assoc. , 50 : 2034 – 2044 . [CSA]
- Lamanna , M. S. and Goldstein , A. H. 1999 . In-Situ Measurements of C2-C10 VOCs Above a Sierra Nevada Ponderosa Pine Plantation . J. Geophys. Res. , 104 ( D17 ) : 21247 – 21262 . [CSA] [CROSSREF]
- Neusüss , C. , Pelzing , M. , Plewka , A. and Herrmann , H. 2000 . A New Analytical Approach for Size-Resolved Speciation of Organic Compounds in Atmospheric Aerosol Particles: Methods and First Results . J. Geophys. Res. , 105 : 4513 – 4527 . [CSA] [CROSSREF]
- NARSTO . 2003 . Particulate Matter Sciences for Policy Makers, A NARSTO Assessment.[INFOTRIEVE] [CSA]
- Noble , C. A. and Prather , K. A. 1996 . Real Time Measurement of Correlated Size and Composition Profiles of Individual Atmospheric Aerosol Particles . Environ. Sci. Technol. , 30 : 2667 – 2680 . [CSA] [CROSSREF]
- Nolte , C. G. , Schauer , J. J. , Cass , G. R. and Simoneit , B. R. T. 1999 . Highly Polar Organic Compounds Present in Meat Smoke . Environ. Sci. Technol. , 33 : 3313 – 3316 . [CSA] [CROSSREF]
- Ramanathan , V. , Crutzen , P. J. , Kiehl , J. T. and Rosenfeld , D. 2001 . Atmosphere—Aerosols, Climate, and the Hydrological Cycle . Science. , 294 ( 5549 ) : 2119 – 2124 . [INFOTRIEVE] [CSA] [CROSSREF]
- Rogge , W. F. , Mazurek , M. A. , Hildemann , L. M. , Cass , G. R. and Simoneit , B. R. T. 1993a . Quantification of Urban Organic Aerosols on a Molecular Level: Identification, Abundance and Seasonal Variation . Atmos. Environ. , 27a : 1309 – 1330 . [CSA]
- Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. , Cass , G. R. and Simoneit , B. R. T. 1993b . Sources of Fine Organic Aerosol: 2. Noncatalyst and Catalyst-Equipped Automobiles and Heavy-Duty Diesel Trucks . Environ. Sci. Technol. , 27 : 636 – 651 . [CSA] [CROSSREF]
- Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. , Cass , G. R. and Simoneit , B. R. T. 1993c . Sources of Fine Organic Aerosol. 3. Road dust, tire debris, and Organometallic Brake Lining Dust: Roads as Sources and Sinks . Environ. Sci. Technol. , 27 : 1892 – 1904 . [CSA] [CROSSREF]
- Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. , Cass , G. R. and Simoneit , B. R. T. 1997a . Sources of Fine Organic Aerosol. 7. Hot Asphalt Roofing Tar Pot Fumes . Environ. Sci. Technol. , 31 : 2726 – 2730 . [CSA] [CROSSREF]
- Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. , Cass , G. R. and Simoneit , B. R. T. 1997b . Sources of Fine Organic Aerosol. 8. Boilers Burning no. 2 Distillate Fuel Oil . Environ. Sci. Technol. , 31 : 2731 – 2737 . [CSA] [CROSSREF]
- Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. and Cass , G. R. 1998 . Sources of Fine Organic Aerosol 9: Pine Oak and Synthetic Log Combustion in Residential Fireplaces . Environ. Sci. Technol. , 32 : 13 – 22 . [CSA] [CROSSREF]
- Rouf , M. A. 1975 . Effect of Separation Distance on the Local Impingement Heat Transfer Coefficient in the Presence of Cross-Flow , MS thesis Department of Mechanical Engineering, University of Minnesota .
- Schauer , J. J. , Kleeman , M. J. , Cass , G. R. and Simoneit , B. R. T. 1999 . Measurement of Emissions from Air Pollution Sources. 2. C1 through C30 Organic Compounds from Medium Duty Diesel Trucks . Environ. Sci. Technol. , 33 : 1578 – 1587 . [CSA] [CROSSREF]
- Schauer , J. J. and Cass , G. R. 2000 . Source Apportionment of Wintertime Gas-Phase and Particle Phase Air Pollutants Using Organic Compounds as Tracers . Environ. Sci. Technol. , 34 : 1821 – 1832 . [CSA] [CROSSREF]
- Schwartz , J. , Dockery , D. W. and Neas , L. M. 1996 . Is Daily Mortality Associated Specifically with Fine Particles? . J. Air Waste Manage. Assoc. , 46 : 927 – 939 . [CSA]
- Simoneit , B. R. T. 1984 . Organic Matter of the Troposphere—III. Characterization and Sources of Petroleum and Pyrogenic Residues in Aerosols Over the Western United States . Atmos. Environ. , 18 : 51 – 67 . [CSA] [CROSSREF]
- Simoneit , B. R. T. 1985 . Application of Molecular Marker Analysis to Vehicular Exhaust for Source Reconciliations . Int. J. Environ. Anal. Chem. , 22 : 203 – 233 . [CSA]
- Simoneit , B. R. T. , Schauer , J. J. , Nolte , C. G. , Oros , D. R. , Elias , V. O. , Fraser , M. P. , Rogge , W. F. and Cass , G. R. 1999 . Levoglucosan, a Tracer for Cellulose in Biomass Burning and Atmospheric Particles . Atmos. Environ. , 33 : 73 – 182 . [CSA] [CROSSREF]
- Stein , S. W. , Turpin , B. J. , Cai , X. , Huang , P.-F. and McMurry , P. H. 1994 . Measurements of Relative Humidity-Dependent Bounce and Density for Atmospheric Particles using the DMA-Impactor Technique . Atmos. Environ. , 28 : 1739 – 1746 . [CSA] [CROSSREF]
- Stolzenburg , M. R. and Hering , S. V. 2000 . A New Method for the Automated Measurement of Atmospheric Fine Particle Nitrate . Environ. Sci. Technol. , 34 : 907 – 914 . [CSA] [CROSSREF]
- Stolzenburg , M. R. , Dutcher , D. D. , Kirby , B. W. and Hering , S. V. 2003 . Automated Measurement of the Size and Concentration of Airborne Particulate Nitrate . Aerosol Sci. Technol. , 37 : 537 – 546 . [CSA] [CROSSREF]
- Sullivan , A. P. , Weber , R. J. , Clements , A. L. , Turner , J. R. , Bae , M. S. and Schauer , J. J. 2004 . A Method for On-Line Measurements of Water-Soluble Organic Carbon in Ambient Aerosol Particles: Results from an Urban Site . Gephys. Res. Lett. , 31 : L13105 [CSA] [CROSSREF]
- Tobias , H. J. and Ziemann , P. J. 1999 . Compound Identification in Organic Aerosols Using Temperature Programmed Thermal Desorption Particle Beam Mass Spectrometry . Anal. Chem. , 71 : 3428 – 3435 . [CSA] [CROSSREF]
- Tobias , H. J. , Kooiman , P. M. , Docherty , K. S. and Ziemann , P. J. 2000 . Real Time Chemical Anlaysis of Organic Aerosols Using a Thermal Desorption Particle Beam Mass Spectrometer . Aerosol Sci. Technol. , 33 : 170 – 190 . [CSA] [CROSSREF]
- Turpin , B. J. , Cary , R. A. and Huntzicker , J. J. 1990 . An in-Situ, Time-Resolved Analyzer for Aerosol Organic and Elemental Carbon . Aerosol Sci. Technol. , 12 ( 1 ) : 161 – 171 . [CSA]
- Waterman , D. , Horsfield , B. , Leistner , F. , Hall , K. and Smith , S. 2000 . Quantification of Polycyclic Aromatic Hydrocarbons in the NIST Standard Reference Material (SRM1649A) Urban Dust Using Thermal Desorption GC/MS . Anal. Chem. , 72 ( 15 ) : 3563 – 3567 . [INFOTRIEVE] [CSA] [CROSSREF]
- Yue , Z. and Fraser , M. 2004a . Characterization of Non-Polar Organic Fine Particulate Matter in Houstion, Texas . Aerosol Sci. Technol. , 38 ( 1 ) : 60 – 67 . [CSA] [CROSSREF]
- Yue , Z. and Fraser , M. P. 2004b . Polar Organic Compounds Measured in Fine Particulate Matter During TexAQS 2000 . Atmos. Environ. , 38 ( 20 ) : 3253 – 3261 . [CSA] [CROSSREF]
- Zhang , Q. , Alfarra , M. R. , Worsnop , D. R. , Allan , J. D. , Coe , H. , Canagaratna , M. R. and Jimenez , J. L. 2005 . Deconvolution and Quantification of Hydrocarbon-Like and Oxygenated Organic Aerosols Based on Aerosol Mass Spectrometry . Environ. Sci. Technol. , 39 : 4938 – 4952 . [INFOTRIEVE] [CSA] [CROSSREF]