Abstract
A new instrument to measure the size-resolved hygroscopic growth of both sub- and super-micron atmospheric aerosol is described. It consists of two white-light optical particle counters measuring the same sample aerosol simultaneously at two different controlled relative humidities. Calibration with aerosols of different refractive index confirms the expected relative insensitivity of the instrument to index of refraction. Data obtained in the field from airborne sampling support the utility of the instrument in measuring differences in size-resolved hygroscopicity in the marine boundary layer and also in addressing the issue of kinetic limitations to aerosol condensational growth.
1. INTRODUCTION
The interaction of aerosols with water in the atmosphere is of fundamental importance in determining the physical and chemical characteristics of the aerosols. Because atmospheric aerosols, particularly marine aerosols, are largely soluble, or have soluble coatings, condensation of water vapor upon them takes place as relative humidity increases and, conversely, evaporation takes place as relative humidity decreases. Hence, their mass, volume or size, mean density, mean refractive index, and derivative radiative properties all depend on the atmospheric relative humidity. This is particularly important in marine environments where humidities are high and gradients large. In light of this, it is not surprising that the degree of aerosol hydration has been shown to be of great significance to the direct impact of aerosols on the atmospheric radiative balance (e.g., CitationCharlson et al. 1992; CitationHegg et al. 1997). Additionally, the interaction of aerosols and water subsumes the activation of such aerosols as cloud condensation nuclei (CCN); indeed this phenomenon is simply an extension of aerosol hydration to humidities at or greater than 100%. Hence, the important and established relationship between aerosol and cloud drop number concentration, with its implications for indirect radiative forcing, is also a facet of aerosol-water interactions (cf., CitationHenning et al. 2002). Finally, aerosol hygroscopicity is also an important factor in the chemical reactivity of aerosols, modulating the interaction of the aerosol surface with numerous gas phase species (CitationRossi 2003; CitationRudich 2003).
An important aspect of aerosol hygroscopicity is its size dependence. This size dependence derives largely from size-dependent chemistry and, given the strong interrelationship between solubility and aerosol lifetime, and vertical distribution (CitationRodhe 1983), plays an important role in the life cycle of various chemical species in the atmosphere. It can, furthermore, be an important factor in determining which aerosol types impact the atmospheric radiation balance. For example, super-micron sea salt can dominate aerosol light scattering in the MBL, in substantial part because of its high hygroscopicity.
Another facet of the issue of aerosol hygroscopicity and clouds encompasses the question of the impact of organics on the activation of aerosol particles as CCN, specifically delaying such activation. The concept of retardation of droplet growth by organic films was broached many years ago (cf., CitationPodzime and Saad 1975; CitationGill et al. 1983). More recently, a number of theoretical assessments have made rather startling claims about the impact of organics on CCN activation and consequently on cloud microphysics and the associated radiative properties of clouds (e.g., CitationChuang et al. 1997; CitationFeingold and Chuang 2002; CitationFacchini et al. 1999). Evidence from laboratory studies is mixed, with some providing support (e.g., CitationHegg et al. 2001; CitationShantz et al. 2003; CitationXiong et al. 1998) but others showing little impact (e.g., CitationCruz and Pandis 1998). The issue is really a question of time scales. Certainly, with sufficient time, water can penetrate even rather thick films of some organic coatings (cf., CitationThomas et al. 1999). As can be inferred from the study of CitationHegg et al. (2001), if the cloud supersaturation field were constant at some value for even a few minutes, the plausible growth delays would have negligible impact. In actual clouds, the time variation of the saturation ratio in an ascending parcel (or, more generally, a cooling parcel) can be acute. The peak supersaturation may only persist for a few seconds after the first droplets activate. Hence, delays of a few seconds in the growth of droplets might preclude their growth to their activation radius prior to the collapse of the supersaturation field due to vapor depletion. In fact, this is precisely the analysis undertaken by CitationShantz et al. (2003), consisting of measured activation delays at various supersaturations in the laboratory followed by incorporation of these delays into a cloud parcel model that predicted supersaturation as a function of time. Significant impacts were found when comparison was made with the typical model formulation that assumes that droplets activate when they reach their critical radius under diffusional growth of inorganic haze particles. All studies agree, however, that the magnitude of the effect will depend on the nature of the organics present and their distribution with size, as well, of course, as on the cloud dynamics. Clearly, only atmospheric measurements can decisively address the issue and here, in contrast to the laboratory and theoretical work, very few analyses are in hand on growth time delays.
Work by CitationBigg (1986) suggested substantial delays in the activation of atmospheric CCN in a static diffusion chamber, with delay times of perhaps as much as 30 seconds being cited. Several more recent studies have suggested activation delays induced by organics as an explanation for discrepancies between predicted and observed CCN concentrations (e.g., CitationChuang et al. 2000). However, the only study of which we are aware that has actually quantified delay times in droplet growth for atmospheric aerosols is that of CitationChuang (2003). In this study, comparison of droplet diameters for various growth times to the measured equilibrium diameters of various sized particles revealed growth delays on the order of three seconds or more for a small subset (ca. 2% by number) of the ambient aerosol sampled. The measurements were made at the surface in Mexico City in the fall of 2000. An interesting aspect of the results was the different temporal patterns observed for delays as a function of the particle size; the sizes examined being 50 and 100 nm. The 100 nm particles most frequently showed delays in the 8–11 a.m. time frame while the 50 nm particles most frequently showed such delays in the afternoon. Although only a small percentage of the measured particles appeared to exhibit growth delays ≥3 s, the study showed that such delays could in fact occur in atmospheric aerosols. Furthermore, delays of one to three seconds were likely for a larger fraction of the particles. The restricted particle size range, venue and season examined preclude any definitive assessment of the incidence of such delays in the atmosphere as a whole. Furthermore, in the MBL there is the issue of aging of potential organic layers prior to encounters with clouds. Organic layers have in fact been found on marine aerosol near the surface (CitationTervahattu et al. 2002) but not collated with observed changes in aerosol hygroscopicity. Such films would be expected to oxidize as the aerosol are transported upwards to cloud levels but the time scale for such oxidation, and thus changes in hygroscopicity, is unclear.
While the above issues are clearly important, few data are available with which to address them. Size-dependent aerosol hygroscopicity was first investigated using aerosols deposited on substrates and latter hydrated in laboratory settings (cf., CitationWinkler and Junge 1972). Such data are difficult to acquire and there is always concern about the impact of the substrate on the results. More recent work has generally involved the use of Tandem Differential Mobility Analyzers (TDMAs), which have the advantage of measuring in situ. However, nearly all such TDMA data only measures hygroscopic growth up to around 0.5 μ m dry diameter due to the upper size limit of most DMAs (e.g., CitationSwietlicki et al. 2000; CitationSantarpia et al. 2004).
One obvious methodology to get around the DMA limit would be to use optical particle counters (OPCs) as the particle sizing technique. There have in fact been a number of such studies addressing the issue of aerosol hygroscopicity that have utilized OPCs. CitationTang (1980) used a Carl Zeiss OPC to measure the change in size of particles of known composition as a function of RH in a laboratory setting. CitationSchumann (1990) used a modified Climet CI-8060 OPC, switching between humidified and non-humidified inlets, to obtain hygroscopic growth data on aerosols at a ground site in the Alps. However, quantitative assessment of size-dependent hygroscopic growth was not made. Similarly, Citationten Brink et al. (2000) used a PMS LAS-X laser OPC to examine the wet and dry size distributions of ambient aerosols at a ground site in the Netherlands. Once again, quantitative assessment of hygroscopic growth was not done with the OPCs (though excellent results were obtained on overall growth with humidity-controlled nephelometers), due in part to reported problems with sample heating. More quantitatively, Kreisberg et al. (2001) employed a High Flow DMA followed by an optical particle detector in conjunction with relative humidity control. This system permitted hygroscopicity measurements up to 1.1 μ m diameter but little ambient data was reported in this study and further work has not, to our knowledge, been reported. Finally, CitationHand et al. (2000) used PMS ASASP-X laser spectrometers as detectors in measurement of particle size as a function of RH. However, the ASASP-X response to particles is a strong function of their refractive index, necessitating an elaborate and non-deterministic correction process. Furthermore, once again no size-resolved hygroscopicity data are reported.
None of the instruments discussed in the above studies have been used in airborne sampling and all require rather long sampling times. Very little actual ambient data on size-resolved aerosol hygroscopicity have been reported. There thus appears to be a need for a field instrument that can measure aerosol hygroscopicity in situ at supermicron sizes, particularly for airborne use. For marine air, these measurements should encompass most of the sea salt mode and thus extend out to 3–4 μ m diameter (CitationReid et al. 2006). Furthermore, an instrument that would also permit assessment of the kinetic limits to equilibration of particles at ambient RH would be very useful.
2. THE AEROSOL HYDRATION SPECTROMETER
2.1. Hardware
We have fabricated an instrument to address the above issues that we call the Aerosol Hydration Spectrometer (AHS). It is relatively portable and can be deployed on aircraft. It can be configured in three distinct ways: (1) measure the nonhydrated (or less hydrated) number-size distribution from 0.2 to 8 μ m at ca 50% RH, (2) measure the hydrated size distribution at 85 to 95% RH, and (3) measure differences in condensational growth between the standard growth time of ∼ 2 s and a growth time ∼ 42 s longer, thus permitting assessment of kinetic limits to the aerosol growth to equilibrium with higher RHs. is a schematic of the system that forms the basis for the following discussion of the system's details. (Note that the entire system as shown is in a vertical plane to minimize sedimentation problems.) The configuration permits several control measurements with particle-free air or monodisperse calibration particles, and identical RH and time delays in each particle sensor to insure identical operation on which the differential analysis of the data depends.
FIG. 1 Schematic of the AHS.
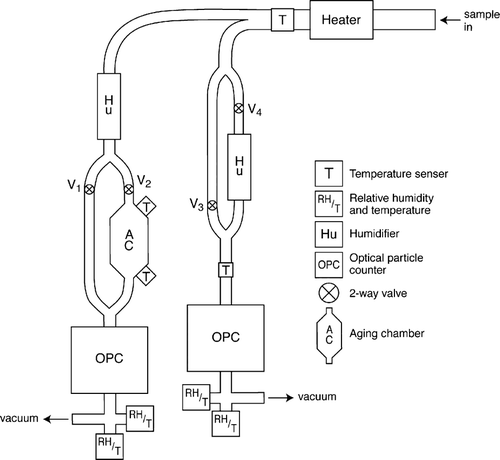
One aspect of the design which should be noted ab initio is the use of white light 90° scattering Optical Particle Counters (OPCs). The white light is supplied by Xenon short arc lamps (75 W) and the scattered pulses registered by a 4096 channel pulse height analyzer. The advantage of the white-light, 90° scattering OPCs is quite simple. The Mie resonance peaks in the critical sub-10 μ m size range are much reduced, essentially washed-out by the superposed multiple wavelengths of white light. This results in a detector response that is linear and monotonic with particle size. This, in turn, renders determination of the aerosol size distribution in the 0.1–10 μ m size range much more certain. Recent advances in the design of white-light OPCs have yielded several attractive commercial instruments. From these, we have selected the Welas 1200 optical systems manufactured by Palas GmbH (Greschbachstr. 3b, D-76229, Karsruhe, Germany). These high-resolution instruments are specified to cover the size range from 0.18 to 40 μ m diameter and have a dynamic range up to 105 cm− 3. Though our own measurements suggest an upper concentration limit substantially lower than this (∼ 2 × 104 cm− 3), the range is more than adequate for most applications in ambient air. The sensor volumes are quite small and permit very short measurement time constants (< 1 s). However, one modification to the standard instrument was necessary to meet our measurement goals. When tested upon receipt from the manufacturer, it was found that the flow constriction (essentially a jet) just upstream of the scattering volume, designed to focus the flow and decrease residence time in the sensed volume, was insufficient to permit differentiation of the smallest particle pulses from noise. We therefore redesigned the jet to more narrowly focus the sample air and also to reduce turbulence by means of a more gradual transition to the jet orifice. The new orifice narrows the flow channel from 0.6 cm diameter to 0.3 cm diameter over a distance of 0.4 cm, the profile being a hyperbolic section. The new jet channel is 0.8 cm in length, identical to the original jet length. After this modification, the detection efficiency of the two channels was assessed by monodisperse aerosols of various sized generated by a DMA and compared to a TSI 3010 CN counter. The efficiency was unity in both channels down to 0.5 μ m. At 0.26 μ m, close to the lower detection limit, the efficiencies had dropped to 0.74 for the delay channel and 0.68 for the direct. From this data, corrections were made to render the channels equivalent.
While the manufacturer supplies clear calibration curves using latex spheres (showing the expected single-valued relationship between particle size and output pulse height), we have made our own calibrations with a variety of particle sizes and compositions similar to those to be encountered in the atmosphere. The test aerosol calibration will be applied to the sensors' pulse height data at RHs representative of the atmosphere to assess discrimination of sizes under conditions of varying index of refraction.
For aircraft use, air enters the aircraft and AHS system through an isokinetic inlet whose transmission efficiency has been established through extensive tests (CitationHegg et al. 2005). The 50% cutoff size is ∼ 8 μ m. After the inlet, the sample air is heated as necessary ca. 10°C to obtain a nominal RH in the 40–50% range. This “reference” RH has been selected to ensure that the aerosol has not effloresced, i.e., it is still on the upper branch of any potential hysteresis loop. The particles are thus always droplets, whose sizes as determined by the OPCs are easily interpretable. Furthermore, this represents the hydration degree and changes therein experienced by marine boundary layer aerosols and also allows hygroscopicity data to be fit by simple functions to interpolate to various desired RHs (cf., CitationKotchenruther et al. 1999). After the initial drying, the flow is then divided; the first, left, branch in passes into a humidifier which actively controls the sample stream RH to a nominal value of 85–90%. The humidifiers shown are microporous tubes with a temperature-controlled water jacket (CitationGasso et al. 2000). This stream then passes through one of two possible routes depending on the valve settings. In the first, it passes directly into the first OPC detector. In the second it first passes through an aging volume before entering the detector. In both instances, just after leaving the detector, the RH is monitored with a Vaisala RH sensor, which acts as a controller for the humidifier. The ageing volume is a stainless steel chamber of ∼ 15 times the diameter of the flow tubing (see below), appreciably increasing the time spent by the sample aerosol at the control RH prior to entering the detector. This assures that the aerosol equilibrates with this RH prior to measurement. As shown, temperature sensors are located at two points in the aging chamber. Coupled with an assumption of no appreciable vapor loss, this permits us to verify the flow RH remains at the nominal value throughout the flow path. We currently follow CitationChuang (2003) in estimating a time interval of > 30 seconds should suffice to reach equilibrium.
The second, right, branch in divides again into two routes. The first flows through a humidifier to the second OPC detector. The second route simply bypasses the humidifier to permit measurement of the nonhydrated aerosol size distribution. Though not shown explicitly, the valving, heating and humidification are all monitored and controlled by a computer-based system, which permits the operation of this system in one of three different configurations.
Before turning to discussion of the three flow configurations of the AHS, we wish to note several aspects of the instrument structure that are likely unclear from the schematic alone. The tubing used in the AHS is 3/8 inch stainless steel (0.95 cm ID). Given the necessary aspiration flow for the OPCs, this results in an internal flow velocity of ∼ 1 m s− 1. All bends in the system have radii of curvature of 1 cm or greater, implying Stokes numbers of 0.04 or less (0.04 for 8 μ m diameter particles). Hence, transmission efficiencies in the bends are ∼ 97% for 8 μ m particles and exceed 99% for 1 μ m particles. However, perhaps the most critical component of the AHS is the aging chamber. This is a vertically oriented cylindrical volume 33.7 cm in overall length with a maximum diameter of 10.8 cm. The first 8.9 cm constitute an expansion cone to smooth the flow transition to the larger channel diameter, as is the final 8.9 cm to smooth the transition back to the sample line. With the vertical orientation, sedimentation of the larger particles (maximum settling velocity of 0.3 cm s− 1) is not an issue in the chamber. Similarly, aspiration from the chamber back into the tubing leading to the OPCs takes place at close to 100% efficiency due to the very low Stokes numbers of the chamber flow (< 10− 3). The effect of this chamber is to produce a substantial additional delay in the residence time of the aerosol at the RH of the branch containing it (compared to the other, direct branch). However, the chamber acts as a mixing chamber due to its large size and there is thus a distribution of particle residence times associated with it. Extensive laboratory tests with step function aerosol input have established the shortest delay time (relative to the direct flow branch) as 12 s while the median delay time is 42 s. Thus, the delay branch has a total residence time of at least 14 s and a median residence time of ∼44 s.
In the first and simplest configuration of the system, valves 1 and 3 are open, and 2 and 4 are closed. The right branch of the system acts to measure the aerosol size distribution at relatively low RH with the OPC. The left branch in this configuration is measuring the aerosol in humidified air. This mode thus allows a comparison of the size distribution at ∼ 85% to that at ∼ 40% RH (the nominal RHs used in the two pathways). It is important to note that comparison of the two branches is predicated on their equivalent response to the same input. Hence, prior to each set of samples (or aircraft flights for typical deployments), a filter is placed just behind the inlet to test zero equivalency of the sensors for a period of several minutes. Periodically, a test aerosol with narrow size dispersion and independently known concentration is sampled by the AHS to determine the counting efficiency of each OPC and permit derivation of the counting efficiency correction factor necessary to render the OPCs equivalent in efficiency.
The second configuration has valves 1 and 4 open, and 2 and 3 closed. In this mode, the two flow paths are equivalent, permitting a comparison of the individual channel responses at either high or low RH. This is essentially a test mode to ensure that the channel responses are equivalent for actual ambient aerosols, particularly at high RH.
The third and most elaborate configuration of the system has valves 2 and 4 open, with 1 and 3 closed. On the right branch of the flow, valve 4 routes the flow once again through the humidifier. In this configuration, the OPC measures the humidified aerosol size distribution with a time scale for droplet growth similar to that usually employed, i.e., assuming a growth time on the order of two seconds or less. OPC 1, however, measures the hydrated size distribution after a median growth period of a nominal 44 s and a minimum growth time of 14 s. Hence, the aerosol sizes measured are the equilibrium sizes for the given RH (again, 85–95%). Comparison of the size distributions measured by the two detectors permits a quantitative assessment of the fraction of equilibrium size attained by the measured aerosols in typical measurement systems. It is important to note that, in a changing aerosol field, comparison of the size distributions involves the time collation of the two OPC signals by lagging the measurements from the right branch by the 42-second delay built into the left side of the system. Because of this necessity, the time response of the overall system is essentially a nominal 44 seconds. This dictates that the system is not capable of discriminating short time or spatial scale events with respect to growth delay. However, for the relatively horizontally homogeneous MBL scenarios where we employ it, this is not an issue. The delays in droplet growth measured by the system permit estimation of the impact of growth delays on, for example, CCN activation.
For most uses of the AHS, it is vital that the two OPC sensors have an identical response with the same atmospheric aerosol input, or can be corrected to such equivalence. A baseline test in the first configuration with the humidifiers off verifies equivalency at lower RH. To determine equivalency at higher RH, the system is checked by running in configuration 2, as discussed above. This test is conducted for several minutes at some point during each flight.
2.2. Analysis Methodology
The analysis procedure utilized is similar to the “Descriptive Hygroscopic Growth Factor” (DHGF) methodology developed by CitationNowak (2005). First, the number-size distributions of both the “wet” and “dry” channels are integrated downward in size. For any particular dry diameter, the wet diameter at which the cumulative wet number concentration is equivalent to the cumulative dry number concentration defines the diameter to which the dry particles have grown for the given change in RH. The DHGF is simply the ratio of these wet to dry diameters and a DHGF spectrum a plot of these ratios versus the dry diameter.This approach assumes that, above a certain large size, there are no particles sampled. The rapid fall off in atmospheric aerosol number concentration with size renders this a safe assumption, and it has several advantages. First, it avoids ambiguities concerning possible multiple dry sizes contributing to a given wet size due to different hygroscopicities. Second, the integration procedure itself acts as a smoothing filter, enhancing the signal to noise ratio. On the other hand, it does preclude the differentiation of aerosol particles with differing hygroscopicities within size ranges as analyzed, for example, by CitationSvenningsson et al. (1992). Furthermore, because of this limitation, in some scenarios it will be difficult to differentiate between changes in aerosol hygrospocitiy induced by chemical changes in aerosol particles and changes due to a differing external mix of more and less hygroscopic particles. What the DHGF methodology always permits is the operational recovery of the “wet” aerosol size distribution from the dry, or vice versa. This is the key relationship for assessing an impact of RH on, for example, aerosol optical properties for which only the particle distributions themselves are important, not how they are achieved.
2.3. Calibration
The two OPCs require a calibration, both to ensure the proper sizing and, in the AHS, to ensure size equvalency between the two channels for the same input. Hence, calibration aerosol for each channel of the AHS was introduced at the aerosol inlet of the instrument system rather than closer to the two OPCs. Four different calibration aerosols were employed. For sizes below ∼600 nm, a DMA was used to generate monodisperse salt aerosols with compositions—and indices of refraction—similar to those encountered in the atmossphere. We used NaCl and NH4NO3 aerosols at various RHs to encompass a range of indices of refration from 1.4 to 1.48. To calibrate at larger sizes, above the range of most DMAs, we used silica spheres produced by Duke Scientific up to the largest available size of 1.6 μ m. For sizes larger than this, polystyrene-latex (PSL) spheres, also from Duke Scientific, were employed. However, these particles had an index of refraction of 1.59, sufficiently different from our target aerosol to require adjustment of the OPC signal to compensate for the apparent size change produced by the higher index of refraction. We utilized the relationship between index of refraction and scattering intensity reported by Citationvan de Hulst (1957) for this adjustment. We also used PSL spheres at sizes below 1 μ m to verify the equivlence of the signal between the PSL spheres and the other calibration aerosols.
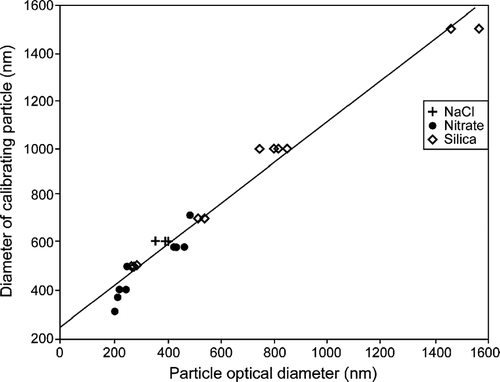
The calibration data were used to linearly regress the calibration particle sizes unto the OPC pulse height analyzer (PHA)—size calibration provided by the manufacturer. Since the factory calibration is completely linear, this is equivalent to calibrating the PHA channels. The regression line for aerosols with indices of refration between 1.4 and 1.48 is shown in . The R2 for the regession line is 0.95. When PSL spheres are included in the analysis over the same size range, the slope of the regression line is within 1% of that shown and the intercepts differ by less than 15%, neither difference being significant. The regression analysis encompassing the larger sizes (and thus necessarily PSL spheres) is shown in . The regression R2 is 0.99. While both regressions provide excellent fits, they have slightly, but significantly, different slopes (0.87 for the lower indices of refraction only and 0.99 for all particles). We use the first regression for the analysis shown below to calibrate the PHA channels and to provide the best fit in the 0.5 to 2.0 μ m size range which is of most interest for these cases, i.e., a calibration based on the data shown in was applied to the data obtained in this study¨ Based on the regession analysis, we also derived the effective size resolution of the OPCs. The resolution varies from 29 nm at the small end of the size range to 58 nm at the large end. This size resolution, while adequate for our needs, is much less than that associated with TDMA-based hygroscopic growth measurements at smaller sizes and is the major source of uncertainty in our DHGF measurements for particle diameters below 2 μ m. Above this size, Poisson counting statistics begin to dominate the overall uncertainty for most aerosol samples.
FIG. 2 Linear regression of calibration aerosols with indices of refraction from 1.40 to 1.48 against AHS factory calibration.
Another critical aspect of the instrument calibration is verification that the DHGFs for aerosols of known composition are in fact as expected from previous laboratory and theoretical studies. To this end, both NH4NO3 and NaCl aerosols were generated and the measured DHGFs compared to literature values. For ammonium nitrate aerosols of 300 nm diameter, the measured DHGF was 1.34 ± 0.04 for the RH increment from 10 to 72%. This is entirely consistent with literature values (e.g., 1.32; CitationTang 1980, Citation1996). Similarly, 600 nm NaCl particles had a measured DHGF of 1.96 ± 0.21 for the RH range of 10 to 89% as compared to a literature value of 2.1 ± 0.2 (CitationTang 1996, Citation1997).
In addition to tests at specific particle sizes, polydisperse, NH4NO3 aerosol were measured to assess the AHS response for a constant composition but polydisperse aerosol. The DHGF for an RH couple of 25 and 81% ranged from 1.31 to 1.47 with a mean value of 1.38 ± 0.06 over a size range from 0.3 to 2.0 μ m diameter. This compares quite well with the value of 1.39 ± 0.03 derived from CitationTang (1980).
3. EXAMPLES OF AHS DATA ANALYSIS FROM THE CARMA STUDY
3.1. Venue and the CARMA Program
In fact, some preliminary measurements using the AHS have already been reported (CitationHegg et al. 2006) but dealt mostly with the relationship between DHGFs and chemical composition. The additional analyses presented here are more general and meant to illustrate the capabilities of the AHS. They are based on data from the CARMA-III field campaign in August of 2005. This field study was part of the Cloud-Aerosol research in the Marine Atmosphere (CARMA) program supported by the Office of Naval Research (ONR). The objective of this program is to quantify the impact of aerosols on clouds in the MBL and the impact of these clouds on the aerosols processed by them. The venue for CARMA III was the region of high-frequency stratocumulus off of the central California coast. This is one of the three most intensive stratocumulus areas in the world (CitationWarren et al. 1988) and the clouds here have been shown to be susceptible to albedo modulation both by remote sensing studies (CitationPlatnick and Twomey 1994) and by a number of in-situ observational studies (cf., CitationDurkee et al. 2000 and references therein). More relevant to the current analysis, it is an area where aerosol characteristics, both physical and chemical, vary widely due to the proximity of strong anthropogenic and biogenic emissions from California coupled with winds that can advect this air into the operational area but commonly also advect it from the pristine marine background airshed to the west. It is thus an excellent location for evaluating a new instrument to measure aerosol hygroscopicity.
3.2. Ancillary Instrumentation
The instrument platform that was utilized is the Center for Interdisciplinary Remotely Piloted Aircraft Studies (CIRPAS) Twin Otter research aircraft. The instrumentation aboard this aircraft, while variable according to mission, can be parsed from a number of recent publications (e.g., CitationHegg et al. 2001; CitationWang et al. 2002; CitationSchmid et al. 2003). For particle size, optical properties and moisture analysis, the key standard instruments are summarized as follows.
| • | For fast (40 Hz) measurement of the water vapor mixing ratio and static air temperature Lyman alpha hygrometers and fast response Rosemont probes are utilized (CitationFriehe et al. 1986; CitationFriehe and Khelif 1992). | ||||
| • | For aerosol optical properties and scattering hygroscopicity, a TSI 3563 three-wavelength nephelometer and the University of Washington passive humidograph, respectively, are employed (cf., CitationGasso et al. 2000; CitationGao et al. 2003). | ||||
| • | For measurement of aerosols in the submicron size range most relevant to cloud drop nucleation, and thus cloud processing, the PMS/DMT PCASP (Passive Cavity Aerosol Spectrometer)-100 (size range from 0.2 to 3.2 μ m diameter) is employed (cf., CitationHegg and Jonsson 2000). | ||||
| • | Larger aerosols, including cloud drops, are measured by two instruments, the PMS/DMT FSSP (Forward Scattering Spectrometer)-100 and the DMT CAPS (Cloud Aerosol Particle Spectrometer) probe (which measure from 2 to 40 and from 0.5 to 50 μ m diameter, respectively). However, these probes are both laser-illuminated (monochromatic) and forward-scattering spectrometers. The FSSP was designed to measure cloud drops and the CAPS, though supposedly designed for aerosol in general utilizes essentially the same principles. Their failings as unactivated aerosol probes are well established and one of the motivations for our usage of white light sensors in the AHS. They nevertheless provide useful if only semi-quantitative data on large aerosol concentrations. | ||||
| • | For aerosol composition, 47 mm Teflon membrane filters (Teflo) were used to collect bulk aerosol samples. A number of bulk chemical analytical techniques can be used to determine composition. The techniques which we utilized are well described in CitationGao et al. (2003a). | ||||
3.3. Vertical Variations in Descriptive Hygroscopic Growth Factor (DHGF) Spectra
The surface layer of the world's oceans is highly enriched in organics (cf., CitationSchneider and Gagosian 1985) and, during the bubble-bursting process which creates sea-salt aerosol, these organics, comprised largely of insoluble or only sparingly soluble lipids, fatty acids, fatty alcohols, etc. (as well as numerous soluble organics such as neutral sugars), adsorb onto the salt particle surfaces as surfactants (e.g., CitationOppo et al. 1999; CitationCini and Loglio 1997). It is thus scarcely surprising that recent work in the clean marine atmosphere has found sea salt and organics to be commonly co-existent in the marine aerosols, with the organics constituting ∼ 10% of the sea salt mass (cf., CitationMiddlebrook et al. 1998). More detailed studies have suggested a relative concentration of organics in fine particles under clean conditions (e.g., CitationNeususs et al. 2000), a phenomenology in accord with the adsorption hypothesis (CitationOppo et al. 1999). It is also interestingly congruent with the relative enrichment of “less hygroscopic” particles at smaller sizes observed in recent H-TDMA analyses in marine air (e.g., CitationSwietlicki et al. 2000). These aspects of the marine organic aerosol phenomenology have recently led to a novel conceptual model of sea-salt aerosol as “inverted micelles,” essentially sea-salt solution droplets coated by hygrophobic organic surfactants (CitationEllison et al. 1999). While the structure of the aerosol is similar to previous concepts, Ellison et al. point out explicitly that, while the hygrophobic coating will render the aerosol initially less capable of taking on water, this coating will likely be oxidized by OH radicals in the atmosphere as the aerosol ages and thus the hygroscopicity of the sea salt should vary with age. Hence, the rate of evolution of the surfactant coating will be an important element in the evolution of the aerosol optical properties as well as its water activity (CitationVaida et al. 2000). One might, on the basis of this model, expect an increase in marine aerosol hygroscopicity with altitude, with near surface hygroscopicities substantially lower than that expected for sea salt (inorganic) itself. Additionally, the well-known production of secondary sulfate aerosol mass, which might be expected to coat, in turn, any organic surface layer, could also render the aerosol more hygroscopic with age. On the other hand, if the aerosol does not have a complete organic surface layer, its hygroscopicity could be quite similar to that of inorganic sea salt, and could actually decrease with altitude if secondary sulfate displaces the more hygroscopic sea salt. Rendering the situation even more complex, it is conceivable that these factors could vary in impact for different size ranges. For example, one could have complete hygrophobic surface layers for small particles but incomplete layers for larger particle, resulting in increases in hygroscopicity in some ranges with aging simultaneous with decreases in other size ranges.
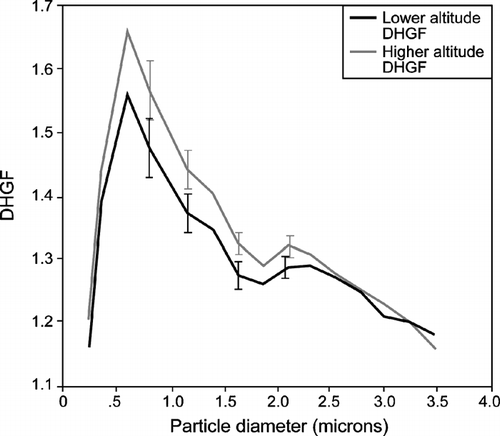
With this discussion as background, the observations of DHGF spectra obtained with the AHS during CARMA-III are interesting. Seven flights had coupled pairs of “high” and “low” altitude DHGF spectra for comparison. The low altitude spectra were uniformly at 30 m MSL while the high altitude spectra were obtained at altitudes varying from 200 m (flight 822) to 470 m (flight 825). Average DHGF spectra for all of the high and low altitude samples are shown in . The high and low altitude passes were selected from each flight in pairs to ensure that the averages were made over the same domains. The mean spectra show an increase in hygroscopicity with altitude over much of the particle size range examined, though it is only significant in the range from approximately 1 to 2 μ m. This can easily be rationalized in terms of the previous discussion by oxidation of insoluble organics and/or secondary formation of soluble inorganics. However, examination of individual cases reveals several more complex scenarios. For example, (flight 825) suggests a substantial increase in hygroscopicity with altitude for particles in the ∼ 0.5 to 2 μ m size range, no significant difference between 2 and 2.5 μ m followed by another significant increase between ∼ 2.5 and 3.2 μ m. This contrast is no doubt due to differences in chemical composition with size. But, as discussed in Section 2.2, one must always consider the possibility that one is dealing with an external mixture of more and less hygroscopic particles and that changes in number of either particle type have produced the observed changes in DHGFs. Nevertheless, for the scenario examined here, a well-mixed MBL, this seems unlikely. For example, for flight 825, which has the largest altitude difference between samples, the particle concentrations only differ by about 6% between the high and low samples. The observed changes in DHGF almost certainly must therefore involve changes in the composition of individual particles. Clearly, however, the question is worth pursing in future work.
FIG. 3 Linear regression of all calibration aerosols against the AHS factory calibration.
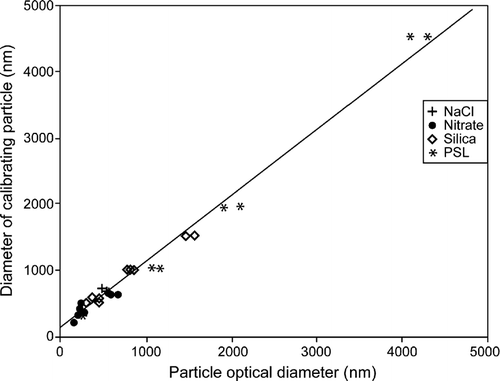
FIG. 4 Mean DHGF spectra for both the higher and lower altitude samples taken during CARMA-III. The higher altitudes ranged from 200 to 470 m MSL while the lower altitude samples were all at 30 m MSL. The error bars represent the standard errors of the mean values.
3.4. Delayed Particle Hydration
Only three entirely credible samples were obtained with the AHS in its delay-analysis mode during CARMA-III due to the need to perform the more basic growth measurements and the constraints of level flight legs and profiles. Nevertheless, they present some interesting features as well as verifying the performance of the instrument in this mode. Two of these samples were of marine aerosol near the sea surface (30 m MSL) but the second was in the plume from a biomass fire sampled just above the MBL inversion. shows the normalized DHGF spectrum for the fire plume together with the normalized spectrum for the average of the two marine samples, which were very similar. Normalization consists of dividing the observed DHGFs with the AHS in the delay mode, which gives the diameter ratio with and without the aging chamber in line, by the size corresponding DHGFs from the AHS with both channels at the same RH but with the aging (delay) chamber off-line. This normalization then accounts for any anomalies in the instrument response along the two sample paths that is not associated with the longer growth period of the delay pathway. More concisely, perhaps, the normalized DHGF, or delay ratio, can be expressed as:
FIG. 5 High and low altitude DHGF spectra for flight 825.
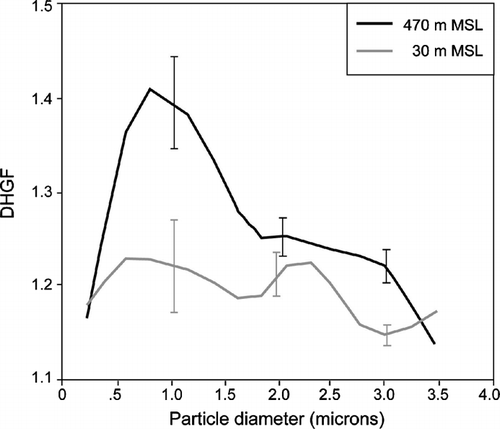
FIG. 6 Normalized DHGF spectra (ratio of delayed to non-delayed channel) from flight 813 showing the difference in growth rate for the fire plume and sea salt aerosols.
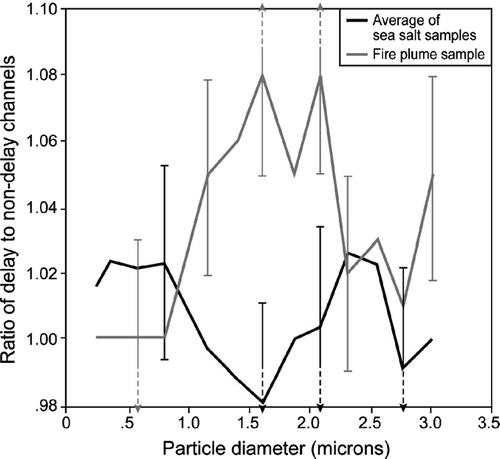
The normalized spectra for the marine aerosol show no evidence of enhanced growth associated with the longer growth time of the delay pathway within the uncertainty of the measurements. Indeed, they show some modest negative growth, i.e., evaporation, likely associated with fluctuating and on average slightly lower RH in the delay pathway. The fire plume aerosol, on the other hand, does show evidence of enhanced growth with the longer time of the delay pathway. It is noteworthy that the fire plume aerosol was over 80% organic by mass while the marine aerosols were 12 and 3% organic by mass, respectively. It is also interesting to note that the fire plume aerosol showed no evidence of a kinetic effect at the smaller sizes, consistent with the data of CitationChuang (2003) who made his measurements at particle sizes of 50 and 100 nm.
4. CONCLUSIONS
A new instrument for measuring size-resolved aerosol hygroscopicity has been described. It utilizes white-light OPCs to extend the range of hygroscopic growth measurement from ∼ 0.2 to 3.5 μ m diameter, a considerably broader range than has previously been accessible with a continuous, in situ instrument. Laboratory size calibration with spheres of different diameter and refractive index, and tests of hygroscopic growth of known monodisperse and polydisperse hygroscopic aerosols, verify the instrument design and operation. Preliminary ambient results, which verify the utility of the measurement, suggest that aerosol hygroscopicity in general increases with altitude in the MBL but that some size ranges can actually decrease in hygroscopicity with altitude. In the delay configuration, The AHS has also demonstrated an ability to detect kinetic effects on the condensational growth of aerosols, suggesting that for some aerosol types, such as largely organic aerosol from biomass combustion, a substantial time interval may be necessary for them to equilibrate at ambient humidities.
Acknowledgments
This research was supported by grant number N0014-97-1-0132 from the Office of Naval Research. We thank two anonymous reviewers and Prof. Rick Flagan for useful comments.
Related Research Data
REFERENCES
- Bigg , E. K. 1986 . Discrepancy Between Observation and Prediction of Concentrations of Cloud Condensation Nuclei . Atmos. Res. , 20 : 80 – 86 .
- Charlson , R. J. , Schwartz , S. E. , Hales , J. M. , Cess , R. D. , Coakley , J. A. Jr. , Hansen , J. E. and Hoffmann , D. J. 1992 . Climate Forcing by Anthropogenic Aerosols . Science, , 255 : 423 – 430 .
- Chuang , P. Y. 2003 . Measurement of the Timescale of Hygroscopic Growth for Atmospheric Aerosols . J. Geophys. Res. , 108 : AAC 5-1 – AAC 5–13 .
- Chuang , P. Y. , Charlson , R. J. and Seinfeld , J. H. 1997 . Kinetic Limitations on Droplet Formation in Clouds . Nature , 390 : 594 – 596 .
- Chuang , P. Y. , Collins , D. R. , Pawlowska , H. , Snider , J. R. , Jonsson , H. H. , Brenguier , J. L. , Flagan , R. C. and Seinfeld , J. H. 2000 . CCN Measurements During ACE-2 and their Relationship to Cloud Microphysics . Tellus , 52B : 843 – 867 .
- Cini , R. and Loglio , G. 1997 . Adsorption and Pollutant Transport by Marine Aerosol . Mar. Pollut. Bull. , 34 : 501 – 504 .
- Cruz , C. N. and Pandis , S. N. 1998 . The Effect of Organic Coatings on the Cloud Condensation Nuclei Activation of Inorganic Atmospheric Aerosol . J. Geophys. Res. , 103 : 13111 – 13123 .
- Durkee , P. A. , Noone , K. J. , Ferek , R. J. , Johnson , D. W. , Taylor , J. P. , Garrett , T. J. , Hobbs , P. V. , Hudson , J. G. , Bretherton , C. S. , Innis , G. , Frick , G. M. , Hoppel , W. A. , O'Dowd , C. D. , Russell , L. M. , Gasparovic , R. , Nielsen , K. E. , Tessmer , S. A. , Ostrom , E. , Osborne , S. R. , Flagan , R. C. , Seinfeld , J. H. and Rand , H. 2000 . The Impact of Ship-Produced Aerosols on the Microstructure and Albedo of Warm Marine Stratocumulus Clouds. A Test of the MAST Hypothesis Li and Lii . J. Atmos. Sci. , 57 : 2554 – 2569 .
- Ellison , G. B. , Tuck , A. F. and Vaida , V. 1999 . Atmospheric Processing of Organic Aerosols . J. Geophys. Res. , 104 : 11633 – 11641 .
- Facchini , M. C. , Mircea , M. , Fuzzi , S. and Charlson , R. J. 1999 . Cloud Albedo Enhancement by Surface Active Organic Solutes in Growing Droplets . Nature , 401 : 257 – 259 .
- Feingold , G. and Chuang , P. Y. 2002 . Analysis of the Influence of Film-Forming Compounds on Droplet Growth). Implications for Cloud Microphysical Processes and Climate . J. Atmos. Sci. , 59 : 2006 – 2018 .
- Friehe , C. A. , Grossman , R. L. and Pann , Y. 1986 . Calibration of an Airborne Lyman-Alph Hygrometer and Measurement of Water Vapor Flux Using a Thermoelectric Hygrometer . J. Atmos. Ocean. Tech. , 3 : 299 – 304 .
- Friehe , C. A. and Khelif , D. 1992 . Fast-Response Aircraft Temperature Sensors . J. Atmos. Oceanic Technol. , 9 : 784 – 795 .
- Gao , S. , Hegg , D. A. , Hobbs , P. V. , Kirchstetter , T. W. , Magi , B. J. and Sadilek , M. 2003a . Water-Soluble Organic Components in Aerosols Associated with Savanna Fires in Southern Africa: Identification, Evolution, and Distribution . J. Geophys. Res. , 108 : SAF 27-1 – SAF 27–16 .
- Gao , S. , Hegg , D. A. , Covert , D. S. and Jonsson , H. 2003 . Aerosol Chemistry, and Light-Scattering and Hygroscopicity Budgets During Outflow form East Asia . J. Atmos. Chem. , 46 : 55 – 88 .
- Gasso , S. , Hegg , D. A. , Covert , D. S. , Collins , D. , Noone , K. J. , Ostrom , E. , Schmid , B. , Russell , P. B. , Livingston , J. M. , Durkee , P. A. and Jonsson , H. 2000 . Influence of Humidity on the Aerosol Scattering Coefficient and its Effect on the Upwelling Radiance During ACE-2 . Tellus , 52B : 546 – 567 .
- Gill , P. S. , Graedel , T. E. and Weschler , C. J. 1983 . Organic Films on Atmospheric Aerosol-Particles, Fog Droplets, Cloud Droplets, Raindrops, and Snowflakes . Rev. Geophys. , 21 : 903 – 920 .
- Hand , J. L. , Ames , R. B. , Kriedenweis , S. M. , Day , D. E. and Malm , W. C. 2000 . Estimates of Particle Hygroscopicity during the Southeastern Aerosol and Visibility Study . J. Air & Waste Manage. Assoc. , 50 : 677 – 685 .
- Hegg , D. A. , Covert , D. S. , Jonsson , H. and Covert , P. A. 2005 . Determination of the Transmission Efficiency of an Aircraft Aerosol Inlet . Aerosol Sci. Technol. , 39 : 1 – 6 .
- Hegg , D. A. , Covert , D. S. , Crahan , K. K. , Jonsson , H. H. and Liu , Y. 2006 . Measurements of the Aerosol Size-Resolved Hygroscopicity at Sub and Supermicron Sizes . Geophys. Res. Lett. , 33 : L21808 doi. 10.1029/2006GL026747
- Hegg , D. A. , Gao , S. , Hoppel , W. , Frick , G. , Caffrey , P. , Leaitch , W. R. , Shantz , N. , Ambrusko , J. and Albrechcinski , T. 2001 . Laboratory Studies of the Efficiency of Selected Organic Aerosols as CCN . Atmos. Res. , 58 : 155 – 166 .
- Hegg , D. A. and Jonsson , H. 2000 . Aerosol Number-to-Volume Relationship and Relative Humidity in the Eastern Atlantic . J. Geophys. Res. , 105 : 1987 – 1995 .
- Hegg , D. A. , Livingston , J. , Hobbs , P. V. , Novakov , T. and Russell , P. 1997 . Chemical Apportionment of Aerosol Column Optical Depth off the Mid-Atlantic Coast of the United States . J. Geophys. Res. , 102 : 25293 – 25303 .
- Henning , S. , Weingartner , E. , Schmidt , S. , Wendisch , M. , Gaggeler , H. W. and Baltensperger , U. 2002 . Size-Dependent Aerosol Activation at High-Alpine Site Junfraujoch (3580 m ASL) . Tellus , 54B : 82 – 95 .
- Kotchenruther , R. A. , Hobbs , P. V. and Hegg , D. A. 1999 . Humidification Factors for Atmospheric Aerosols Off the Mid-Atlantic Coast of the United States . J. Geophys. Res. , 104 : 2239 – 2251 .
- Kreisberg , N. M. , Stolzenberg , M. R. , Hering , S. V. , Dick , W. D. and McMurry , P. H. 2003 . A New Method for Measuring the Dependence of Particle Size Distributions on Relative Humidity, with Application to Southeastern Aerosol and Visibility Study . J. Geophys. Res. , D14 : 14935 – 14949 .
- Middlebrook , A. M. , Murphy , D. M. and Thomson , D. S. 1998 . Observations of Organic Material in Individual Marine Particles at Cape Grim during the First Aerosol Characterization Experiment (ACE-1) . J. Geophys. Res. , 103 : 16,475 – 16,484 .
- Neususs , C. , Pelzing , M. , Plewka , A. and Hermann , H. 2000 . A New Analytical Approach for Size Resolved Speciation of Organic Compounds in Atmospheric Aerosol Particles: Methods and First Results . J. Geophys. Res. , 105 : 4513 – 4527 .
- Nowak , A. 2005 . Das Feuchte Partikelgroessenspktrometer: Eine Neue Messmethode Zur Bestimmung Von Partikelgroessenverteilung (< 1 μ m) und Groessenaufgeloesten Hygroskopischen Wachsumsfaktoren Bei Definierten Luftfeuchten , Doctoral Thesis Leipzig, , Germany : Leibnitz Institute for Tropospheric Research . Permoserstr. 15, D-04303
- Oppo , C. , Bellandi , S. , Degli Innocenti , N. , Startini , A. M. , Loglio , G. , Schiavuta , E. and Cini , R. 1999 . Surfactant Components of Marine Organic Matter as Agents for Biogeochemical Fractionation and Pollutant Transport Via Marine Aerosols . Mar. Chem. , 63 : 235 – 253 .
- Platnick , S. and Twomey , S. 1994 . Determining the Susceptibility of Cloud Albedo to Changes in Droplet Concentration with the Advanced very High-Resolution Radiometer . J. Appl. Meteor. , 33 : 334 – 347 .
- Podzimek , J. and Saad , N. 1975 . Retardation of Condensation Nuclei Growth by Surfactant . J. Geophys. Res. , 80 : 3386 – 3392 .
- Reid , J. S. , Brooks , B. , Crahan , K. K. , Hegg , D. A. , Eck , T. F. , O'Neill , N. , de Leeuw , G. , Reid , E. A. and Anderson , K. D. 2006 . Reconciliation of Coarse mode Sea-Salt Aerosol Particle Size Measurements and Parameterizations at a Subtropical Ocean Receptor Site . J. Geophys. Res. , 111 : D02202
- Rodhe , H. 1983 . “ Precipitation Scavenging and Tropospheric Mixing ” . In Precipitation Scavenging, Dry Deposition, and Resuspension , Edited by: Pruppacher , H. R. , Semonin , R. G. and Slinn , W. G. N. 719 – 730 . New York : Elsevier .
- Rossi , M. J. 2003 . Heterogeneous Reactions on Salts . Chem. Rev. , 103 : 4823 – 4882 .
- Rudich , Y. 2003 . Laboratory Perspectives on the Chemical Transformations of Organic Matter in Atmospheric Particles . Chem. Rev. , 103 : 5097 – 5124 .
- Santarpia , J. L. , Runjun , L. and Collins , D. R. 2004 . Direct Measurement of the Hydration State of Ambient Aerosol Populations . J. Geophys. Res. , 109 : D18209
- Schmid , B. , Hegg , D. A. , Wang , J. , Bates , D. , Redemann , J. , Russell , P. B. , Livingston , J. M. , Honsson , H. H. , Welton , E. J. , Seonfeld , J. H. , Flagan , R. C. , Covert , D. S. , Dubovik , O. and Jefferson , A. 2003 . Column Closure Studies of the Lower Tropospheric Aerosol and Water Vapor during ACE-Asia Using Airborne Sunphotometer, Airborne In-Situ and Ship-Based Lidar Measurements . J. Geophys. Res. , 108 : 8656 doi:10.1029/2002JD003361
- Schneider , J. K. and Gagosian , R. B. 1985 . Particle Size Distribution of Lipids in Aerosols off the Coast of Peru . J. Geophys. Res. , 90 : 7889 – 7898 .
- Schumann , T. 1990 . On the Use of a Modified Clean-Room Optical Particle Counter for Atmospheric Aerosols at High Relative Humidity . Atmos. Res. , 25 : 499 – 520 .
- Shantz , N. , Leaitch , W. R. and Caffrey , P. 2003 . Effect of Organics of Low Solubility on the Growth Rate of Cloud Droplets . J. Geophys. Res. , 108 : AAC 5-1 – 5-9 .
- Svenningsson , I. B. , Hansson , H-C. , Wiedensohler , A. , Ogren , J. A. , Noone , K. J. and Hallberg , A. 1992 . Hygroscopic Growth of Aerosol Particles in the Po Valley . Tellus , 44B : 556 – 569 .
- Swietlicki , E. , Zhou , J. , Covert , D. S. , Hameri , K. , Busch , B. , Vakeva , M. , Dusek , U. , Berg , O. H. , Widensohler , A. , Aalto , P. , Makela , J. , Martinsson , B. G. , Papaspiropoulos , G. , Mentes , B. , Frank , G. and Stratmann , F. 2000 . Hygroscopic Properties of Aerosol Particles in the Northeastern Atlantic during ACE-2 . Tellus , 52B : 201 – 227 .
- Tang , I. N. 1980 . “ Deliquescence Properties and Particle Size Change of Hygroscopic Aerosols ” . In Generation of Aerosols , Edited by: Willeke , K. 153 – 170 . Ann Arbor, MI : Ann Arbor Science .
- Tang , I. N. 1996 . Chemical and Size Effects of Hygroscopic Aerosols on Light Scattering Coefficients . J. Geophys. Res. , 101 : 19245 – 19250 .
- Tang , I. N. 1997 . Thermodynamic and Optical Properties of Mixed-Salt Aerosols of Atmospheric Significance . J. Geophys. Res. , 102 : 1883 – 1893 .
- ten Brink , H. M. , Khlystov , A. , Kos , G. P. A. , Tuch , T. , Roth , C. and Kreyling , W. 2000 . A High-Flow Humidograph for Testing the Water Uptake by Ambient Aerosol . Atmos. Environ. , 34 : 4291 – 4300 .
- Tervahattu , H. , Juhanoja , J. and Kupianen , K. 2002 . Identification of an Organic Coating on Marine Aerosol Particles by TOF-SIMS . J. Geophys. Res. , 107 : ACH18-1 – 18-7 .
- Thomas , E. , Rudich , Y. , Trakhtenberg , S. and Ussyshkinm , R. 1999 . Water Adsorption by Hydrophobic Organic Surfaces). Experimental Evidence and Implications to Atmospheric Properties of Organic Aerosols . J. Geophys. Res. , 104 : 16053 – 16059 .
- Vaida , V. , Tuck , A. J. and Ellison , G. B. 2000 . Optical and Chemical Properties of Atmospheric Organic Aerosols . Phys. Chem. Earth (c) , 25 : 195 – 198 .
- van de Hulst , H. C. 1957 . Light Scattering by Small Particles , 470 New York : Dover .
- Wang , J. , Flagan , R. C. , Seinfeld , J. H. , Jonsson , H. H. , Collins , D. R. , Russell , P. B. , Schmid , B. , Redemeann , J. , Livingston , J. M. , Gao , S. , Hegg , D. A. , Welton , E. J. and Bates , D. 2002 . Clear-Column Radiative Closure during ACE-Asia: Comparison of Multiwavelength Extinction Derived from Particle size and Composition with Results from Sunphotometry . J. Geophys. Res. , 107 ( D23 ) : 4688 doi: 10.1029/2002JD002465
- Warren , S. G. , Hahn , C. J. , London , J. , Chervin , R. M. and Jenne , R. L. 1988 . Global Distribution of Total Cloud Cover and Cloud type Amounts Over the Ocean NCAR Technical Note NCAR/TN-317+STR
- Winkler , P. and Junge , C. 1972 . The Growth of Atmospheric Aerosol Particles as a Function of Relative Humidity . J. Rech. Atoms. , 6 : 617 – 638 .
- Xiong , J. Q. , Zhong , M. , Fang , C. , Chen , L. C. and Lippmann , M. 1998 . Influence of Organic Films on the Hygroscopicity of Ultrafine Sulfuric Acid Aerosol . Environ. Sci. Technol. , 32 : 3536 – 3541 .