Apportionment of primary and secondary pollutants during a July 2001 intensive study at the National Energy Technology Laboratory is reported. PM2.5 was apportioned into primary and secondary contributions using PMF2, and results were compared with apportionment based on UNMIX 2.3. Input to PMF2 included PM2.5 mass data from four per 24 hour PC-BOSS filters and TEOM, NOx, NO2, O3, non-volatile, semi-volatile, and volatile organic material, elemental carbon, sulfate, and PIXE determined trace metals. Nine factors were identified in the PMF analysis. Six factors were associated with primary particles from crustal, mobile (gasoline and diesel), and three local sources high in trace metals. Three factors were associated with secondary sources. Two were associated with local emissions dominated by organic material, one was dominated by transported ammonium sulfate. UNMIX was able to identify the two major mobile sources, major local secondary source and transported secondary source. The three major sources of PM2.5 were identified as secondary transported material (dominated by ammonium sulfate) from west and southwest (46%), secondary material formed during mid-day photochemical processes (21%), and primary emissions from diesel (10%) and gasoline (8%) mobile sources. The other five sources accounted for the remaining 15% of the PM2.5. These findings are consistent with the majority of secondary ammonium sulfate in the Pittsburgh area resulting from distant transport, and so decoupled from local activity involving organic pollutants in the metropolitan area. In contrast, the major local secondary sources were dominated by organic material.
INTRODUCTION
Organic carbonaceous material and ammonium sulfate are major components of ambient fine particulate material, and can be emitted directly in particulate form (primary aerosol) or formed in the atmosphere from products of gas-phase transformation reactions (secondary aerosol). It is important to be able to identify the composition and concentrations of PM2.5 associated with these primary and secondary sources in order to develop effective air quality control strategies. The estimation of primary and secondary contributions of ammonium sulfate in ambient air using a variety of receptor apportionment techniques has been documented. For the estimation of primary and secondary organic carbonaceous pollutants in ambient atmospheres, investigators have used the ratio of the ambient concentrations of particulate organic carbon (OC) to elemental carbon (EC) to investigate the extent of secondary formation (CitationTurpin et al. 1991; Cabada et al. 2005) for organic aerosols. The OC/EC ratio method for the determination of secondary and primary contributions to carbonaceous aerosols assumes that the primary OC to EC ratio is similar for all significant sources and that the OC/EC ratio in the primary emissions is conserved with time. However, variations of source strengths, the presence of different sources with changing time and meteorology are expected to change the OC/EC ratio, and so likely to introduce uncertainties in the ultimate primary and secondary estimations (CitationAnderson et al. 2005). Chemical mass balance receptor modeling methods have also been developed that employ fine particulate organic compounds as tracers to apportion the primary source contributions to atmospheric fine particulate organic material (OM) concentrations (CitationSchauer and Cass 2000).
This article reports the estimation of primary and secondary contributions of fine particulate mass, organic carbonaceous material and ammonium sulfate in the ambient atmosphere using a combination of the PMF2 (CitationPaatero 1997) and the EPA UNMIX (CitationHenry and Norris 2002) multivariate receptor modeling and analysis software packages. The analysis is made using PM2.5 composition including PIXE determined elemental content and tracer species usually monitored in ambient studies, e.g., EC, NO X , NO2, and O3. Both PMF and UNMIX are ambient-based models that do not require source profiles to estimate source contributions. Variability in the ambient data is key to success with both UNMIX and PMF2.
The NETL Sampling Site
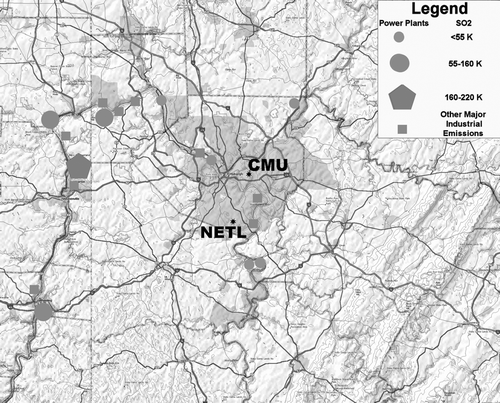
This site is located at 40.31N latitude and 79.98W longitude at an elevation of 325 m on an open hill at the NETL facility. Two experimental combustors, located at the NETL facility just 200 m east of the site occasionally burn a variety of fuels (230 kg/hr of coal and about 20 kg/hr of various other fuels). About 600 m ESE is a coal-fired steam plant which burns up to 860 kg/hr of coal to provide heating for the NETL facility. Considering the infrequent occurrence of winds from the direction of these two facilities, the short time period when these winds are present, and observations of PM2.5 mass during periods of easterly winds, the contribution of these nearby sources to PM2.5 at the NETL site appears to be negligible (CitationAnderson et al. 2006). Major coal-fired power plants, iron and coke processing facilities and chemical manufacturing plants are located along the Ohio River Valley to the west and southwest of the NETL site. The closer of these sites and other significant stationary sources are shown relative to the NETL sampling site in . A major incinerator facility and mid-size power plant, along with several small chemical and metallurgical industries are located along the Ohio River close to the Ohio–Pennsylvania border to the northwest of the NETL site. Smaller coal-fired power plants and a coke processing facility are between the incinerator facility and Pittsburgh, and two smaller (with respect to emissions) coal-fired power plants are a few km to the southeast. Some coke and iron processing facilities are also located along the Monongahela River to the east of the facility. In addition, the sampling site is at the southern portion of the Pittsburgh metropolitan area. These are all potential sources of local and transported pollutants at the site.
FIG. 1 Map showing the location of the NETL sampling site in the Pittsburgh metropolitan area and the location of major point sources in the vicinity.
EXPERIMENTAL TECHNIQUES
Sampling Protocols
Four sample sets were collected each day during a July 2001 intensive study at the NETL site using a PC-BOSS sampler (CitationLewtas et al. 2001). The sampling time periods (EDT) were: 5:00 AM to 10:00 AM; 10:00 AM to 2:00 PM; 2:00 PM to 7:00 PM; and 7:00 PM to 5:00 AM the next day.
The PC-BOSS Sampler Results
The PC-BOSS sampler and analysis of the collected samples at the NETL sampling site has been previously described (CitationLewtas et al. 2001; CitationModey and Eatough 2004). PM2.5 components were collected on two filter packs after a PM2.5 inlet, a particle concentrator (lower cut-point of less than 0.1 μ m) and a diffusion denuder comprised of parallel strips of charcoal-impregnated cellulose fiber filter (CIF, Schleicher and Schuell, Keene, NH) to remove gas phase organic material, SO2, HNO3, O3, and NO x .
Upstream of the particle concentrator, a side flow manifold held a single filter pack containing a 47-mm diameter Whatman Nuclepore filter (0.4 μ m pore, Whatman Inc., Clifton, NJ) for sulfate and PIXE determination, and a 47-mm diameter charcoal-impregnated glass fiber filter (CIG) (Schleicher and Schuell, Dassel, Germany) for gas phase volatile organic material (VOM) determination. The sulfate determination on the side flow Nuclepore filter allowed for the determination of the particle concentrator efficiency and losses (CitationLewtas et al. 2001; CitationModey and Eatough 2004). Samples collected on the Nuclepore filters were concentrated in a 4-cm2 area on the filter using a stainless steel masking disk, positioned underneath the Nuclepore filter. These samples were analyzed for elemental content by proton induced x-ray emission analysis (CitationMangelson et al. 1979).
The denuder was followed by two parallel filter packs. One contained a 47-mm Pallflex quartz filter (Gelman Sciences, Ann Arbor, MI) followed by a 47-mm diameter CIG filter. The quartz filter was used to determine fine particulate elemental carbon (EC), nonvolatile organic material (NVOM), and sulfate concentrations. The CIG filter was used to capture semi-volatile organic material (SVOM) evolved from particles collected on the quartz filter. In this article, NVOM and SVOM are identified based on whether the organic material was collected on the quartz or CIG filter, respectively. Particulate samples collected on both of these filters were concentrated on a 4-cm2 area on the filters using the mask described earlier for the Nuclepore filter.
The other parallel filter pack contained a 47-mm diameter ring-supported Teflon filter (Whatman Inc.) followed by a 47-mm diameter Nylasorb filter (Gelman Sciences). Unlike sample collection on the quartz and CIG filter pack, collection on the Teflon and Nylasorb filters was over the entire filter surfaces. The Teflon filters were analyzed for sulfate and nonvolatile nitrate, and the Nylasorb filters were analyzed for nitrate lost from particles during sampling. With these combination of sample analysis techniques on both parallel channels of the minor flow of the sampler, negative and positive sampling artifacts were minimized. Additional details on filter pre-treatment, blank corrections and analysis for major PM2.5 constituents have been previously given (CitationModey et al. 2006)
Gas Phase Data
NO x , NO2, and O3 data were obtained through the NETL air-monitoring program in Pittsburgh (CitationAnderson et al. 2006).
Filter Pre-treatment
All quartz filters were baked in an oven at 800°C for 10 hours before use. The CIG sorbent filters used in the study were baked in an oven at 340°C for 24 hours in an atmosphere of nitrogen. Such treatments were to ensure that the filters contained minimal residual organic material prior to being used for sampling. The Nuclepore, Teflon, and Nylasorb filters and the denuder CIF strips were used as received from the manufacturers without any pre-treatment.
Analysis of Collected Carbonaceous Material
A 2-cm2 portion from the center of the quartz filter and similarly sized portions from the centers of the CIG filters were analyzed for EC and OM by thermal desorption of the collected materials using temperature programmed volatilization (CitationEatough et al. 1993; CitationEllis and Novakov 1982, CitationModey et al. 2006). Both EC and OM were converted to CO2 over a barium chromate combustion catalyst (UIC Inc., IL) at 800°C and the CO2 concentration was then determined using non-dispersive infrared (NDIR) detection on an ULTRAMAT 3—Gas Analyzer (Siemens Inc.). The NDIR detector was calibrated using three certified CO2 standards which spanned the analysis concentration range. The CIG filters were heated from 50°C to 340°C at a ramp rate of 15°C/min in a stream of nitrogen. The maximum temperature for the CIG analysis was imposed by the fact that degradation of the filter charcoal began 10–20°C below the maximum analysis temperature. Collected SVOM lost from the particles were evolved from the CIG between 220 to 320°C.
The quartz filters were heated from 50°C to 800°C at a ramp rate of 28°C/min in a stream of N2/O2 (70:30 % v/v). Elemental carbon was estimated from the high temperature (usually above 440°C) peak (CitationEllis and Novakov 1982) on the thermogram obtained from the quartz filter analysis. Because of the presence of high concentrations of nonvolatile organic material that evolve at a temperature just below that for elemental carbon in some of the samples, the uncertainty in this EC estimate varied from about 20–50%. However, the uncertainty in NVOM was typically 5–10%. The shape of the thermograms indicated that all collected organic compounds and EC were evolved from the collection matrix at the maximum temperatures used. No evidence for the high-temperature evolution of CO2 from inorganic carbonates was seen. Field handled blank determinations were made for both quartz and CIG filters, and used for background correction in the sample thermograms. No corrections are made for pyrolysis in the analysis, however the constant temperature rise procedure does minimize pyrolysis (CitationEllis and Novakov 1982).
Analysis for Sulfate and Nitrate
Sulfate and nitrate analysis was by ion chromatography. as previously detailed (CitationModey et al. 2006). All ion chromatographic analyses were performed using a Dionex LC20 Chromatograph equipped with a CD20 conductivity detector. The eluent was a 3.5 mM Na2CO3/1.0 mM NaHCO3 solution used at a flow rate of 1.2 mL/min. An anion self-generating suppressor was used. One 2-cm2 punch (O-ring shaped) of the quartz filters was extracted by ultrasonication with deionized water in a monovette (Sarstedt, Newton, NC). The extracted solution was analyzed for sulfate and nitrate. Whole Teflon filters and half of each Nuclepore filter were also extracted by ultrasonication in deionized water in a monovette and analyzed for sulfate and nitrate. Each Nylasorb filter was extracted with eluent and analyzed for nitrate. Field handled blank determinations were made for all filters, and used for background correction on the field sampled filters.
PIXE Data
Nuclepore filters from the NETL site were analyzed for elements with Z ≥ 13(Si) by proton-induced x-ray emission analysis (CitationMangelson et al. 1979).
RESULTS AND DISCUSSION
Assumptions and Calculations
Sulfate determined on the NETL samples by ion chromatography was assumed to be present as ammonium sulfate based on the measured ammonium ion to sulfate ratio in the NETL PM characterization program (CitationAnderson 2002). Organic material (NVOM, SVOM and VOM) was assumed to be 61% carbon (CitationTurpin and Lim 2001). The carbonaceous material determined from the analysis of the quartz filter is called nonvolatile organic material (NVOM) while that identified from analysis of the CIG filter after the quartz filter is called semi-volatile organic material (SVOM). PM2.5 concentrations were calculated as the sum of measured NVOM, SVOM, EC, ammonium sulfate, ammonium nitrate, and crustal material was estimated from the PIXE Al and Si data. A non-volatile PM2.5 mass (labeled “V Mass or NVM” n subsequent figures) was also calculated from the sum of the PC-BOSS components except SVOM and semi-volatile (measured on the Nylasorb filter) nitrate. The TEOM mass listed in subsequent Figures is missing SVOC and VOM, due to the 50°C operating temperature. Details on the results have been published (CitationModey et al. 2005).
UNMIX Apportionment of PM2.5
The sources and composition of fine particulate matter in the Pittsburgh urban atmosphere has been previously explored by UNMIX analysis of the PM2.5 mass determined with the PC-BOSS sampler (CitationAnderson et al. 2005). The PIXE results were not used in these analysis. However, inclusion of the PIXE data in the UNMIX analysis does not significantly alter the results. The results of these analyses given in CitationAnderson et al. (2005) are summarized in and . As indicated, all potential markers other than the PIXE data were used in the UNMIX analysis. Four source solutions were identified. The various sources were identified as associated with emissions from primary mobile gasoline emissions, primary mobile diesel emissions, secondary material locally produced, and secondary material transported into the urban region.
FIG. 2 UNMIX apportionment of PM2.5 mass at the NETL site during July 2001. The first tick mark on the x-axis for each date represents the sample from 5:00 AM to 10:00 AM.
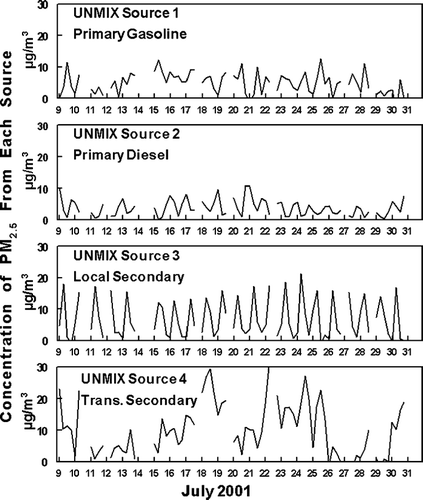
TABLE 1 UNMIX Source Solution Profiles for PM2.5 at the NETL Site. Tracer Concentrations (in μ g/m3 for particulate species and OM and ppb for all other gases) were Normalized Against PM2.5 Concentrations, μ g/m3. R2 ≥ 0.71; Signal/noise ≥ 1.2; Strength = 1.7
The average composition of each the various PM2.5 components associated with each identified source can be calculated from a combination of the average PM2.5 concentrations for a source given in and the source profile also given in the table. The majority of the ammonium sulfate is associated with material transported into the urban region. The results of the PM2.5 apportionment indicate that only 2% of the NVOM and 6% of the SVOM is associated with the PM2.5 transported to the urban area. The majority of the SVOM (83%) was secondary material produced locally. In addition, the majority of the NVOM was associated with primary emissions from the mobile gasoline source (60%).
PMF Analysis of the Data
The entire data set analyzed by UNMIX, plus trace elements determined with average concentrations at least four times the limit of quantitation were included in the PMF2 analysis. PM2.5 trace elements used included Al, Si, Cl, K, Ca, Sc, Ti, V, Cr, Mn, Fe, Ni, Cu, Zn, As, Se, Br, Rb, Sr, Y, Zr, and Pb. Protocols for and details of a typical analysis using the PMF2 program have been given (CitationPaatero 1997; CitationPaatero et al. 2002; CitationPolissar et al. 1998). Positive matrix factorization uses realistic estimated uncertainties of the individual data points to provide optimum data point scaling in the least squares solution of a factor analysis problem. The estimated uncertainties were based on the expected precision and limits of quantitation of the data from several field studies comparing collocated PC-BOSS samplers (CitationCui et al. 1998) and comparing PC-BOSS and other samplers (CitationEatough et al. 1993; CitationLewtas et al. 2001; CitationModey and Eatough 2004; CitationPang et al. 2001; CitationTang et al. 1994). These values are summarized in .
TABLE 2 Precision of the Various Data Points was calculated as LOD + [] P, where the Limits of Detection (LOD) are as given here and P was 0.080 for PM2.5 and TEOM and 0.10 for all other species
Quality of the PMF Analysis
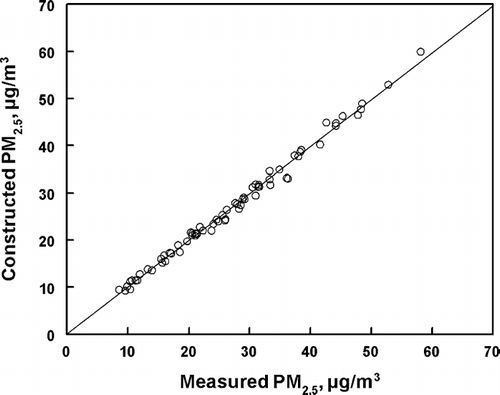
A solution with nine factors were identified as the optimal solution for the NETL data set. Solutions with less than nine factors had some factors which were clearly combinations of the nine factor solution, with a missing factor(s) (Factor 4 in the case of an 8 source solution) and with poorer overall description of the data. Solutions with more than nine factors added an addition factor with very little mass and poor mass closure. Thus, a nine source solution was considered to be optimal. The analysis of the Q value (CitationPolissar et al. 1998), the frequency distribution of the scaled residuals, and the results of the linear regression analysis of the constructed and experimental PM2.5 fine mass concentrations allow an evaluation of the quality of the fitted nine factor solution model. The “loss function,” Q, should be approximately equal to the degrees of freedom. In the data set analyzed here, there were nine factors obtained from the input data with a total of 33 input data parameters each for 71 separate sample sets. Thus the theoretical degrees of freedom are 1,148. The calculated Q value for the PMF2 nine source solution was 959, in good agreement with the theoretical value. The data set is well defined by the solution. As would be expected from this agreement of the calculated and theoretical Q values, the distribution of residuals were narrow and close to normal. Evaluation of the results by the data point screening of both species and factor concentrations (CitationPaatero and Hopke 2003) indicated there were no variables with unexpected noise in the final results. It should be noted that the experimental uncertainty in PM2.5 mass was used in the PMF analysis. We did investigate the effect of increasing the uncertainty by a factor of two and three. The PMF results were little changed by this increase in the uncertainty of the measured PM2.5 mass. The identified factors were also unchanged with respect to nature of the species present and the time pattern of the factor PM2.5 concentrations. The comparison of experimental and constructed PM2.5 mass concentrations obtained from the PMF2 solution are shown in . The linear regression comparison of the experimental PM2.5 mass concentrations and those calculated from the nine factor PMF2 solution gave (N = 71, r2 = 0.993) a slope of 0.997 ± 0.010 and an intercept of 0.00 ± 0.99 μ g/m3. The data are well described by the PMF2 solution.
FIG. 3 Comparison of PC-BOSS measured and PMF constructed concentrations of PM2.5 for the various samples included in the PMF analysis. The solid line is the regression slope.
Nature of the Resolved Sources
Since the identification of the nine factors in the PMF analysis is based only an ambient, and not source data, it is to be expected the factors may contain contributions from more than one specific source. An evaluation of apparent contributors to each factor and the probably principal source contributing to each factor follows. Thereafter, the factors are identified by that major source for better clarity. Results of the positive matrix factorization analysis are shown in , , . contains the factor profiles, normalized to the amount of material per gram of PM2.5 in the factor. gives a graphical comparison of concentrations and major PM2.5 species for each factor and gives the time pattern of the concentration of PM2.5 associated with each factor. As shown in , Factor 1 is rich in the expected soil elements Al, Si, and Fe. In addition, , the concentration of this source is generally low for all samples, as is expected for crustal material. Based on these observation, Factor 1 is identified as including crustal material. However, as shown in , in addition to the presence of the expected crustal elements, ammonium sulfate and SVOM (shown below to both be products of secondary formation processes) were major components of the PM2.5 associated with this factor. Thus Factor 1 is identified as a crustal source which includes significant associated secondary material.
FIG. 4 Factor profiles associated with the various PMF identified factors. The Y axis is in units of g species/g PM2.5 for EC, NVOM, SVOM, VOM, ammonium sulfate TEOM, and constructed NV mass (NVM); ppm/μ g PM2.5 for NO2, O3, and NO x ; and g/g PM2.5 for the elemental species. The identification of the major source contributor associated with each factor is described in the text.
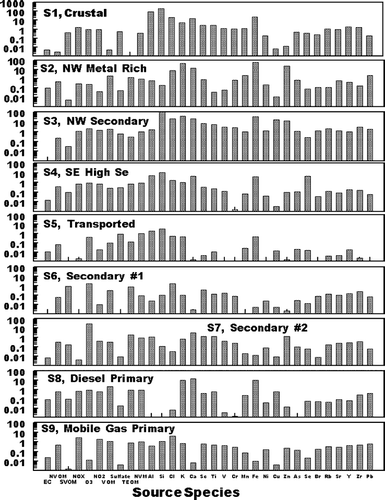
FIG. 5 Composition of the PM2.5 associated with the various PMF identified sources, see text. The area of the various pie charts is related to the average concentrations of PM2.5 from each source. The average concentration for the PM2.5 from each source is given with the caption for each pie chart. The concentrations of the various components in each pie chart are in μ g/m3.
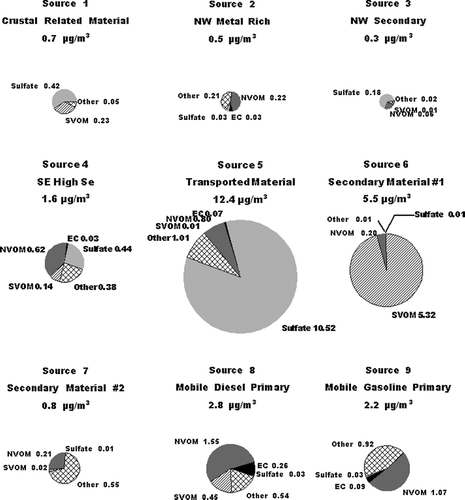
FIG. 6 Concentrations of PM2.5 associated with the various PMF identified sources. The first tick mark on the x-axis for each date represents the sample from 5:00 AM to 10:00 AM.
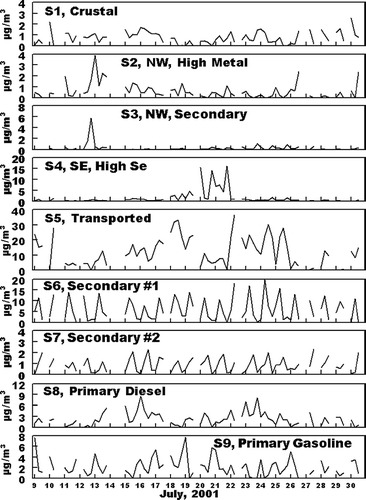
Factors 2 and 3 were both present in the highest concentrations on July 12 through 14, and were present in lower concentrations during all other sampling periods, . July 12 through 14 was the only time period with persistent transport from the northwest, as identified from HYSPLIT (CitationNOAA 2002; CitationModey and Eatough 2004, CitationModey et al. 2005) calculated back trajectories. The higher concentrations of Factors 2 and 3 at the start of this period may be associated with the transport of pollutants built up in a source area to the northwest during the relative higher pressure time period which preceded this transport. Possible sources could include the major industrial emission sources, (coal fired power plants, chemical and metallurgical industries, including the largest secondary Zn smelter in the U.S.) and a large commercial incinerator) located along the Ohio River in the vicinity of East Liverpool, Ohio, 60 km northwest of the NETL site and close to the Ohio—Pennsylvania border. Factor 2 is rich in Si, Cl, K, and transition, metals, especially Fe, Cu, and Zn. These elements are consistent with those expected for emissions from the large incinerator operation in the East Liverpool area and the secondary Zn smelter located 10 km east of the incinerator facility. The second northwest related factor, Factor 3, has comparable significant contributions from these elements and, in addition has higher contributions from O3 and NO2, . The PM2.5 from this factor is also enriched in sulfate and SVOM, relative to the concentrations seen in Factor 2. Based on these observations, Factor 2 is identified as a primary, metal rich source with a combination of pollutants from the cluster of industries to the northwest and Factor 3 as additional primary material from the same area but with significant material associated with secondary processes in emissions from these primary sources. These are referred to NW Metal Rich and NW Secondary sources, respectively.
Factor 4 was also present during a limited number of days, with high concentrations of PM2.5 from the source seen through July 19–21. This was the only time period during the study with persistent transport to the site from the south to southeast (CitationModey and Eatough 2004; CitationModey et al. 2006). The high concentrations of PM2.5 present during this time period, , suggest the source of these emissions must be relatively close. The composition of the PM2.5 in the factor ( and ) indicate emissions associated with this factor was the major contributor to Se, and a significant contributor to Al, Si, Ca, and Fe. Major nearby sources to the east including both a steel mill and large coke processing facility along the Monongahela River about 10 km to the northeast and east, respectively. However, the composition of emissions from these sources is not consistent with the profile shown in for Factor 4 (CitationWietkamp et al. 2003). A more probable source for these emissions are two coal-fired power plants located about 12 km to the southeast of the NETL sampling site. The presence of high concentrations of Si, Ca, Se, and Fe in the source profile is consistent with emissions from a coal-fired power plant with wet scrubbers. However, the Factor 4 profile also has significant amounts of sulfate, NVOM, and SVOM, and . This would be consistent with a mixture of primary emissions from coal-fired power plants, primary emissions from mobile sources or small industries in the area and secondary products formed during transport being present in the Factor 4 profile. This factor is referred to as the SE High Se Source.
Factor 5 is the dominant source of ammonium sulfate, . In addition, the occurrence of PM2.5 associated with this factor is also consistent with transport to the NETL site from the west or southwest (CitationModey and Eatough 2004; CitationModey et al. 2006). The sulfate in this source is expected to have originated from secondary processes resulting in the conversion of SO2(g) to ammonium sulfate during transport of emissions from sources to the west and southwest (CitationAnderson et al. 2004). This would include the large number of SO2 sources from coal-fired power plants, coke precessing industries and steel mills located along the Ohio River from Stubenville, 50 km west of the NETL site and other sources to the south and west of Stubenville (CitationModey and Eatough 2004; CitationAnderson et al. 2004). The presence of NVOM, EC. and trace elements in lower concentrations in the source profile are consistent with lower concentrations of primary emissions from these sources, mixed with the high concentrations of secondary ammonium sulfate formed during transport to the NETL site. Therefore, the factor is referred to as the Transported Source. As indicated in , this source is the major contributor to PM2.5 at the NETL site during the time of the study. The PM2.5 concentrations identified for Source 5 from the PMF analysis are in agreement with the concentrations for the corresponding Transported Source identified in the UNMIX analysis, see below.
The PM2.5 from the remaining factors (e.g., Factors 6, 7, 8, and 9) are present at the NETL sampling site on essentially all sampling days. It seems probable that these factors represent material from the local area. Two of the factors (Factors 6 and 7) are associated with ozone and VOM, but the factor profiles contain little in the way of elemental carbon and transition or crustal elements. This would suggest that these factors represent secondary formation products from the local photochemistry of emitted material. As indicated in , , , Factor 6 is dominated by SVOM and is present at high concentrations during the 10:00 AM to 2:00 PM sample collected each day. The PM association with this factor is dominated by SVOM with a lower concentration of NVOM. Thus, the PMF analysts suggests that the majority of the SVOM is secondary in nature and is probably formed locally from emitted VOCs. This is in agreement with previous results obtained using UNMIX (CitationAnderson et al., 2005). PM associated with Factor 6 is referred to as a Secondary #1 Source. Factor 7 is present each day during both the 10:00 AM to 2:00 PM and the 2:00 PM to 7:00 PM sampling times. Thus the SVOM and NVOM present in lower concentrations associated with this factor may represent slower conversion processes which give rise to secondary carbonaceous material throughout the midday and afternoon photochemical processes. Thus material is referred to as Secondary #2. The PMF Source 6 and Source 7 mid-day production of secondary material is comparable to the Local Secondary source identified by UNMIX as discussed below.
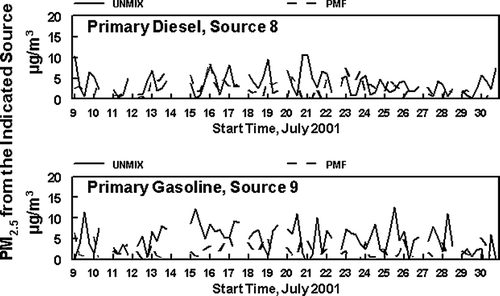
Factor 8, is attributed to Primary Mobile Diesel emissions and will include both the primary emissions and any material associated with these emissions such as road dust or rapidly formed secondary material. EC, NVOM, SVOM, and NO x are significant components in the profile for this source, , suggesting it contains diesel combustion emissions. The EC to NVOM ratio is 0.21. However, the concentrations of mobile diesel emissions identified in the PMF Factor is only about half that seen in the UNMIX analysis and the time patterns for the two apportionments are somewhat different, . Possible reasons for these differences are discussed below. Finally, Factor 9 is attributed to Primary Mobile Gasoline emissions and associated pollutants. This assignment is supported by the importance of NVOM and NO x and the lower contribution of EC, as compared to that for Factor 8. The EC to NVOM ratio for this source is 0.08, suggesting there may be some, but not major mixing of the diesel and gasoline sources in the factors. In addition, the highest concentrations of this source are routinely seen in the early morning samples, consistent with the expected contribution of the morning rush hour traffic, coupled with a lower mixing height in the morning. As was the case with the diesel source, Factor 9 also appears to contain some resuspended dust and other material which coexists with the gasoline combustion emissions. Again, the concentration of the Primary Mobile Gasoline emissions calculated with PMF are about half that seen for the UNMIX analysis and the time patterns for the two analyses are different, .
FIG. 7 Concentrations of PM2.5 associated with the UNMIX and PMF identified Mobile Gasoline and Mobile Diesel Primary Sources. The differences in the two results reflect the inability of the UNMIX analysis to identify all sources seen in the PMF analysis.
Diurnal Time Pattern and Weekday vs. Weekend for the Primary Gasoline and Diesel Sources
Primary emissions from both the gasoline and diesel source are expected to show two patterns (CitationLewis et al. 2003). Emissions should be highest during the morning and evening weekday commute time periods and lower during the midday period. The actual concentrations observed should be highest during the morning commute period because of the lower mixing height present during this time period. Because of the influence of the night-time inversion, concentrations may be higher during the night, even if emissions are lower. These expected trends are clearly seen in the PMF results for primary automobile emissions with the average weekday concentrations being 4.1, 1.6, 1.1, and 2.8 μ g PM2.5/m3 for the morning, midday, evening and night periods, respectively, . However, this expected pattern is completely lacking in the UNMIX results, the corresponding concentrations being 4.2, 4.8, 8.1, and 3.5 μ g PM2.5/m3, indicating that the PMF results are the more reasonable. A reasonable pattern during the weekday is also seen for primary diesel emissions in the PMF analysis, the average weekday concentrations being 2.5, 2.7, 1.8, and 2.7 μ g PM2.5/m3 for the morning, midday, evening and night periods, respectively, . The higher concentrations of primary diesel emissions during the midday period probably reflects the increased truck traffic in the suburban location during the midday work period, e.g., see location of NETL sampling site in .
FIG. 8 Diurnal variation of the concentrations of Primary Gasoline and Primary Diesel Sources as a function of time of day and day of the week.
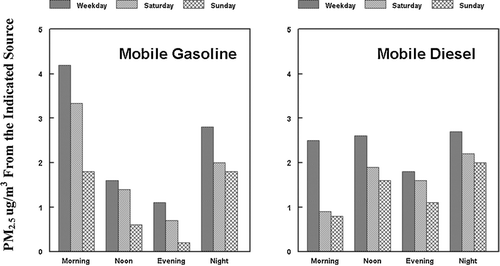
Changes in the morning concentrations of primary gasoline emissions are also consistent in the PMF results for the weekend, as compared to the weekday periods at the suburban NETL location, average concentrations being 4.2, 4.3, and 1.8 μ g PM2.5/m3 for the weekday, Saturday and Sunday results, respectively. The high Saturday morning concentration is somewhat unexpected. However, regular decreases are seen in the midday and evening comparisons in going from the weekday, Saturday to Sunday concentrations, with very low Sunday concentrations for the primary gasoline emission concentrations at NETL, . Similar, but less pronounced trends are seen in the primary diesel emission data, .
Contributions to the Measured Aerosol
The three major PM2.5 components associated with the apportioned PM2.5 mass are ammonium sulfate, SVOM, and NVOM, . The average concentration of ammonium sulfate during the July intensive study period was 11.7 μ m/m3. Of this total, a maximum of only 7.8% (0.92 μ g/m3) was primary. However, the sulfate associated with three of the possible primary sources (0.42 μ g/m3 from Crustal Related Material, 0.44 μ g/m3 from the SE High Se Source, expected to be from coal-fired power plants, and 0.21 μ g/m3 from the two NW Sources) is probably dominated by secondary material formed after emission from an SO2 source. Thus, only the 0.03 μ g/m3 from the Diesel Source and 0.03 μ g/m3 from the Gasoline Source (0.4% of the total) are clearly primary. The major source of sulfate was the secondary material transported from the west and southwest, 10.5 μ g/m3, or 90% of the total.
NVOM averaged 4.7 μ g/m3. An average of 2.6 μ g/m3 (55% of the total) was primary material from gasoline and diesel combustion emissions, with the contribution being about the same from gasoline, 1.1 μ g/m3, or diesel, 1.5 μ g/m3, primary emissions. The major secondary source was the transported material from the west and southwest, Source 5, which averaged 0.8 μ g/m3, or 35% of the secondary NVOM.
In contrast, the great majority of the SVOM was secondary. SVOM averaged 6.3 μ g/m3. An average of 5.3 μ g/m3 (84% of the total SVOM) was associated with the local secondary production of SVOM during the middle of the day. An additional 6% of the total was from other local secondary formation processes. Only 0.6 μ g/m3 (9% of the total) was attributed to primary emissions, with the most important being the Mobile Diesel Primary Source, which could contain some secondary SVOM material formed rapidly after emission.
The combination of ammonium sulfate, NVOM, and SVOM accounted for an average of 16.5 μ g/m3 of secondary material, over 90% of the total secondary attributed mass. In contrast, primary material accounted for an average of 7.1 μ g/m3, with 70% of the total being attributed to primary gasoline and diesel combustion engine emissions. An average of 4.7 μ g/m3 of the primary material was organic material. An additional average of 0.5μ g/m3 was attributed to EC. These species make up 73% of the primary material identified by the PMF analysis. An average of 26% appears to be associated with the metals and associated oxides, etc. identified by the PIXE analysis.
Comparison with the UNMIX Analysis Results
The results obtained by the UNMIX and PMF analyses can be compared in a few cases.
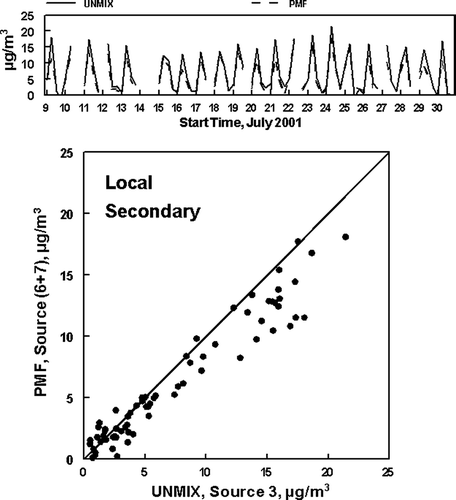
The first is the comparison of the local secondary sources. UNMIX was able to identify only one such source, UNMIX Source 3 in and . In contrast, the PMF analysis identified two such sources with different time patterns, Source 6 and Source 7. The concentrations of UNMIX Source 3 are compared to the sum of the PMF Source 6 and Source 7 results in . As indicated, there is quite reasonable agreement between the two results with the UNMIX solution occasionally giving higher concentrations. Occasional higher UNMIX concentrations would be expected since fewer sources were identified by UNMIX. Comparison of the results given in and reveals that some of the days for which UNMIX gave the higher concentrations included all the days when appreciable concentrations were seen for either Sources 2, 3, or 4 (e.g., July 13, 20, and 21), the sources seen only under specific wind direction conditions. This comparison suggests that UNMIX was not able to identify these sources from the limited days when they were present at the site and that the sources were included in the identified secondary material.
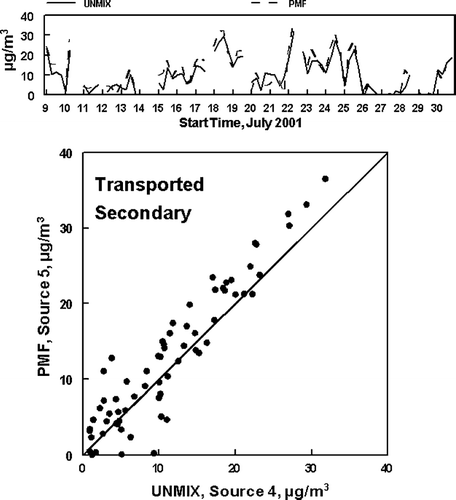
FIG. 9 Concentrations of PM2.5 associated with the UNMIX and PMF identified Transported Secondary sources. The first tick mark on the x-axis for each date in the top graph represents the sample from 5:00 AM to 10:00 AM. The lower graph is the x−y comparison of the data. The line is the slope = 1 line.
The second comparison which can be made is of the transported source, UNMIX Source 4 and PMF Source 5. The concentrations calculated for these two sources are compared in . As shown in , the temporal agreement between the two methods is very good. However, there is a general tendency for a bias between the two calculated concentrations, with PMF giving the higher result. Comparison of the results given in and indicate this is a result of the generally higher attribution of ammonium sulfate, and to a lesser extent NVOM and EC to the Transported PM2.5 in the PMF analysis. The average concentration of ammonium sulfate was 10.5 μ g/m3 for the PMF analysis and only 9.3 μ g/m3 for the UNMIX result, accounting for some of the PM2.5 mass difference in the two analyses. The much smaller concentrations of EC and NVOM were higher for the PMF analysis by about a factor of two but the SVOM inferred by the UNMIX analysis was the higher.
FIG. 10 Concentrations of PM2.5 associated with the UNMIX identified Local Secondary source and the sum of the PMF two PMF identified Local secondary sources. The first tick mark on the x-axis for each date in the top graph represents the sample from 5:00 AM to 10:00 AM. The lower graph is the x−y comparison of the data. The line is the slope = 1 line.
The two remaining UNMIX sources which can be compared to the PMF results is UNMIX diesel and gasoline sources, . The average concentrations for the diesel source obtained from the UNMIX analysis was 4.9 μ g/m3, compared to the much lower PMF value of 2.8 μ g/m3. Likewise, the concentrations for the gasoline source obtained from the UNMIX analysis was 3.7 μ g/m3, compared to the lower PMF value of 2.2 μ g/m3. Both the concentrations and diurnal patterns obtained with PMF and UNMIX are different for these sources. This probably reflects the inability of UNMIX to identify the additional sources found in the PMF analysis.
Attribution of EC and OM
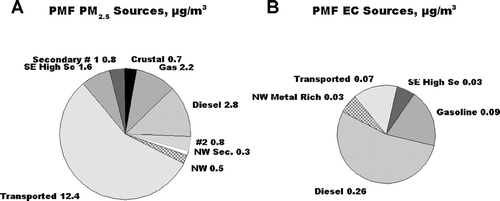
An additional interesting observation which can be made with respect to the PMF analysis result is the attribution of EC in the analysis. It is sometimes assumed that elemental carbon is a unique tracer for primary diesel emissions. Other sources of EC are assumed to be negligible. The PMF analysis results give an opportunity to examine that assumption for the July Pittsburgh special study. Pie charts giving the average PM2.5 and average EC attributed to each of the various sources are given in . As shown, the Diesel source is a minor source, being only 10% of the total PM2.5, but it is associated with 52% of the total EC. This reflects that the EC:PM2.5 ratios for the four identified major sources of EC are 0.090 g EC/g PM2.5 for the Mobile Diesel Primary source, compared to 0.043, 0.005, 0.055, and 0.018 g EC/g PM2.5, for the four other major contributors to EC, NW, Mobile Gas Primary, Transported Material, NW Metal Rich and SE high SE sources, respectively. Attempts were made using PMF control parameters to reduce EC in the Mobile Gas Primary Source. However, this results in an increase in the EC in the Transported Source, and not in the Diesel Source. The Diesel profile given in of 0.090 g EC/g PM2.5 is the maximum EC that can be attributed to Mobile Diesel Primary emissions using PMF. The largest contributor to the total EC is the Diesel source. Mobile Gas Primary emissions have a lower EC:PM2.5 ratio, however, the concentration of PM2.5 from this source is comparable to that for Diesel, hence, the Gasoline source is a significant contributor to the total EC (19%). The ratio of EC to PM2.5 is even lower in the Transported Material Source (0.005 g EC/g PM2.5), but the high concentration of PM2.5 from this source results in 13% of the EC being from this source. EC would be expected in this source because of the presence of coke processing facilities in addition to the normal mobile sources in the Ohio River Valley which is the point of origin of this material. The ratio of EC to PM2.5 is quite high in the NW Metal Rich source, 0.055 g EC/g PM2.5, but the lower concentration of PM2.5 from this source results in only 6% of the EC being from this source. Again, coke oven emissions are expected to be present in this source. An important point to be made is that the assumption that EC could be used as a tracer of diesel emissions would have over-estimated the diesel source contribution by about a factor of two.
FIG. 11 Average contribution of the various identified sources to A. PM2.5 and B. EC.
An additional point to consider is the NVOM/EC ratio for the two major primary sources of carbonaceous material These ratios are 6 and 12 g NVOM/ g EC for the Mobile Diesel Primary and Mobile Gasoline Primary sources, respectively. While the first ratio is comparable to that used in OM/EC based calculation of primary and secondary carbonaceous material (CitationCabada et al. 2004; CitationTurpin et al. 1991), the second value is more comparable to ratios assumed to represent secondary material. The results obtained using the OM/EC approach would be quite different from that obtained in the PMF analysis of the data set.
Sources of VOM
An additional parameter important to the source apportionment reported here is the composition of VOM in various combustion emissions, since this VOM will contribute to the formation of secondary particulate material. This contribution is expected to be important in Primary Gasoline Source emissions, but was not identified in the UNMIX analysis. The sources of VOM were estimated using the PMF model with VOM as the fitted parameter. Because VOM was not identified in the crustal material, , while at the same time, significant fractions of many of the species in the analysis originated from crustal material, the data were first corrected for all crustal material contributions using the PM2.5 PMF results. PMF analysis of VOM sources was then done using the adjusted data. The results of this analysis are given in . An attempt was made in both the PM2.5 and VOM analysis to see if a VOM source could be identified which contained only VOM with minor contributions from any other fine particulate species. Such a source would be expected, for example, for vegetative gas phase organic material emissions. However, such a source could not be identified in the various analyses. Rather, eight sources whose time patterns were consistent with each of the non-crustal PM2.5 sources were identified. The Q value obtained for the PMF apportionment of VOM for the eight source solution summarized in was 1148, just slightly higher than the value of 959 found for the nine source PM2.5 apportionment. Linear regression analysis of the VOM measured and PMF calculated results gave R2 = 0.72 with a slope of 0.95 ± 0.02. The sigma value for the comparison was ± 1.5 ppb (± 11%), comparable to the expected precision of the VOM data of ± 10%. Calculated and measured VOM were in agreement and the results were within the precision of the VOM data.
TABLE 3 Sources of VOM as identified by the PMF analysis of PM2.5 and of VOM. Average concentrations are in ppm VOM
The results of the apportionment of VOM are given in . As indicated, with the exception of the crustal material, all sources of PM2.5 could be identified as sources of VOM. In addition, the amount of VOM predicted to be present from each source from the VOM and the PM2.5 PMF analyses were in reasonable agreement. The major sources of VOM were the Mobile Primary Diesel (30% of the total), Primary Gasoline (23% of the total) and the Local Secondary # 1 (16% of the total). The VOM from each of the anthropogenic sources followed the same diurnal pattern as PM2.5 from that source. In addition, total VOM identified by the two analyses were also comparable, . Likewise, total PM2.5 identified by the two analyses were also in agreement, , the difference between the total PM2.5 for the PM and VOM analyses given in of 0.8 μ g/m3 being the correction applied to the PM data for the crustal contribution in the VOM PMF analysis.
FIG. 12 Concentrations of VOM associated with the various PMF identified sources. The first tick mark on the x-axis for each date represents the sample from 5:00 AM to 10:00 AM.
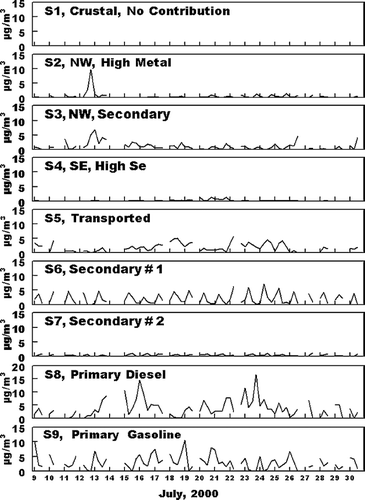
TABLE 4 Sources of PM2.5 as identified by the PMF analysis of PM2.5 and of VOM. Average concentrations are in μ g/m3
CONCLUSIONS
Four per day samples of PM2.5 concentration and composition, combined with appropriate gas phase compounds have been used in the PMF based attribution of PM2.5 during the July 2001 summer intensive study at the NETL PM characterization site. The PMF analysis was able to identify nine sources, compared to a total of four sources which could be identified by UNMIX analysis of the same data set. The sum of the PM2.5 concentrations for these various sources were in excellent agreement with the measured PM2.5 concentrations for all samples. Three local primary sources were identified, Crustal, Mobile Diesel and Mobile Gas. The mobile sources were comparable in concentration (2.8 and 2.2 μ g/m3 for Diesel and Gas, respectively) and higher by about a factor of three to four than the crustal source, which averaged 0.7 μ g/m3. However, other major local sources were two secondary sources which appeared to be associated with photochemical processes, 5.5 μ g/m3 during the mid-day period (dominated by SVOM) and 0.8 μ g/m3 during the afternoon (dominated by NVOM). Two minor sources from the northwest, which appear to be associated with incinerator, secondary zinc smelter, chemical industry and power pant emissions, and one from the southeast, from coal-fired power plant emissions, were identified. These sources were only present in significant concentrations on a few time periods when transport was from these direction. The major source identified, an average of 12.4 μ g/m3, was associated with transport of both primary and secondary material from sources along the Ohio River Valley (CitationAnderson et al. 2004). A total of 85% of this long-distance transported PM2.5 was secondary ammonium sulfate. Both ammonium sulfate and SVOM were dominated by secondary processes. Non-volatile organic material was about equally divided between primary and secondary sources.
The results of this PMF source apportionment analysis indicate that local primary anthropogenic emissions (including the coal-fired power plant emissions which appear to originate from the southern portion of the Pittsburgh metropolitan area) averaged only 6.1 μ g/m3. Secondary material associated with these primary emissions averaged 6.6 μ g/m3. Of the total average of 12.9 μ g/m3 PM2.5 from local emissions and atmospheric conversion processes, 5.8 μ g/m3 is semi-volatile organic material which would not be monitored with a PM2.5 Federal Reference Method monitor. Thus, if only local contributions were considered, the new PM2.5 24-h standard would be easily met at the NETL site during the July study period (CitationMusick 2000). This is the period when highest PM2.5 concentrations are typically seen (CitationModey and Eatough, 2004). However, primary and secondary pollutants which originated from outside the Pittsburgh Metropolitan area averaged 13.2 μ g/m3. Any protocol for the attainment of the PM2.5 standard must take both local and transported sources of PM2.5 into account.
The participation of Brigham Young University in this program was funded under University Coal Research Grant DE-FG26-99FT40581 from the U.S. Department of Energy, National Energy Technology Laboratory, to Brigham Young University, and by an Oak Ridge Institute for Scientific Exchange appointment for Delbert J. Eatough. Reference in this report to any specific commercial product, process, or service is to facilitate understanding and does not necessarily imply its endorsement or favoring by the U.S. Department of Energy.
Notes
a Units are all ng/m3.
Related Research Data
REFERENCES
- Anderson , R. R. , Irdi , G. , Lynn , R. , Martello , D. V. , Rohar , P. C. , Tamilia , J. P. , Veloski , G. A. , Waldner , K. , White , C. M. and Eatough , D. J. 2006 . The DOE NETL OST PM2.5 Characterization Program . Environ. Sci. Technol. , Submitted[CSA]
- Anderson , R. R. , Modey , W. K. , Martello , D. V. , Lucas , L. J. , Davidson , C. I. and Eatough , D. J. 2005 . Apportionment of Ambient Primary and Secondary Pollutants during a 2001 Summer Study in Pittsburgh Using EPA UNMIX . J. Air & Waste Manage. Assoc. , in press[CSA]
- Anderson , R. R. , Martello , D. V. , White , C. M. , Crist , K. C. , Kuruvilla , J. , Modey , W. K. and Eatough , D. J. 2004 . The Regional Nature of PM2.5 Episodes in the Upper Ohio River Valley . J. Air & Waste Manage. Assoc. , 54 : 971 – 984 . [CSA]
- Anderson , R. R. 2002 . Pittsburgh, PA : National Energy Technology Laboratory . 2002, unpublished data
- Cabada , J. C. , Pandis , S. N. , Subramanian , R. , Robinson , A. L. , Polidori , A. and Turpin , B. 2004 . Estimating the secondary organic aerosol contribution to PM2.5 using the OC to EC ratio method . Aerosol Sci. Technol. , 38 : 140 – 155 . [CROSSREF] [CSA]
- Cui , W. , Eatough , D. E. and Eatough , N. L. 1998 . Fine particulate organic material in the Los Angeles Basin—I: assesment of the high-volume Brigham Young Univeristy organic sampling system, BIG BOSS . J. Air & Waste Manage. Assoc. , 48 : 1024 – 1037 . [CSA]
- Eatough , D. J. , Wadsworth , A. , Eatough , D. A. , Crawford , J. W. , Hansen , L. D. and Lewis , E. A. 1993 . A multi-system, multi-channel diffusion denuder sampler for the determination of fine particulate organic material in the atmosphere . Atmos. Environ. , 27A : 1213 – 1219 . [CSA]
- Ellis , E. C. and Novakov , T. 1982 . Application of thermal analysis to the characterization of organic aerosol particles . Sci. Total Environ. , 23 : 227 – 238 . [CROSSREF] [CSA]
- Henry , R. C. and Norris , G. A. 2002 . EPA UNMIX 2.3 analysis software, http://www.epa.gov/ttnamti1/files/ambient/pm25/workshop/unmix2.pdf
- Lewtas , J. , Booth , D. , Pang , Y. , Reimer , S. , Eatough , D. J. and Gundel , L. 2001 . Comparison of sampling methods for semi-volatile organic carbon (SVOM) associated with PM2.5 . Aerosol Sci. Technol. , 34 : 9 – 22 . [CROSSREF] [CSA]
- Lewis , C. W. , Norris , G. A. , Conner , T. L. and Henry , R. C. 2003 . Source Apportionment of Phoenix PM2.5 Aerosol with the UNMIX Receptor Model . J. Air & Waste Manage. Assoc. , 53 : 325 – 338 . [CSA]
- Mangelson , N. F. , Nielson , K. K. , Eatough , D. J. , Christensen , J. J. , Izatt , R. M. and Richards , D. O. 1979 . Proton Induced X-Ray Emission Analysis of Pima Indian Autopsy Tissues . Analytical Chemistry , 51 : 1187 – 1194 . [INFOTRIEVE] [CROSSREF] [CSA]
- Modey , W. K. , Eatough , J. and Eatough , D. J. 2004 . Twenty-Four-Hour PC-BOSS Air-Monitoring Results from the NETL Fine-Particulate Sampling Site in Pittsburgh, Pennsylvania: An Annual Perspective . Aerosol Sci. Technol. , 38 : 194 – 204 . [CROSSREF] [CSA]
- Modey , W. K. , Anderson , R. R. , Martello , D. V. , Takahama , S. , Lucas , L. J. , Davidson , C. I. and Eatough , D. J. 2006 . Meteorological influence on, and patterns in ambient fine particulate chemical composition at two sampling sites in metropolitan Pittsburgh: A 2001 intensive summer study . Atmos. Environ. , Special Issue on Supersite and Related Studies, in press[CSA]
- Musick , D. 2000 . A summary of the ambient air program for PM2.5 . Environ. Manager , February 2000[CSA]
- NOAA . 2002 . http://www.arl.noaa.gov/ready/hysplit4.html
- Paatero , P. 1997 . Least squares formulation of robust, non-negative factor analysis . Chemomet. Intell. Lab. Syst. , 37 : 13 – 35 . [CROSSREF] [CSA]
- Paatero , P. and Hopke , P. K. 2003 . Discarding or Downweighing High-Noise Variables in Factor Analysis Models . Anal. Chim. Acta , 490 : 277 – 289 . [CROSSREF] [CSA]
- Paatero , P. , Hopke , P. K. , Song , X. -H. and Ramadan , Z. 2002 . Understanding and controlling rotations in factor analytic models . Chemomet. Intell. Lab. Syst. , 60 : 253 – 264 . [CROSSREF] [CSA]
- Pang , Y. , Eatough , N. L. and Eatough , D. J. 2002 . PM2.5 Semivolatile Organic Material at Riverside, California: Implications for the PM2.5 Federal Reference Method Sampler . Aerosol Sci. Technol. , 36 : 277 – 288 . [CROSSREF] [CSA]
- Polissar , A. V. , Hopke , P. , Malm , W. C. and Sisler , J. F. 1998 . Atmospheric aerosol over Alaska. 2. Elemental composition and sources . J. Geophys. Res. , 103 : 19,045 – 10,057 . [CSA]
- Schauer , J. J. and Cass , G. R. 2000 . Source apportionment of wintertime gas-phase and particle-phase air pollutants using organic compounds as tracers . Environ. Sci. Technol. , 34 : 1821 – 1832 . [CROSSREF] [CSA]
- Tang , H. , Lewis , E. A. , Eatough , D. J. , Burton , R. M. and Farber , R. J. 1994 . Determination of the Particle Size Distribution and Chemical Composition of Semi-volatile Organic Compounds in Atmospheric Fine Particles with a Diffusion Denuder Sapling System . Atmos. Environ. , 28 : 939 – 947 . [CROSSREF] [CSA]
- Turpin , B. J. and Lim , H. -J. 2001 . Species contributions to PM2.5 mass concentrations: Revisiting common assumptions for estimating organic mass . Aerosol Sci. Technol. , 35 : 602 – 610 . [CSA]
- Turpin , B. J. , Huntzicker , J. J. , Larson , S. M. and Cass , G. R. 1991 . Los Angeles summer midday particulate carbon: Primary and secondary aerosol . Environ. Sci. Technol. , 25 : 1788 – 1793 . [CROSSREF] [CSA]
- Wietkamp , E. , Lipsky , E. , Robinson , A. , Anderson , N. , Leifeste , H. , Subramanian , R. , Cabada-Amaya , J. , Khlystov , A. , Stanier , C. , Lucas , L. , Takahama , S. , Wittig , B. , Davidson , C. , Pandis , S. , Polidori , A. , Lim , J. -J. , Turpin , B. , Pancras , P. and Ondov , J. Fenceline sampling adjacent to a large coke production facility in Pittsburgh, PA . Presented at the Association of Aerosol Research meeting, Particulate Matter: Atmospheric Sciences, Exposure, and the Fourth Colloquium on PM and Human Health . March 2003 , Pittsburgh, PA.