Abstract
Heterogeneous reactions of trace gases with mineral dust aerosol not only impact the chemical balance of the atmosphere but also the physicochemical properties of the dust particle and the ability of the particle to act as a cloud condensation nuclei (CCN). Recent field studies have shown that carbonate minerals are preferentially associated with nitrates whereas aluminum silicates (i.e., clay minerals) are preferentially associated with sulfates. To better understand how this association can impact the climate effects of mineral dust particles, we have measured the CCN activity of a number of pure and internal mixtures of aerosols relevant to these recent field studies. The CCN activity of CaCO 3 -Ca(NO 3 ) 2 aerosol, simulating the activity of mineral dust aerosol that has been partially processed by nitrogen oxides in the atmosphere, is significantly enhanced relative to CaCO3 aerosol of the same diameter. Similar results are obtained for a clay mineral, kaolinite, internally mixed with (NH 4 ) 2 SO 4 . For example, at 0.3% supersaturation, a 200 nm particle containing a soluble nitrate or sulfate component is 2 to 4 times more active than an unreacted particle. The results presented here show that when determining the contribution of mineral dust aerosol to the overall impact of the aerosol indirect effect on radiative forcing, changes in chemical composition due to atmospheric processing cannot be ignored.
INTRODUCTION
As aerosols are transported in the atmosphere, they can undergo heterogeneous reactions that lead to changes in the chemical composition of the particle surface (CitationBauer et al. 2004; CitationBian and Zender 2003; Citationde Reus et al. 2005; CitationLiao et al. 2004; CitationMartin et al. 2003; CitationSong and Carmichael 2001; CitationTang et al. 2004; CitationUnderwood et al. 2001). Often, tropospheric aerosols become internal mixtures of both water-soluble and insoluble components as a result of processing or coagulation (CitationKorhonen et al. 2003). For mineral dust aerosol, it has been observed in both field and modeling studies that dust particles are frequently coated with soluble compounds such as sulfates or nitrates. Specifically, nitrate is commonly associated with calcium-rich dust, while sulfate is usually observed with aluminosilicate- and iron-rich dusts (CitationOoki and Uematsu 2005; CitationSong and Carmichael 2001; CitationSullivan et al. 2006).
As a result of the heterogeneous chemistry that can occur on the particle surface, the physicochemical properties of the transported aerosol are expected to be different than those of the freshly emitted aerosol. These potential changes will significantly impact the role an aerosol has on climate forcing. Atmospheric aerosols affect the Earth's climate either through the scattering and absorption of solar radiation, direct radiative forcing, or by acting as cloud condensation nuclei (CCN), indirect radiative forcing. Currently, the aerosol indirect effect has the largest amount of uncertainty when estimating the impact of aerosols on climate and as such has become a focus of a number of field and laboratory studies (CitationAbbatt et al. 2005; CitationDinar et al. 2006; CitationHuff Hartz et al. 2006; CitationMircea et al. 2005; CitationPetzold et al. 2005; CitationRamaswamy et al. 2001). There has been much discussion as to what characteristics of an aerosol govern its CCN activity (size, chemical composition, etc.) (CitationDusek et al. 2006a; CitationMcFiggans et al. 2006). While recent reports suggest that in most cases composition plays a secondary role to size in establishing the ability of aerosol particles to act as CCN (CitationDusek et al. 2006a; CitationKaufman and Koren 2006), it is important to establish for insoluble particles such as mineral dust how chemical changes of the dust, through heterogeneous reactions, influence CCN activity (CitationDusek et al. 2006b; CitationFan et al. 2004; CitationLevin et al. 2005).
The carbonate component of mineral dust (calcite, CaCO3, and dolomite, CaMg(CO3)2) has been shown to be particularly reactive with trace atmospheric gases such as HNO3 and N2O5 (CitationKrueger et al. 2003; CitationMogili et al. 2006). Single particle analysis measurements have shown that carbonates can be completely converted to nitrates in the atmosphere (CitationLaskin et al. 2005; CitationMatsuki et al. 2005). Consequently, it is expected that in the atmosphere freshly emitted, partially processed, and fully reacted CaCO3 will be present. Recent laboratory studies have shown that the physicochemical properties of Ca(NO3)2 are markedly different than those of CaCO3 particles (CitationGibson et al. 2006a; CitationGibson et al. 2006b). The cloud condensation nuclei activity of pure 100 nm Ca(NO3)2 (representative of fully processed mineral dust) is nearly two orders of magnitude greater than that of pure 100 nm CaCO3 (freshly emitted mineral dust aerosol) at supersaturations less than 0.225% (CitationGibson et al. 2006a). In the atmosphere it is expected that the carbonate component of mineral dust will initially be nearly 100% carbonate and then, as it undergoes aging in the atmosphere through heterogeneous chemistry, it will be partially processed containing a carbonate core and a soluble nitrate coating. Although the association of sulfates with clay minerals will occur through a different mechanism, including particle aggregation with an already existing ammonium sulfate particle or through SO2 oxidation on the surface of the clay, the “aged” dust particle will have a similar, albeit different composition, soluble coating. In order to gain a better understanding of the role these processed particles play in climate forcing, quantitative, composition dependent measurements of the aerosol physicochemical properties are required.
Here, we discuss the capability of the University of Iowa's Multi-Analysis Aerosol Reactor System (MAARS) to generate and characterize the physicochemical properties of internally mixed particles representative of mineral dust aerosol that has been transported and aged in the troposphere. To simulate internally mixed, aggregated, or coated aerosols consisting of soluble and insoluble components in a laboratory setting, particles can be generated through the use of several different techniques. These methods include vaporization-condensation (CitationAbbatt et al. 2005; CitationGarland et al. 2005; CitationHan and Martin 2001; CitationKatrib et al. 2004; CitationLiao and Roberts 2006; CitationPerry et al. 2004) or direct atomization from a mixed solution of the aerosol components (CitationBuzorius et al. 2002; CitationDusek et al. 2006b; CitationHameri et al. 2002). In the vaporization-condensation method, an aerosol is atomized, dried, and size-selected to produce a monodisperse aerosol stream. The core aerosol is then coated via the condensation of another material onto the aerosol surface. For example, CitationKatrib et al. (2004) have used this method to generate polystyrene latex spheres (PSLs) coated with oleic acid to better understand product formation in ozone reactions with organic aerosols. To coat aerosol particles in this manner typically requires the use of a heated flow reactor and/or furnace to obtain an air flow saturated with vapor of the coating material. Alternatively, internally mixed or coated aerosols can be atomized directly from aqueous mixtures of the aerosol components. This technique is commonly used to generate organic aerosols internally mixed with a soluble salt like (NH4)2SO4 (CitationHameri et al. 2002). One caveat that must be considered when using this aerosol generation technique is the potential for forming an externally mixed aerosol as particles comprised solely of the coating material that may be generated (CitationBuzorius et al. 2002).
In this study, internally mixed aerosol particles are generated from suspensions of an insoluble compound with an added amount of a soluble material. Monodisperse PSL spheres mixed with (NH4)2SO4 are used to verify the formation of internally mixed particles from the atomizer. By using PSLs of a known diameter as the core of the mixed aerosol particles, the ammonium sulfate mass fraction in each particle can be quantified. Through careful control of the concentration of the aqueous salt in solution, mineral dust aerosol containing only trace amounts of soluble material can also be generated. Additionally, we show that the CCN activity of insoluble mineral dust components is enhanced dramatically when internally mixed with a small amount of an aqueous salt. Specifically, the CCN potential of CaCO3 and kaolinite with and without, respectively, Ca(NO3)2 and (NH4)2SO4 coatings are compared. The impact of a soluble component on the CCN activity of mineral dust aerosol and the resulting implications for indirect climate forcing are discussed.
EXPERIMENTAL METHODS
CCN measurements were carried out using MAARS, which has been discussed in great detail elsewhere (CitationGibson et al. 2006b). Briefly, aerosols are generated using a commercially available constant output atomizer (TSI, Inc. Model 3076) that has been modified to decrease the solution volume by using a Pyrex test tube (Fisher Scientific, 18 × 150 mm) in place of the one liter container supplied by the manufacturer. The Pyrex test tube, containing ca. 20 ml of a suspension of approximately 2 g of an insoluble powder such as CaCO3 in water (Optima, Fisher Scientific), is positioned in the bottle so that the liquid feed tube can access any insoluble powder that has settled out on the bottom of the test tube. A drain tube (1/4″ OD Teflon tubing) has been added to the atomizer lid to ensure continuous recirculation of the sample solution.
The output of the atomizer passes through a series of diffusion dryers typically reaching a relative humidity (RH) of ca. 10%. The resulting dried, polydisperse aerosol is then fed into a flow reactor/hydration chamber where it is mixed with a dry air stream. The particles are size-selected to produce a monodisperse aerosol flow with a differential mobility analyzer (DMA-1; TSI, Inc. electrostatic classifier Model 3080), before mixing with the dry air stream. Upon exiting the flow reactor, the aerosol size distribution is measured with a scanning mobility particle sizer (SMPS; TSI, Inc. Model 3936), which consists of a second DMA (DMA-2), and a condensation particle counter (CPC; TSI, Inc. Model 3025A). SMPS size distributions are measured using a 210 s scan time and flow rates of 3.0 (sheath) and 0.3 (sample) lpm. MAARS is equipped to measure the CCN activity of aerosol particles with a continuous flow streamwise thermal-gradient cloud condensation nuclei counter (Droplet Measurement Technologies, Model CCN-2) (CitationRoberts and Nenes 2005). In CCN measurements, the number of particles that act as cloud condensation nuclei (#CCN) relative to the total number of cloud nuclei (#CN) counted by the CPC is determined. The supersaturation generated in the CCN counter is calibrated to a temperature gradient across the diffusion chamber based on the ammonium sulfate activity data presented in CitationTang and Munkelwitz (1994). As previously discussed in the literature (CitationPradeep Kumar et al. 2003), due to counting inefficiencies, the maximum fraction of activated particles that is measured is in the range of 0.7 to 0.9 and not unity. In our system, the maximum fraction is typically near 0.9, but has been observed to range from 0.7 to 0.9. Therefore, measured #CCN/#CN ratios have been scaled by the reciprocal of the maximum ratio for each data set so that the vertical scale for these measurements goes from zero to one.
In this work, our goal is to produce internally mixed aerosol particles consisting of an insoluble (CaCO3, clay, or PSLs) and soluble (nitrate or sulfate) component that can then be used to further study the impact of heterogeneous processing of mineral dust aerosol on climate. The individual mineral dust components and soluble salts under investigation in this work are commercially available and were used as received. Ammonium sulfate (Fisher Scientific, ACS) and calcium nitrate (ACS, 99.0–103.0%) aerosols are generated from an aqueous solution of the salt, ranging in concentration from 0.01–1 weight percent by volume (wt%). CaCO3 (OMYA) and kaolinite (Al2Si2O5(OH)4; Source Clays Repository, KGa-1, low-defect) were atomized from a suspension of the insoluble powder in water. To generate internally mixed aerosol particles, the insoluble mineral dust component was atomized from a suspension of the powder in a solution of the soluble material. The fraction of soluble material included in the internally mixed particle is dependent on the concentration of the solution from which the aerosol is generated. Parameters for the lognormal size distributions of pure calcium nitrate and ammonium sulfate generated from solutions ranging in concentration from 0.01–0.1 wt%, equivalent to a molality range of 0.6–8 × 10− 3 m, are provided in .
TABLE 1 Parameters for lognormal size distributionsFootnote a of (NH4)2SO4 and Ca(NO3)2 aerosols generated from solutions of varying concentration.
The methodology for generating internally mixed aerosols and quantifying the amount of soluble material in each particle is developed using PSLs of varying diameters with manufacturer's stated sizes of 202 ± 10, 356 ± 14, and 535 ± 10 nm (Polysciences, Inc., Cat. # 07304, 07306, and 07307, respectively), aerosolized from a suspension of the PSLs in (NH4)2SO4. As the PSLs are dried, (NH4)2SO4 crystallizes on the particle surface. The resultant aerosol stream from the atomizer does include a certain amount of pure ammonium sulfate particles in addition to the coated PSLs, but at these ammonium sulfate concentrations there is little overlap between the ammonium sulfate and PSLs size distributions. Visual evidence of (NH4)2SO4 aggregated with PSL spheres was obtained using transmission electron microscopy (TEM; JEOL JEM-1230). TEM samples of 535 nm PSL in 0.1 wt% (NH4)2SO4 were prepared by exposing mesh grids to the aerosol stream from a DMA that has been set to size-select particles from 505–583 nm, outside of the range where the pure ammonium sulfate distribution would fall. Size-selection was done to ensure minimal contribution from pure ammonium sulfate to the TEM samples.
RESULTS AND DISCUSSION
Determination of the Soluble Mass Fraction of Internally Mixed Particles
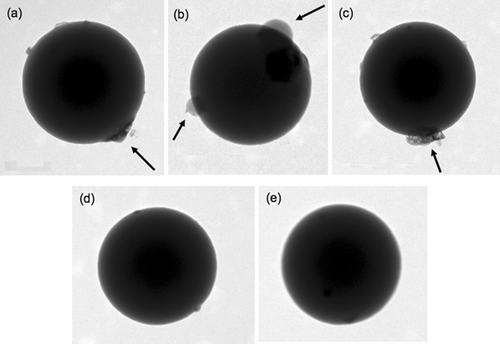
displays TEM images of 535 nm PSL mixed with 0.1 wt% (NH4)2SO4. The images ((a)–(c)) show that, when aerosolized from a solution containing ammonium sulfate, the surface of each PSL sphere has distinct (NH4)2SO4 crystallites (indicated by arrows in ). These are unlike the images of pure PSLs ((d)–(e)) which do show signs of some contamination either from water impurities or remaining surfactant from the original PSL suspension. These impurities, however, do not appear crystalline in nature. Additionally, it should be noted that some of the differences in the appearance of the (NH4)2SO4 crystallites from image to image in can be attributed to electron beam damage from the TEM (CitationHuang and Turpin 1996), which occurs within a few minutes upon exposure to the electron beam.
FIG. 1 TEM images of 535 nm PSL internally mixed with (NH4)2SO4 from 0.1 wt% solutions are shown in (a)–(c). Crystallites of (NH4)2SO4, designated by the arrows, are visible as islands on the surface of the PSLs. TEM images of 535 nm PSLs without (NH4)2SO4 coatings are shown in (d) and (e). The PSLs without (NH4)2SO4 are more spherical compared to the particles with (NH4)2SO4 although the uncoated particle surfaces do show some small “bumps” on the surface that may be due to impurities in the water.
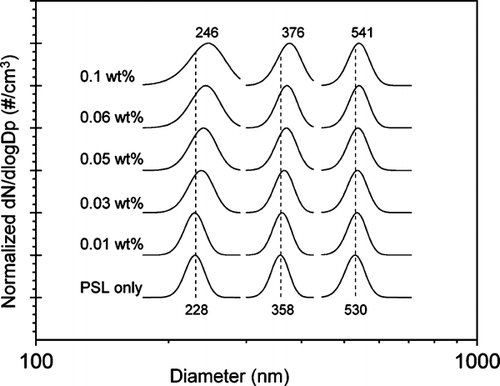
Gaussian fits to the measured SMPS size distributions for the 202, 356, and 535 nm PSLs only and mixed with varying concentrations of ammonium sulfate are shown in . The Gaussian fit is used to determine the peak diameter of the size distribution. First, it can be seen in that the measured mobility diameters are all within the range of the manufacturer's specifications for the PSLs with the exception for the smallest PSL, which shows the particles are actually approximately 10% larger than the specified diameter, 228 nm compared to 202 nm. Second, it can be clearly seen that as the PSLs are mixed with (NH4)2SO4 solutions, the particle diameter increases. Furthermore, as the (NH4)2SO4 concentration in the solution increases, the peak diameter of the distribution shifts to increasingly larger diameters. In general, the change in the measured peak diameter ranges from less than one to approximately 18 nm, and is dependent on the concentration of the (NH4)2SO4 solution. The smallest changes in peak diameter are observed for the larger PSLs, a result that is not unexpected. For example, if a 202 nm and a 535 nm PSL were coated with the same amount of soluble material, there would be a greater change in diameter for the smaller particle. Also, at larger diameters there is lower resolution in the SMPS, as the spacing between size bins increases, making it more difficult to detect smaller changes in particle size. Additionally, as the concentration of the (NH4)2SO4 solution is increased, broadening of the aerosol size distribution is observed for each of the three PSL core sizes, most noticeably for the smallest PSLs. This may be due to the fact that while the core PSL is a constant diameter, the amount of (NH4)2SO4 that can crystallize on the surface is not. As will be discussed in further detail below, the atomizer produces a distribution of droplets that vary in diameter. This results in the same size core PSL particles being aggregated with different quantities of ammonium sulfate which then fall into different SMPS size bins. Thus, a more polydisperse distribution of internally mixed PSL-(NH4)2SO4 particles is measured as the (NH4)2SO4 solution concentration increases. Furthermore, for a given concentration, broadening of the measured size distribution is smaller as particle diameter increases due to the loss of resolution in the SMPS.
FIG. 2 Size distributions of 202, 356, and 535 nm PSLs shown mixed with various concentrations of aqueous (NH4)2SO4. Each size distribution displayed is a Gaussian fit to the measured SMPS distribution. The peak diameter of each distribution has been normalized to one. The distributions have been offset for clarity.
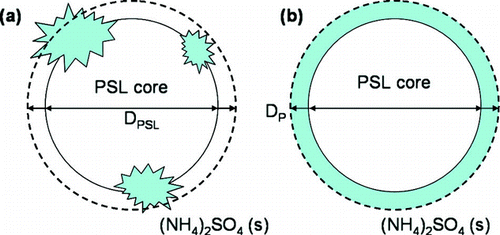
The mass fraction of the ammonium sulfate aggregated to the PSL spheres can be estimated from the change in the peak diameter of the aerosol size distributions. The particle size and composition used to calculate the contribution of (NH4)2SO4 to the particles is shown pictorially in . As the TEMs in show, there is deviation from a spherical shape when the (NH4)2SO4 aggregates on the PSL surface. The mobility diameter measured for the internally mixed PSLs will be of a sphere equivalent in volume to the PSL-(NH4)2SO4 aggregate. This diameter is represented by the dashed line in . Because the total amount of (NH4)2SO4 that crystallizes on the PSL surface will vary from particle to particle, when calculating the mass fraction of (NH4)2SO4 it is assumed that the ammonium sulfate forms a uniform crystalline layer on the PSL surface (). The thickness of this layer is the difference between the measured mobility diameter and the known diameter of the PSL core. The mass of the PSL core (mPSL) is calculated from the peak diameter that is determined from a Gaussian fit to the size distribution of the pure PSL and its known density of 1.05 g/cm3. Assuming sphericity of the internally mixed PSL-(NH4)2SO4 particles as well as volume additivity, the mass of the ammonium sulfate (msolute) is equivalent to:
FIG. 3 A representative schematic of the particle shape and diameter used to determine the mass fraction of (NH4)2SO4 (fsolute) associated with each PSL sphere. A representation of the internally mixed particles based on the TEM images from is shown in (a), where solid crystallites of (NH4)2SO4 are formed on the PSL surface. When the particle is sized by the SMPS, the measured diameter is that of a sphere equivalent in volume to the internally mixed PSL particle. This diameter is represented by the dashed black line. In (b), the particle is assumed to have a uniform, crystalline layer of (NH4)2SO4 coating the PSL surface, where the thickness is the difference between the measured particle diameter, DP, and known PSL diameter, DPSL.
The value of fsolute determined, reported as percent of the total particle mass, will vary as a function of the initial solution concentration. The ammonium sulfate mass fraction calculated using the density ranges from 1–30%, 5–21%, and 5–10% for the 202, 356, and 535 nm PSLs, respectively, from the lowest (0.01 wt%) to the highest (0.1 wt%) ammonium sulfate solution concentrations. (NH4)2SO4 does not crystallize in a uniform coating on the PSL surface under the vacuum conditions of the TEM. The assumed formation of a uniform crystalline layer on the PSL surface overestimates fsolute and therefore, the mass determined using the density of (NH4)2SO4 represents an upper limit to the amount of soluble material internally mixed with each PSL sphere.
Alternatively, the concentration of the solution the particles are atomized from in g/cm3 (csolute) can be used in place of ρsolute in Equation (Equation1) to determine the mass of soluble material in an internally mixed particle. The use of solution concentration in place of density would be appropriate when the soluble material does not crystallize on the insoluble PSL core but instead remains in the aqueous phase. This is the case with nitrate salts such as Ca(NO3)2.
Unlike PSLs, CaCO3, and kaolinite are not monodisperse when aerosolized and as a result it is more difficult to determine how much soluble material is associated with each particle. CaCO3, for example, has a somewhat broad size distribution that is lognormal in nature (192 nm mean diameter, width of 0.96). Therefore, when a 200 nm particle is size-selected from the polydisperse mixed aerosol stream of CaCO3 and Ca(NO3)2, the CaCO3 core will vary in diameter. Additionally, because the distribution of droplets produced from the atomizer is polydisperse as well (0.35 μ m mean diameter, width of 1.9), insoluble CaCO3 particles will be enclosed in solution droplets of varying volume. Thus, two insoluble CaCO3 particles that are equivalent in diameter may have different fractions of soluble nitrate associated with them when internally mixed.
The mass of the CaCO3 core (mcore) and the nitrate component at a given dry particle diameter can be calculated using Equations (3) and (4) (CitationDusek et al. 2006b):
FIG. 4 (a) Calculated core CaCO3 diameter of 200 nm internally mixed CaCO3-Ca(NO3)2 particles generated from 0.01 (filled diamonds), 0.05 (open circles), and 0.1 (filled triangles) wt% Ca(NO3)2 solutions, shown as a function of atomizer droplet diameter (Dd). (b) Calculated mass fraction (fsolute) of Ca(NO3)2 in 200 nm internally mixed CaCO3-Ca(NO3)2 particles as a function of Dd. (c) Estimate of the frequency of occurrence of internally mixed 200 nm CaCO3-Ca(NO3)2 particles with a given fsolute size-selected from carbonate-nitrate mixtures of varying Ca(NO3)2 concentration.
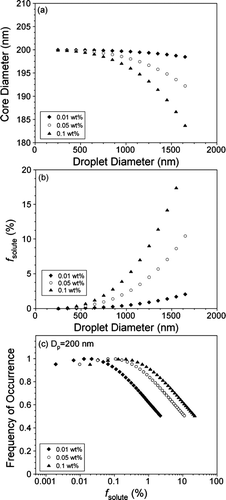
As expected, the fraction of Ca(NO3)2 in each aerosol particle at a given droplet diameter increases with solution concentration. The maximum percentage by mass of nitrate aggregated with CaCO3 is 2% and 23% for 0.01 wt% and 0.1 wt% Ca(NO3)2 solutions, respectively. displays the frequency of occurrence of different values of fsolute that are expected based on the distribution of droplets produced from the constant output atomizer. For the CCN results presented in the following section, the soluble mass fraction is determined to range from < 1 to 11% and < 1 to 5% for CaCO3-Ca(NO3)2 generated from 0.01 and 0.05 wt% Ca(NO3)2 solutions and kaolinite-(NH4)2SO4 generated from 0.01 and 0.02 wt% ammonium sulfate solutions, respectively. Although the fraction of soluble material in each internally mixed particle has a wide range, according to the most likely value of fsolute is less than one percent of the total particle mass. Equations (3) and (4) can also be used to estimate the fraction of ammonium sulfate from a 0.1 wt% solution aggregated to the PSL spheres. Doing so returns maximum calculated values of the (NH4)2SO4 mass as 30, 8, and 3% of the total particle mass for the 202, 356, and 535 nm PSL particles, respectively. These results are in good agreement with those calculated using Equation (Equation1).
Once the composition of the internally mixed aerosol particles is better understood, the implications of trace amounts of soluble material associated with an insoluble mineral dust particle, in terms of indirect climate forcing, can be explored. The measurements of the CCN activity of internally mixed CaCO3-Ca(NO3)2 and kaolinite-(NH4)2SO4 particles discussed here demonstrate that even minimal quantities of a soluble component (< 1%) can dramatically alter the indirect climate forcing of mineral dust aerosol.
CCN Activity of Single Component and Internally Mixed Aerosols
While it is important to understand the behavior of single components of atmospherically aged mineral dust aerosol, this may not accurately represent typical atmospheric conditions. It is possible that the carbonate or aluminosilicate component of mineral dust aerosol will only be partially processed, thus developing a thin coating or inclusion of soluble nitrate or sulfate. Therefore, we have examined the CCN activity of individual mineral dust components and internal mixtures of these components with soluble inclusions, both as a function of percent supersaturation (% SS) and mobility diameter. Internally mixed aerosols are generated from a mixture of CaCO3 in an aqueous solution of either 0.01 or 0.05 wt% Ca(NO3)2 or a mixture of kaolinite with 0.01 and 0.02 wt% (NH4)2SO4. The use of dilute solutions minimizes the contribution of pure Ca(NO3)2 and (NH4)2SO4 aerosol to the measured #CCN/#CN ratio of the internally mixed particles at diameters greater than 150 nm. Below 150 nm, there can be significant contribution of pure Ca(NO3)2 or (NH4)2SO4 to the measured CCN activity.
shows the measured CCN activity of several different single aerosol components and internally mixed aerosols size-selected at 200 nm. Specifically, the CCN activity of calcium carbonate is compared to that of calcium nitrate, and the activity of kaolinite is compared to ammonium sulfate. The critical supersaturation (Sc), where 50% of the particles are activated, for CaCO3 and kaolinite are determined from a sigmoidal fit to the activation data shown in and . The value of Sc for Ca(NO3)2 and (NH4)2SO4 was not measurable and lies outside the range of our instrument (< 0.1% SS). The results displayed in and show that soluble Ca(NO3)2 and (NH4)2SO4 are much more effective CCN than the insoluble mineral dust components CaCO3 and kaolinite. This is most apparent in the 0–0.2% SS region. At 0.2% SS, both Ca(NO3)2 and (NH4)2SO4 are 100% activated while CaCO3 and kaolinite are less than 15 and 2% active, respectively. Comparing CaCO3 to Ca(NO3)2 in shows how chemical reactions in the atmosphere will alter the CCN activity of a particle that is completely converted from a carbonate to a nitrate salt.
FIG. 5 CCN activity as a function of % supersaturation for 200 nm (a) pure CaCO3 (filled circles) and Ca(NO3)2 (open circles); (b) pure kaolinite (filled diamonds) and (NH4)2SO4 (open diamonds); (c) CaCO3 coated with 0.01 wt% (open triangles) and 0.05 wt% (filled triangles) Ca(NO3)2; and (d) kaolinite internally mixed with 0.01 wt% (open inverted triangles) and 0.02 wt% (filled inverted triangles) (NH4)2SO4. Each data point is the average of 2–10 measurements and the error bars represent the standard deviation of these multiple measurements. In (a) and (c), the solid black lines represent sigmoidal fits to the activity of pure Ca(NO3)2 and CaCO3, while the dashed black lines are sigmoidal fits to the internally mixed CaCO3 data. Similarly, the solid black lines in (b) and (d) represent sigmoidal fits to the activity of pure kaolinite and (NH4)2SO4 and the dashed black lines sigmoidal fits to the internally mixed kaolinite data. The Ca(NO3)2 and (NH4)2SO4 fits were calculated using a critical supersaturation value determined from Köhler theory. In each plot, the dashed gray line represents the 50% activation point. The data have all been normalized such that the maximum value of #CCN/#CN is one.
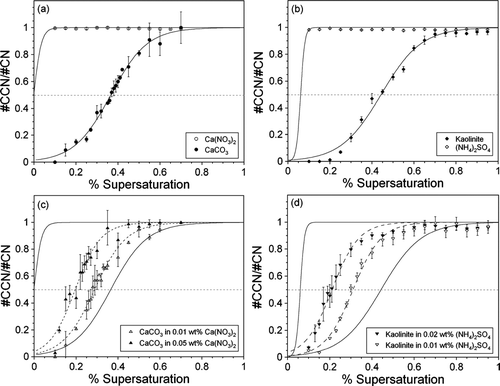
Enhanced CCN activity of CaCO3 and kaolinite is observed for size-selected 200 nm particles when internally mixed with Ca(NO3)2 or (NH4)2SO4 ( and 5d), which are more representative of partially processed particles. At 200 nm, there is almost no contribution (< 5%) from pure Ca(NO3)2 or (NH4)2SO4 to the internally mixed aerosols. The critical supersaturation for CaCO3 decreases by as much as 46%, from 0.37 ± 0.02% SS for the pure CaCO3 aerosol to 0.20 ± 0.02% SS when mixed with 0.05 wt% Ca(NO3)2. The value of Sc for 200 nm kaolinite is reduced from 0.44 ± 0.02 % SS to 0.21 ± 0.02 % SS when mixed with 0.02 wt% (NH4)2SO4. For kaolinite especially, the reported Sc values should be considered an upper limit, as there may be significant contributions to the measured activity from multiply charged particles larger than 200 nm. The values of Scfor all the experiments shown in are summarized in .
TABLE 2 Critical supersaturation values for 200 nm size-selected pure and internally mixed mineral dust components
The influence of soluble material on the CCN activity of mineral dust aerosol is further illustrated in the 0–0.2% SS range, where the efficacy of CaCO3 as a CCN increases by a factor 4 when internally mixed with Ca(NO3)2, determined by comparing the integrated area of this region for each aerosol. In the same range, the presence of (NH4)2SO4 increases the CCN potential of kaolinite by a factor of 6.
displays the CCN activity at 0.3% SS of Ca(NO3)2 in comparison to insoluble CaCO3 as a function of mobility diameter. The critical diameter (Dc), where 50% of the particles are activated, is determined from a sigmoidal fit to the data for each aerosol. It can be seen that for a given supersaturation, Ca(NO3)2 activates at a critical diameter that is ca. 4 times smaller than the insoluble mineral dust aerosol. Although not shown, a similar difference is observed when the CCN activity of another carbonate, dolomite (CaMg(CO3)2), and a 1:1 molar mixture of Ca(NO3)2 and Mg(NO3)2 are compared. A comparison of the CCN activity of kaolinite and (NH4)2SO4 (not shown) yields results analogous to those obtained for CaCO3 and Ca(NO3)2.
FIG. 6 CCN activity at 0.3% SS for (a) Ca(NO3)2 (open circles) and CaCO3 (filled circles) and (b) CaCO3 internally mixed with 0.01 wt% (open triangles) and 0.05 wt% (filled triangles) Ca(NO3)2. The data are plotted as the fraction of particles that can nucleate a cloud (#CCN/#CN) as a function of mobility diameter. These particles represent partially processed mineral dust aerosol. Each data point represents the average of 2–14 measurements, and the error bars are equivalent to the standard deviation of these multiple measurements. The data have been normalized by multiplying each point by the reciprocal of the maximum activation. The dashed gray line indicates the critical diameter where 50% of the particles can activate a cloud. The solid black lines represent sigmoidal fits to the Ca(NO3)2 and CaCO3 data. For CaCO3, data points that are affected by multiple charging of the aerosol particles are not included in the sigmoidal fit. In (a), the points that have been excluded from the fit are shown in gray. The dashed black lines are sigmoidal fits to the internally mixed CaCO3-Ca(NO3)2 data.
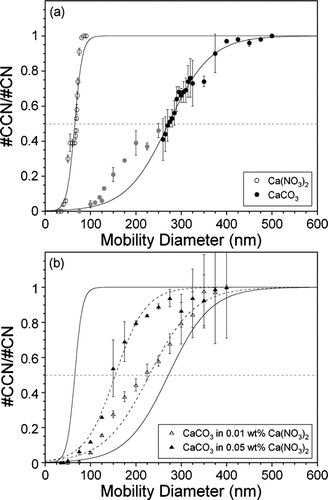
It should be noted that CaCO3 and kaolinite, like most mineral dust components, are non-spherical in nature, unlike the fully processed nitrate or ammonium sulfate aerosol. The actual value of the critical diameter is expected to decrease when particle shape effects are taken into consideration, based on the relationship between mobility and volume equivalent diameter (CitationHinds 1999; CitationKhlystov et al. 2004; CitationZelenyuk et al. 2006; CitationZelenyuk and Imre 2007). As previously mentioned, the presence of multiply-charged particles may also lead to some difficulty in determining Dc. Contributions from multiply-charged particles are more prevalent when the critical diameter and the peak diameter of the particle size distribution are large, i.e., greater than 100 nm. The measured activated fraction will be artificially high due to multiply-charged particles that are greater in diameter than the singly-charged particles that have been size-selected (CitationPetters et al. 2006). This is especially true below 50% activation, and helps to explain the multiple peaks observed in the CCN activation curve for CaCO3 shown in . Consequently, the sigmoidal fit used to determine Dc is only applied to a portion of the data for CaCO3 (above 250 nm) to help account for these contributions. Additionally, particle shape and chemical heterogeneity may also lead to broadening of the measured CCN activation curves for CaCO3 and kaolinite (CitationPetters et al. 2006).
displays the CCN activity at 0.3% SS of CaCO3 aerosol particles internally mixed with 0.01 and 0.05 wt% Ca(NO3)2 as a function of mobility diameter. As was observed in the 200 nm data, these results show that the presence of Ca(NO3)2 enhances the ability of CaCO3 to act as a cloud condensation nuclei. Furthermore, as the concentration of Ca(NO3)2 internally mixed with the CaCO3 aerosol increases, the CCN activity of the CaCO3 increases as well. The value of Dc decreases to smaller diameters by as much as 42% with as dilute as 0.05 wt% Ca(NO3)2 relative to pure CaCO3. The critical diameter for kaolinite at 0.3% SS (not shown) also decreases significantly when mixed with 0.01 wt% ammonium sulfate, and becomes similar to that of pure (NH4)2SO4 aerosol when the sulfate solution concentration is increased to 0.02 wt%. The results obtained in these studies suggest that understanding the amount of chemical processing mineral dust aerosol undergoes as it is transported in the atmosphere will be critical in determining its CCN activity, as it is significantly altered by even minute amounts of soluble components.
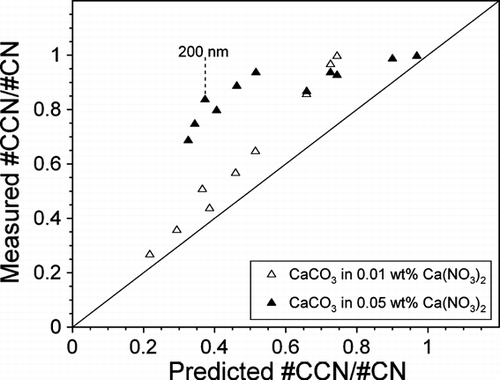
Predicted CCN Activity of Externally Mixed CaCO3 and Ca(NO3)2 Versus Measured CCN Activity
To further verify that the aerosol particles are indeed an internal mixture of mineral dust and soluble material, the measured CCN activity can be compared to the CCN activity that is predicted based on an external mixture. The CCN activity of CaCO3 externally mixed with Ca(NO3)2 can be predicted from the CCN activity and size distributions of the pure components. The individual size distributions of CaCO3 and Ca(NO3)2 can be summed to determine the size distribution of an externally mixed aerosol (not shown). The activity at a specific diameter can be calculated through the use of the following equation:
FIG. 7 Measured #CCN/#CN versus predicted #CCN/#CN for CaCO3 internally mixed with 0.01 wt% (open triangles) and 0.05 wt% (filled triangles) Ca(NO3)2 for diameters ranging from 150–400 nm at 0.3% SS. Moving across the x-axis the diameter of each data point increases by 25 nm. The predicted activities assume an external mixture and are based on the measured SMPS size distributions and CCN activity () of the individual components. The 200 nm point discussed in the text is indicated in the plot.
CONCLUSIONS AND ATMOSPHERIC IMPLICATIONS
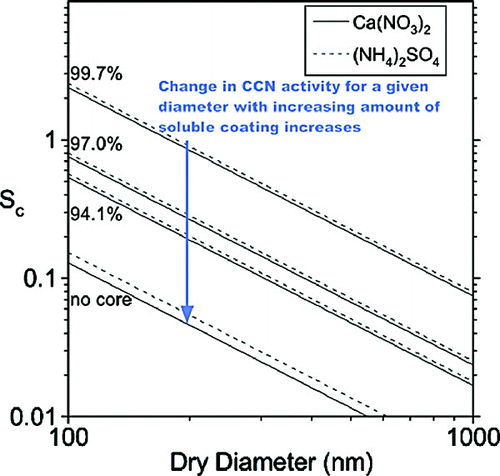
Internally mixed aerosols from aqueous mixtures of insoluble and soluble components have been generated, characterized, and used in CCN activity measurements. The amount of soluble material associated with each PSL, CaCO3, or kaolinite particle can be calculated and is shown to increase as a function of soluble solution concentration. Measurements of the CCN activity of internally mixed CaCO3-Ca(NO3)2 and kaolinite-(NH4)2SO4 show that even trace quantities (calculated as typically less than 1% of the total particle mass for a mixed 200 nm particle) of a soluble component can alter the CCN potential of an insoluble aerosol, in good agreement with previous studies on internally mixed soot particles (CitationDusek et al. 2006b) and Sc values predicted from Köhler theory for aqueous droplets containing different sized insoluble cores. These theoretical values of Sc calculated for Ca(NO3)2 and (NH4)2SO4 droplets containing insoluble cores that comprise various percentages of the total particle volume are shown as a function of diameter in .
FIG. 8 Theoretical Sc, shown as a function of dry particle diameter, for Ca(NO3)2 (solid lines) and (NH4)2SO4 (dashed lines) droplets containing varying size insoluble cores. Sc has been calculated according to Köhler theory and assumes complete dissociation of Ca(NO3)2 and (NH4)2SO4. The series of lines shown are for a droplet containing no core and cores that represent 94.1, 97.0, and 99.7% of the total particle volume (corresponding to 98, 99, and 99.9% of the total particle diameter, respectively).
The data obtained in this work provide important insight into the influence of atmospheric chemistry on the climate impact of mineral dust aerosol through enhanced cloud nucleation activity. The ability of mineral dust aerosol to act as cloud condensation nuclei is strongly dependent on the chemical composition of the aerosol. In general, as mineral dust is transported and processed in the atmosphere it can develop a soluble coating typically composed of sulfates or nitrates, or organic species, consequently changing the physicochemical properties of the aerosol. Because the presence of soluble species on the dust surface increases the particle CCN activity, there will be an enhancement in the concentration of cloud droplets formed from mineral dust aerosol, consequently decreasing precipitation efficiency and changing the radiative properties of the cloud (CitationLevin et al. 2005; CitationRosenfeld et al. 2001; CitationYin et al. 2002). This work shows that CaCO3 that has undergone chemical processing by nitrogen oxides to form calcium nitrate acts as a much more efficient CCN than freshly emitted CaCO3. The enhancement of the CCN activity will depend on the extent of chemical processing. Additionally, this enhancement is dependent on the chemical composition of both the insoluble and soluble species, as the behavior of internally mixed CaCO3-Ca(NO3)2 differs from internally mixed kaolinite-(NH4)2SO4. The results presented here are particularly applicable to submicron mineral dust aerosol, which has been shown to comprise 8–31% of submicron aerosol mass (CitationArimoto et al. (2006)). In this size range, internal mixing of mineral dust with soluble material, especially sulfate, is prevalent (CitationSullivan et al. 2006). The results shown here indicate that there is a large impact on CCN activity for mineral dust aerosol even when trace amounts of soluble material are associated with the particle. Thus, the importance of the chemical reactions leading to these soluble components must be taken into account when determining mineral dust aerosol indirect radiative forcing.
Acknowledgments
This material is based upon work supported by the National Science Foundation under Grant Nos. CHE0503854 and ATM0613124. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. The authors would like to thank Jean Ross of the Central Microscopy Research Facility for her assistance in obtaining TEM images. VHG would like to thank Professor Kim Prather and Ryan Sullivan for helpful discussions.
Notes
a
Lognormal distribution: y = Aexp [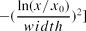 ].
].
a Reported error based on the calculated sigmoidal fit of the data shown in .
b Sc is outside the range of the CCN counter.
Related Research Data
REFERENCES
- Abbatt , J. P.D. , Broekhuizen , K. and Pradeep , Kumar P. 2005 . Cloud Condensation Nucleus Activity of Internally Mixed Ammonium Sulfate/Organic Acid Aerosol Particles . Atmos. Environ. , 39 : 4767 – 4778 .
- Arimoto , R. , Kim , Y. J. , Kim , Y. P. , Quinn , P. K. , Bates , T. S. , Anderson , T. L. , Gong , S. , Uno , I. , Chin , M. , Huebert , B. J. , Clarke , A. D. , Shinozuka , Y. , Weber , R. J. , Anderson , J. R. , Guazzotti , S. A. , Sullivan , R. C. , Sodeman , D. A. , Prather , K. A. and Sokolik , I. N. 2006 . Characterization of Asian Dust During ACE-Asia . Glob. Planet. Change , 52 : 23 – 56 .
- Bauer , S. E. , Balkanski , Y. , Schulz , M. , Hauglustaine , D. A. and Dentener , F. 2004 . Global Modeling of Heterogeneous Chemistry on Mineral Aerosol Surfaces: Influence on Tropospheric Ozone Chemistry and Comparison to Observations . J. Geophys. Res. , 109 : D02304 doi:10.1029/2003JD003868.
- Bian , H. and Zender , C. S. 2003 . Mineral Dust and Global Tropospheric Chemistry: Relative Roles of Photolysis and Heterogeneous Uptake . J. Geophys. Res. , 108 : 4672 doi:10.1029/2002JD003143.
- Buzorius , G. , Zelenyuk , A. , Brechtel , F. and Imre , D. 2002 . Simultaneous Determination of Individual Ambient Particle Size, Hygroscopicity and Composition . Geophys. Res. Lett. , 29 : 1974 doi:10.1029/2001GL014221.
- de Reus , M. , Fischer , H. , Sander , R. , Gros , V. , Kormann , R. , Salisbury , G. , Van Dingenen , R. , Williams , J. , Zoellner , M. and Lelieveld , J. 2005 . Observations and Model Calculations of Trace Gas Scavenging in a Dense Saharan Dust Plume During MINATROC . Atmos. Chem. Phys. , 5 : 1787 – 1803 .
- Dinar , E. , Taraniuk , I. , Graber , E. R. , Katsman , S. , Moise , T. , Anttila , T. , Mentel , T. F. and Rudich , Y. 2006 . Cloud Condensation Nuclei Properties of Model and Atmospheric HULIS . Atmos. Chem. Phys. , 6 : 2465 – 2482 .
- Dusek , U. , Frank , G. P. , Hildebrandt , L. , Curtius , J. , Schneider , J. , Walter , S. , Chand , D. , Drewnick , F. , Hings , S. , Jung , D. , Borrmann , S. and Andreae , M. O. 2006a . Size Matters More Than Chemistry for Cloud-Nucleating Ability of Aerosol Particles . Science , 312 : 1375 – 1378 .
- Dusek , U. , Reischl , G. P. and Hitzenberger , R. 2006b . CCN Activation of Pure and Coated Carbon Black Particles . Environ. Sci. Technol. , 40 : 1223 – 1230 .
- Fan , S.-M. , Horowitz , L. W. , Levy , H. II and Moxim , W. J. 2004 . Impact of Air Pollution on Wet Deposition of Mineral Dust Aerosols . Geophys. Res. Lett. , 31 : L02104 doi:10.1029/2003GL018501.
- Garland , R. M. , Wise , M. E. , Beaver , M. R. , DeWitt , H. L. , Aiken , A. C. , Jimenez , J. L. and Tolbert , M. A. 2005 . Impact of Palmitic Acid Coating on the Water Uptake and Loss of Ammonium Sulfate Particles . Atmos. Chem. Phys. , 5 : 1951 – 1961 .
- Gibson , E. R. , Hudson , P. K. and Grassian , V. H. 2006a . Aerosol Chemistry and Climate: Laboratory Studies of the Carbonate Component of Mineral Dust and Its Reaction Products . Geophys. Res. Lett. , 33 : L13811 doi:10.1029/2006GL026386.
- Gibson , E. R. , Hudson , P. K. and Grassian , V. H. 2006b . Physicochemical Properties of Nitrate Aerosol: Implications for the Atmosphere . J. Phys. Chem. A , 110 : 11785 – 11799 .
- Hameri , K. , Charlson , R. and Hansson , H.-C. 2002 . Hygroscopic Properties of Mixed Ammonium Sulfate and Carboxylic Acids Particles . AIChE J , 48 : 1309 – 1316 .
- Han , J.-H. and Martin , S. T. 2001 . An Aerosol Chemical Reactor for Coating Metal Oxide Particles with (NH4)2SO4-H2SO4-H2O. 1. New Particle Formation . Aerosol Sci. Technol. , 34 : 363 – 372 .
- Hinds , W. C. 1999 . Aerosol Technology: Properties, Behavior, and Measurement of Airborne Particles , New York : Wiley .
- Huang , P.-F and Turpin , B. 1996 . Reduction of Sampling and Analytical Errors for Electron Microscope Analysis of Atmospheric Aerosols . Atmos. Environ. , 30 : 4137 – 4148 .
- Hudson , P. K. , Gibson , E. R. , Young , M. A. , Kleiber , P. D. and Grassian , V. H. 2007 . A Newly Designed and Constructed Instrument for Couples Infrared Extinction and Size Distribution Measurements of Aerosols . Aerosol Sci. Technol. , 41 : 701 – 710 .
- Huff Hartz , K. E. , Tischuk , J. E. , Chan , M. N. , Chan , C. K. , Donahue , N. M. and Pandis , S. N. 2006 . Cloud Condensation Nuclei Activation of Limited Solubility Organic Aerosol . Atmos. Environ. , 40 : 605 – 617 .
- Katrib , Y. , Martin , S. T. , Hung , H.-M , Rudich , Y. , Zhang , H. , Slowik , J. G. , Davidovits , P. , Jayne , J. T. and Worsnop , D. R. 2004 . Products and Mechanisms of Ozone Reactions with Oleic Acid for Aerosol Particles Having Core-Shell Morphologies . J. Phys. Chem. A , 108 : 6686 – 6695 .
- Kaufman , Y. J. and Koren , I. 2006 . Smoke and Pollution Aerosol Effect on Cloud Cover . Science , 313 : 655 – 658 .
- Khlystov , A. , Stanier , C. and Pandis , S. 2004 . An Algorithm for Combining Electrical Mobility and Aerodynamic Size Distributions Data When Measuring Ambient Aerosol . Aerosol Sci. Technol. , 38 : 229 – 238 .
- Korhonen , H. , Napari , I. , Timmreck , C. , Vehkamaki , H. , Pirjola , L. , Lehtinen , K. E.J. , Lauri , A. and Kulmala , M. 2003 . Heterogeneous Nucleation as a Potential Sulphate-Coating Mechanism of Atmospheric Mineral Dust Particles and Implications of Coated Dust on New Particle Formation . J. Geophys. Res. , 108 : 4546 doi:10.1029/2003JD003553.
- Krueger , B. J. , Grassian , V. H. , Laskin , A. and Cowin , J. P. 2003 . The Transformation of Solid Atmospheric Particles into Liquid Droplets Through Heterogeneous Chemistry: Laboratory Insights into the Processing of Calcium Containing Mineral Dust Aerosol in the Troposphere . Geophys. Res. Lett. , 30 : 1148 doi:10.1029/2002GL016563.
- Laskin , A. , Iedema , M. J. , Ichkovich , A. , Graber , E. R. , Taraniuk , I. and Rudich , Y. 2005 . Direct Observation of Completely Processed Calcium Carbonate Dust Particles . Faraday Discuss. , 130 : 453 – 468 .
- Levin , Z. , Teller , A. , Ganor , E. and Yin , Y. 2005 . On the Interactions of Mineral Dust, Sea-Salt Particles, and Clouds: A Measurement and Modeling Study from the Mediterranean Israeli Dust Experiment Campaign . J. Geophys. Res. , 110 : D20202 doi:10.1029/2005JD005810.
- Liao , H. , Seinfeld , J. H. , Adams , P. J. and Mickley , L. J. 2004 . Global Radiative Forcing of Coupled Tropospheric Ozone and Aerosols in a Unified General Circulation Model . J. Geophys. Res. , 109 : D16207 doi:10.1029/ 2003JD004456.
- Liao , Y.-C and Roberts , J. T. 2006 . Self-Assembly of Organic Monolayers on Aerosolized Silicon Nanoparticles . J. Am. Chem. Soc. , 128 : 9061 – 9065 .
- Martin , R. V. , Jacob , D. J. , Yantosca , R. M. , Chin , M. and Ginoux , P. 2003 . Global and Regional Decreases in Tropospheric Oxidants from Photochemical Effects of Aerosols . J. Geophys. Res. , 108 : 4097 doi:10.1029/ 2002JD002622.
- Matsuki , A. , Iwasaka , Y. , Shi , G. , Zhang , D. , Trichkine , D. , Yamada , M. , Kim , Y. S. , Chen , B. , Nagatani , T. , Miyazawa , T. , Nagatani , M. and Nakata , H. 2005 . Morphological and Chemical Modification of Mineral Dust: Observational Insight into the Heterogeneous Uptake of Acidic Gases . Geophys. Res. Lett. , 32 doi:10.1029/2005GL024176
- McFiggans , G. , Artaxo , P. , Baltensperger , U. , Coe , H. , Facchini , M. C. , Feingold , G. , Fuzzi , S. , Gysel , M. , Laaksonen , A. , Lohmann , U. , Mentel , T. F. , Murphy , D. M. , O'Dowd , C. D. , Snider , J. R. and Weingartner , E. 2006 . The Effect of Physical and Chemical Aerosol Properties on Warm Cloud Droplet Activation . Atmos. Chem. Phys. , 6 : 2593 – 2649 .
- Mircea , M. , Facchini , M. C. , Decesari , S. , Cavalli , F. , Emblico , L. , Fuzzi , S. , Vestin , A. , Rissler , J. , Swietlicki , E. , Frank , G. , Andreae , M. O. , Maenhaut , W. , Rudich , Y. and Artaxo , P. 2005 . Importance of the Organic Aerosol Fraction for Modeling Aerosol Hygroscopic Growth and Activation: A Case Study in the Amazon Basin . Atmos. Chem. Phys. , 5 : 3111 – 3126 .
- Mogili , P. K. , Kleiber , P. D. , Young , M. A. and Grassian , V. H. 2006 . N2O5 Hydrolysis on the Components of Mineral Dust and Sea Salt Aerosol: Comparision Study in an Environmental Aerosol Reaction Chamber . Atmos. Environ , 40 : 7401 – 1408 .
- Ooki , A. and Uematsu , M. 2005 . Chemical Interactions Between Mineral Dust Particles and Acid Gases During Asian Dust Events . J. Geophys. Res. , 110 : D03201 doi:10.1029/2004JD004737.
- Perry , K. D. , Cliff , S. S. and Jimenez-Cruz , M. P. 2004 . Evidence of Hygroscopic Mineral Dust Particles from the Intercontinental Transport and Chemical Transformation Experiment . J. Geophys. Res. , 109 : D23S28 doi:10.1029/2004JD004979.
- Petters , M. D. , Prenni , A. J. , Kreidenweis , S. M. and DeMott , P. J. 2006 . On Measuring the Critical Diameter of Cloud Condensation Nuclei using Mobility Selected Aerosol . Aerosol Sci. Technol , submitted
- Petzold , A. , Gysel , M. , Vancassel , X. , Hitzenberger , R. , Puxbaum , H. , Vrochticky , S. , Weingartner , E. , Baltensperger , U. and Mirabel , P. 2005 . On the Effects of Organic Matter and Sulphur-Containing Compounds on the CCN Activation of Combustion Particles . Atmos. Chem. Phys. , 5 : 3187 – 3203 .
- Pradeep , Kumar p. , Broekhuizen , K. and Abbatt , J. P.D. 2003 . Organic Acids as Cloud Condensation Nuclei: Laboratory Studies of Highly Soluble and Insoluble Species . Atmos. Chem. Phys. , 3 : 509 – 520 .
- Ramaswamy , V. , Boucher , O. , Haigh , J. , Hauglustaine , D. , Haywood , J. , Myhre , G. , Nakajima , T. , Shi , G. Y. and Solomon , S. 2001 . “ Radiative Forcing of Climate Change ” . In Climate Change 2001: The Scientific Basis. Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change (IPCC) , Edited by: Houghton , J. T. , Y. , Ding , D. , J. Griggs , M. , Noguer Linden , P. , P.J.V.D. Linden and D. , Xiaosu . 349 – 416 . New York : Cambridge University Press .
- Roberts , G. and Nenes , A. 2005 . A Continuous-Flow Streamwise Thermal-Gradient CCN Chamber for Atmospheric Measurements . Aerosol Sci. Technol. , 39 : 206 – 221 .
- Rosenfeld , D. , Rudich , Y. and Lahav , R. 2001 . Desert Dust Suppressing Precipitation: A Possible Desertification Deedback Loop . Proc. Natl. Acad. Sci. U.S.A. , 98 : 5975 – 5980 .
- Song , C. H. and Carmichael , G. R. 2001 . Gas-Particle Partitioning of Nitric Acid Modulated by Alkaline Aerosol . J. Atmos. Chem. , 40 : 1 – 22 .
- Sullivan , R. C. , Guazzotti , S. A. , Sodeman , D. A. and Prather , K. A. 2007 . Direct Observations of the Atmospheric Processing of Asian Mineral Dust . Atmos. Chem. Phys. , 7 : 1213 – 1236 .
- Tang , I. N. and Munkelwitz , H. R. 1994 . Water Activities, Densities, and Refractive Indices of Aqueous Sulfates and Sodium Nitrate Droplets of Atmospheric Importance . J. Geophys. Res. , 99 : 18801 doi:10.1029/ 94JD01345.
- Tang , Y. , Carmichael , G. R. , Kurata , G. , Uno , I. , Weber , R. J. , Song , C.-H , Guttikunda , S. K. , Woo , J.-H , Streets , D. G. , Wei , C. , Clarke , A. D. , Huebert , B. and Anderson , T. L. 2004 . Impacts of Dust on Regional Tropospheric Chemistry During the ACE-Asia Experiment: A Model Study with Observations . J. Geophys. Res. , 109 : D19S21 doi:10.1029/2003JD003806.
- Underwood , G. M. , Song , C. H. , Phadnis , M. , Carmichael , G. R. and Grassian , V. H. 2001 . Heterogeneous Reactions of NO2 and HNO3 on Oxides and Mineral Dust: A Combined Laboratory and Modeling Study . J. Geophys. Res. , 106 : 18055 – 18066 . doi:10.1029/2000JD900552
- Yin , Y. , Wurzler , S. , Levin , Z. and Reisin , T. G. 2002 . Interactions of Mineral Dust Particles and Clouds: Effects on Precipitation and Cloud Optical Properties . J. Geophys. Res. , 107 : 4724 doi:10.1029/2001JD001544.
- Zelenyuk , A. , Cai , Y. and Imre , D. 2006 . From Agglomerates of Spheres to Irregularly Shaped Particles: Determination of Dynamic Shape Factors from Measurements of Mobility and Vacuum Aerodynamic Diameters . Aerosol Sci. Technol. , 40 : 197 – 217 .
- Zelenyuk , A. and Imre , D. 2007 . On the Effect of Particle Alignment in the DMA . Aerosol Sci. Technol. , 41 : 112 – 124 .