Abstract
Until now, micrometeorological measurements of surface/ atmosphere exchange fluxes of submicron aerosol chemical components such as nitrate, sulfate or organics could only be made with gradient techniques. This article describes a novel setup to measure speciated aerosol fluxes by the more direct eddy covariance technique. The system is based on the Aerodyne quadrupole-based Aerosol Mass Spectrometer (Q-AMS), providing a quantitative measurement of aerosol constituents of environmental concern at a time resolution sufficient for eddy-covariance. The Q-AMS control software was modified to maximize duty cycle and statistics and enable fast data acquisition, synchronized with that of an ultrasonic anemometer. The detection limit of the Q-AMS based system for flux measurements ranges from 0.2 for NO3 − to 15 ng m−2 s−1 for hydrocarbon-like organic aerosol (HOA), with an estimated precision of around 6 ng m−2 s−1, depending on aerosol loading. At common ambient concentrations the system is capable of resolving deposition velocity values < 1 mm s−1, sufficient for measurements of dry deposition to vegetation. First tests of the system in the urban environment (6 to 20 June 2003) in Boulder, CO, USA, reveal clear diurnal, presumably traffic related, patterns in the emission of HOA and NO3 −, with indication of fast production of moderately oxygenated organic aerosol below the measurement height, averaging about 15% of the HOA emission. The average emission factor for HOA was 0.5 g (kg fuel)−1, similar to those found in previous studies. For NO3 − an emission factor of 0.09 g (kg fuel)−1 was estimated, implying oxidation of 0.5% of the traffic derived NOx below the measurement height of 45 m. By contrast, SO4 2− fluxes were on average downward, with deposition velocities that increase with friction velocity from 0.4 to 4 mm s−1.
1. Introduction
1.1. Background
Aerosols are involved in many areas of current environmental concern. They modulate the climate forcing potential of the atmosphere directly through light-scattering and through various indirect effects. For example, they may act as cloud condensation nuclei (CCNs) resulting in increased cloud cover, and deposition of soot aerosols is suspected to decrease the albedo of snow surfaces. High concentrations of particulate matter have been associated with increased mortality rates, especially in densely populated urban areas, and are responsible for poor visibility, a particular concern for transportation safety, and for aesthetic reasons in many National Parks across the United States (e.g., CitationCharlson et al. 1992; CitationDockery et al. 1993; CitationRamanathan et al. 2001; CitationWatson 2002). Because of their relatively slow deposition compared with reactive gases, aerosol particles can have a long atmospheric lifetime and often dominate the long-range transport of acidifying compounds containing nitrogen (N) and sulfur (S), as well as the trans-boundary air pollution fluxes of toxic metals, persistent organic compounds and radioactive materials. Aerosols often dominate the deposition of N and S into sensitive ecosystems at low precipitation sites. Climate effects, air quality, transport, and deposition can only be modelled accurately, if the emissions, chemical and physical transformation and deposition processes are fully understood. At present, although emissions from individual large stacks may be monitored accurately, estimates of area sources of particles (e.g., due to transport sources, cooking, heating, and re-suspension) are highly uncertain. Similarly, few reliable measurements exist of the deposition rates of particles and how they change with land cover, particle size, and composition as well as meteorology.
1.2. Existing Micrometeorological Approaches to Measure the Surface/Atmosphere Exchange of Atmospheric Particles
Micrometeorological flux measurement approaches enable vertical surface/exchange fluxes to be measured directly at the urban canopy scale, averaged over typically 100s or 1000s of m2. While fluxes of CO2 and H2O are measured routinely within large international networks, flux measurements of particles are comparably rare and, with few exceptions, limited to campaign-based measurements (cf. reviews by CitationGallagher et al. 1997; CitationGallagher et al. 2002). This is in part due to the constraints imposed by available aerosol instrumentation.
The most direct flux measurement approach, which is also applied in the CO2 flux networks, is the eddy covariance approach, in which the turbulent flux (F χ) of a tracer is derived as the covariance in the tracer's concentration (χ) and the vertical wind component (w):
Both entities must be measured sufficiently fast to resolve all turbulent eddies that contribute to the flux (typically several Hz). This approach has been used during several studies to measure particle number fluxes, without information on the chemical composition of the particles. Particle number concentration were measured either with size-resolving instrumentation such as optical or aerodynamic particle spectrometers to derive size-segregated fluxes (typically from 0.1 μ m upwards) (e.g., CitationGallagher et al. 1997; CitationNemitz et al. 2002a), or with total particle counters such as condensation particle counters (CPC), deriving the total particle flux, which includes particle sizes down to typically 10 nm, depending on the CPC used (CitationBuzorius et al. 1998; CitationBuzorius et al. 2001; CitationDorsey et al. 2002; CitationNemitz et al. 2002a). These types of measurements have been used to derive deposition velocities (Vd) as a function of particle diameter (dp) for a range of vegetation types, which may be multiplied by the size-distribution of individual aerosol components such as SO4 2−, NO3 −, NH4 +, organics or toxic metals, to estimate dry deposition fluxes of the different chemical species. However, this assumes that the net behavior of all particle types is representative for the behavior of the particular compound of interest, as it combines information on different turbulent moments (i.e., concentrations with fluxes). In addition, an increasing number of flux measurements indicates that particle fluxes over vegetation may be bi-directional with both apparent emissions and deposition occurring. This has been attributed to effects of growth/evaporation due to interactions with water or reactive precursor gases (e.g., CitationKowalski 2001; CitationNemitz and Sutton 2004). Chemically resolved flux measurements are required to fully resolve these interactions and thus derive more reliable parameterizations of Vd.
Chemical analyzers that can determine the chemical composition of particles are usually too slow for use in eddy-covariance. Instead, chemically resolved surface/atmosphere exchange fluxes have been measured with the flux-gradient approach that relates Fχ to the vertical concentration gradient (dχ/dz) through an, essentially empirical, eddy-diffusivity (K H ), which depends on the height (z) above the zero-plane displacement height (d) and the Monin-Obukhov stability length (L):
In this approach, concentration gradients were either measured by filter-pack (e.g., CitationDuyzer 1994; CitationSutton 1990; CitationWyers and Duyzer 1997) or, more recently, with automated instruments that sample aerosol in the liquid phase, which is analyzed online for major ions, e.g., by selective ion membrane/conductivity or ion chromatography (e.g., CitationNemitz et al. 2000a; CitationNemitz et al. 2004).
In a third approach, the relaxed eddy-accumulation technique (REA), up- and down-draughts are conditionally sampled into two separate storage reservoirs or filters from which the flux is calculated as
Both gradient methods and REA have the disadvantage of relying on empirical parameterizations and/or similarity theory, while gradient techniques also require access to a range of heights, which can cause difficulties above forests and urban areas and makes gradient methods inapplicable for aircraft platforms.
One of the few studies that have attempted eddy covariance (EC) flux measurements of individual aerosol chemical species has measured fluxes of SO4 2− over grassland, using a pyrolyzer together with a modified SO2 analyzer (CitationWesely et al. 1985). Recent developments in aerosol mass spectrometry provide a range of new aerosol chemical analyzers with fast response times. CitationHeld et al. (2003) recently evaluated the applicability of Laser-Ablation Time-of-Flight Aerosol Mass Spectrometers for disjunct eddy covariance flux measurements. CitationGonzales et al. (2003)reported initial EC measurements with an Aerodyne Quadrupole Aerosol Mass Spectrometer (Q-AMS) for one single species at a time over forest. In this article, we report the development of a similar system, optimized for concurrent measurements of typically up to 10 groups of compounds. The article first outlines the general principles of this new flux measurement system, and then explains the details of the implementation to measure fluxes above a city (Boulder, CO, USA), where fluxes are large. These measurements serve to test the performance of the system and to provide initial information on chemically speciated aerosol fluxes in the urban environment.
2. METHODS
2.1. The Aerodyne Aerosol Mass Spectrometer
A detailed description of the standard Q-AMS instrument and its modes of operation was provided by CitationJayne et al. (2000) and CitationJimenez et al. (2003). In brief, a tightly focused beam of particles contained in a sample stream of 1.5 cm3 s−1 enters a differentially pumped vacuum chamber through an aerodynamic lens. Depending on the choice of the position of a moveable chopper, the particle beam may be allowed to pass, it may be fully blocked out, or chopped, with a photodiode detecting the time of particles passing through the 2% chopper opening. The particle beam passes a series of skimmers while the gas-phase is differentially pumped, which results in an enrichment of the particles in the sample stream with respect to the airbeam, by a factor of about 107. After passing a particle time-of-flight region, the particles are flash-vaporized at a temperature of typically 550°C. The resulting gaseous species are ionized by standard 70 eV electron impact ionization and the ions extracted into a quadrupole mass spectrometer (QMS, Balzers QMA 410) for detection. The Q-AMS measures the non-refractory components (operationally defined as those compounds that evaporate in a few milliseconds at the vaporizer temperature) contained in submicron particles, determined by the transmission characteristics of the aerodynamic lens (approx PM1). The lens used here has unity transmission in the size range 60 to 600 nm, with some transmission over the range 35 to 1500 nm (cf. below). Up to now, the Q-AMS has been operated in one of two main modes, as described in the following sections.
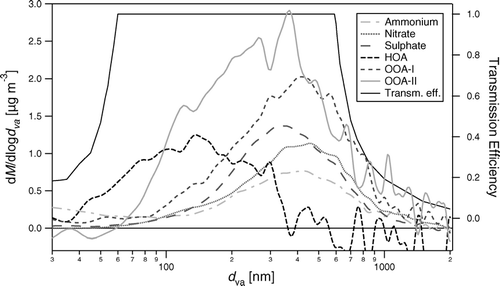
FIG. 11 Averaged size-spectra of ammonium, nitrate, sulfate and organic aerosol (divided into HOA and OOA), in relation to the transmission function of CitationJayne et al. (2000). Towards the small size end, the size distribution of ammonium and OOA-I are affected by the contribution of O+ and CO2 + ions (from gas-phase O2 and CO2 molecules respectively behaving like small particles for the PToF measurement).
2.1.1. Mass Spectrum (MS) Mode
In the mass spectrum (MS) mode the QMS is controlled to scan over the entire range of mass/charge ratios (m/z) of 1 to 300, every 300 ms. The sample beam is sequentially opened and blocked for subtraction of the instrument's background signal. This mode provides the full composition of the non-refractory aerosol components. Routines have been developed to deconvolve the mass spectrum of the main inorganic species and the total organics (CitationAllan et al. 2004), and to extract the main components of the organic spectrum at a given location (CitationZhang et al. 2005a; CitationZhang et al. 2005c). These are being further refined as information becomes available from field studies and from laboratory studies on the fragmentation patterns of individual aerosol sources and/or species.
2.1.2. Particle Time-of-Flight (PToF) Mode
In the particle time-of-flight (PToF) mode, the sample stream is chopped and the particle time-of-flight between chopper opening and detection by the QMS are used to infer the vacuum aerodynamic diameter (d va ; DeCarlo et al. 2004) of the detected particle mass distribution. Individual particles can be detected, if the particle contains an amount of the species currently analyzed by the QMS sufficient to be distinguished from the background noise. Typically, the Q-AMS steps through 10 to 20 selected m/z to accumulate size-distributions of the associated aerosol components. In this mode, particles and air molecules can be distinguished by the large differences in their times-of-flight. Additional data from the MS Mode may be needed, however, to distinguish the contribution of different parent compounds to the ion counts at certain m/z and to quantify the fragmentation pattern. In general, the m/z's are selected to minimize ambiguities.
Typically, the Q-AMS is operated in “alternate” mode, in which the MS and PToF modes are alternated every 20 seconds or so, while averages of the size-distributions and mass spectra are accumulated for typically 5 minutes.
2.2. Software Mode for Eddy-Covariance
The error in the eddy-covariance flux measurement of particles is related to the number of particles detected per averaging time (CitationFairall 1984), making it important to maximize the number of particles detected by maximizing the duty cycle of the measurement, i.e., the fraction of time spent on obtaining the mass concentration of one particular m/z. In the MS Mode, the QMS scans over 300 m/z, hereby spending equal amounts of time on mass peaks and between mass peaks. Considering that the background is analyzed for half of the time, this equates to a duty cycle of only 1/(300 × 2 × 2) = 0.083% per m/z. Similarly, in the PToF Mode the chopper opening of 2% removes 98% of all particles resulting in a duty cycle of ∼ 0.2% per m/z if a total of ten m/z are monitored sequentially (CitationBahreini et al. 2003; CitationDeCarlo et al. 2006). In addition, due to switching between m/z's in PToF Mode and between background and signal in MS Mode, these standard modes do not allow a continuous fast measurement of the concentration required for EC calculations.
To provide robust eddy covariance fluxes, the analytical technique needs to average over a large number of particles and the signal needs to be clearly distinguishable from the background noise. shows the Q-AMS signal recorded at 16.7 Hz while sampling ambient air in Boulder, measured at selected m/z while the chopper position changed from closed to open on a 1 Hz cycle. This plot demonstrates the excellent signal/ noise ratio of the Q-AMS when sampling only selected ions. The spikes during in the open chopper position reflect the contribution from individual larger particles, while the signal variation in the closed position is clearly lower. The non-zero signal at m/z 16 in the closed chopper position is due to the O+ background in the instrument, which is due to ionization of O2 and H2O from the gas-phase.
FIG. 1 Raw measurement signal at 16.7 Hz comparing blocked to open beam (switched every 5 s), demonstrating the excellent signal/noise ratio of the Q-AMS instrument when sampling selected ions.
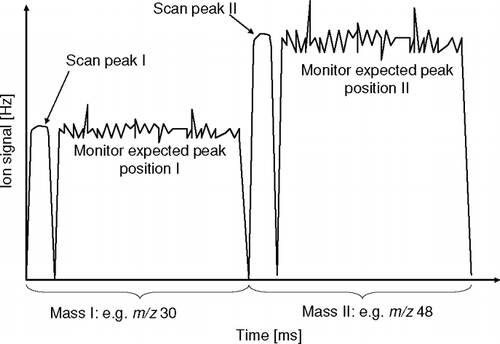
Clearly, the Q-AMS has the potential to provide good signal to noise for averaging times relevant to eddy covariance measurements, if operated with a suitable protocol. This, together with similar needs for aircraft-based measurements, sparked the development of a new software operating mode, which is comparable with selective ion monitoring (SIM) in quadrupole-based GC-MS. The new Jump Mass Spec (JMS) Mode has been described in detail by CitationCrosier et al. (2007), and it will only be briefly reviewed here as it pertains to EC flux measurements. The JMS mode works similarly to the MS Mode. However, instead of scanning the whole range of m/z, the QMS steps between N individual selected m/z. For each m/z the measurement consists of three stages: first, the QMS jumps to the anticipated start of the peak. Some time (t setscan = 2 ms) is provided for the QMS power supplies to settle, before it is scanned across the peak at the same rate as used in the MS mode, at t scan = 1 ms / peak. Finally, the QMS is set to the anticipated position of the peak for a further settling time (t setpeak = 0.75 ms) and some measurement time (t meas) (). The duration of t meas depends on the number of m/z selected. This measurement is repeated for each m/z in the selection. For fast measurement at a precise data rate, two continuous waveforms are composed and programmed in continuous computer analogue output voltage channels, which cycle through the QMS DC and RF control voltages of the whole measurement cycle.
FIG. 2 Signal from the selective ion monitoring as implemented in the JMS mode (conceptual schematic). For each selected m/z, the instrument scans the peak and the settles on the anticipated peak position.
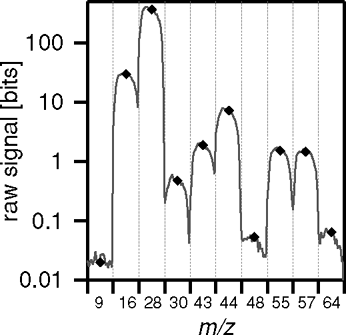
The data at the peak-position is averaged to provide the fast data for the JMS mode. The peak shapes are averaged over 1 minute and saved for quality control. The difference between the height of the scanned peak and the concentration derived from the assumed peak position provides a measure for the drift of the mass calibration and can form the basis for correction procedures. An example of such comparison is shown in , averaged over 1 minute.
FIG. 3 Example peak shapes obtained during the JMS measurement cycle, averaged over 1 min. The symbols indicate the concentration derived during t meas, prior to subtraction of the DC signal level (derived from m/z = 9).
If the JMS mode is used for concentration measurements, the chopper is used to alternate between open and blocked beam to derive a continuous background, similar to the MS Mode (e.g., as shown in ). Typically, in the so-called General Alternation Mode (GENALT) the JMS Mode is alternated with the PToF and MS Modes to provide information on (a) the full aerosol composition (MS Mode), (b) the size distribution of selected compounds (PToF Mode), and now (c) the concentration of these compounds with much increased signal-to-noise ratio (JMS). This is of particular value when averaging times need to be short, e.g., during aircraft or other mobile measurements, or when concentrations are very low. In this alternation mode the m/z calibration is automatically optimized from the MS Mode measurements after each averaging period of typically 5 min. A more detailed description of this JMS Mode and its merits compared with the MS Mode was provided by CitationCrosier et al. (2007), who use this mode during fast airborne measurements.
For eddy-covariance flux measurements, however, a variant of this mode (EC-JMS Mode) was implemented, in which the beam is initially blocked for typically 1 min, before it is opened for a user-defined averaging period (e.g., 29 minutes). The individual concentrations are derived with high time-resolution as the average signal over t meas. The peak scans (which are averaged over 1 min and saved) may be used to identify and, if necessary, correct for drifts in the m/z calibration of the system with time, since these drifts cannot automatically be corrected during the sampling period to avoid introducing gaps in the measurement time series.
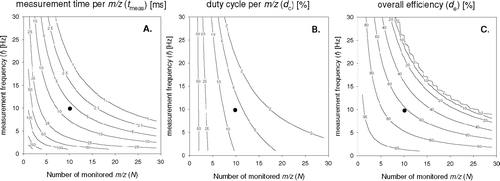
The time t meas and the duty cycle per m/z (d c ) depend on both the number of m/z monitored (N m/z) and the measurement frequency (f):
From these parameters, the overall efficiency of the measurement (d e), defined as the fraction of time spent on actual measurement, rather than on switching between m/z's and peak shape determination, is given by:
These parameters are presented in for the current system (t setscan = 2 ms; t scan = 1 ms; t setpeak = 0.75 ms), which, in Boulder, was operated with 10 m/z's at 10 Hz, resulting in t meas = 6.25 ms and a duty cycle per m/z of d c = 6.3%. For comparison, for a traditional continuous chemical sensor for eddy-covariance application d c = 100% and t meas = 1/f.
FIG. 4 Dependence of (a) averaging time and (b) duty cycle per m/z on sampling frequency (f), (c) effective overall efficiency and number of selected m/z (N m/z). The marker (•) illustrates the conditions as used during this study (10 m/z at 10 Hz).
The three wind components (u, v, and w) as well as the temperature (T) from the anemometer are recorded at the same frequency as the Q-AMS concentration data through the use of analogue inputs, which are repetitively acquired while the program waits for buffers containing the electron multiplier signal of the QMS to be returned. It should be noted that the analogue signals are updated at the native frequency of the anemometer, which was set to 10 Hz for the anemometer used during these measurements. A serial output was implemented, through which the Q-AMS calibration parameters and the raw eddy covariance data (concentrations and anemometer data) may be transferred to a second computer for online flux calculations and display.
Unlike the modified EC-PToF mode that was used for initial flux measurements of individual compounds by CitationGonzales et al. (2003), in the EC-JMS mode, no information is obtained on the sizes of the particles; nor is it possible to monitor all fragments of the compounds of interest in the EC mode. It is thus necessary to interpret the EC measurements in the light of the PToF and MS measurements, which may be provided by a second AMS instrument. Alternatively, the instrument can enter the GENALT for some time after each EC averaging period. In practice, operation so far consisted either of 30 min of GENALT followed by 30 minutes of EC-JMS mode, or, alternatively, by 5 minutes of GENALT followed by 25 minutes of EC-JMS mode, depending on the degree to which the concentration should be characterized by the EC Q-AMS.
2.3. Methods: Calculation of Concentrations
The ionization efficiency (IE) of the Q-AMS is calibrated with pure NH4NO3 particles of a known mobility diameter. The quantitative determination of chemically resolved aerosol concentration by the Q-AMS relies on the knowledge of the ionization efficiencies of these compounds relative to NO3 − and the fragmentation pattern of the target compounds. The concentration of a chemical species i (χ i ) is calculated as (e.g., CitationAllan et al. 2004; CitationJimenez et al. 2003):
In general, the organic aerosol fraction is composed of thousands of different compounds and each m/z may represent several fragments from a range of parent compounds. The AMS is therefore usually used to infer the total organic concentration or to detect individual characteristic markers (such as m/z 60 for levoglucosan and similar species from biomass burning). Analysis of AMS data from many urban sites around the world now indicate that the organic aerosol component in cities can be decomposed into ∼ 2–5 distinct groups of species, which can be quantified from the entire organic mass spectrum by multi component analysis (MCA) or positive matrix factorization (PMF) (CitationZhang et al. 2005a; CitationZhang et al. 2005c; CitationZhang et al. 2007; CitationLanz et al. 2007). The most frequently identified components, which are also the relevant ones for this study, are:
| 1. | Hydrocarbon-like aerosol (HOA) is often dominated by primary organic aerosol (POA), which is mainly emitted by combustion sources. The mass spectrum of HOA found in several urban areas closely resembles the mass spectrum of fresh diesel particles and lubrication oil, but also contains particles from gasoline exhaust and other reduced POA sources such as meat cooking (CitationMohr et al. 2008). A significant fraction of HOA is often contained in a small aerosol size mode of 50–200 nm, with some contribution in the accumulation mode. | ||||
| 2. | Oxygenated organic aerosol (OOA) is highly oxygenated aerosol with a high O/C ratio, as indicated in a large contribution of m/z 44 (CO2 +) (CitationAiken et al. 2007; CitationAiken et al. 2008). Mass spectra from remote sites tend to resemble each other and also the mass spectrum of fulvic acid. Similar to sulfate, OOA is predominantly found in the accumulation mode (300–900 nm), consistent with secondary organic aerosol (SOA) formation over regional scales. | ||||
| 3. | In many environments, including Boulder, a second oxygenated aerosol type can be identified and thus OOA is sub-divided into OOA-I (representing aged, more highly oxidized aerosol) and OOA-II, a less oxidized class which is characterized by high m/z 43 (see Section 3.1) (CitationLanz et al. 2007; CitationUlbrich et al. 2008). | ||||
2.4. Methods: Flux Calculations
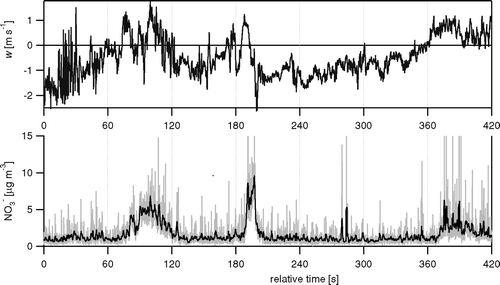
Fluxes were calculated using standard eddy-covariance methodology (e.g., CitationAubinet et al. 2000), from the time-series of the three wind components and the concentrations, using an analysis program written in LabView. demonstrates for an example time series how, above Boulder, updrafts (periods of w > 0) tend to be associated with increased concentrations in aerosol NO3 −, resulting in a positive (upward) flux according to Equation (Equation1). The data of the figure imply a time lag of about 4 s, between w and χ (NO3 −), caused by the delay due to transport in the sample line, which is consistent with the time-lag in the main inlet calculated from inlet volume and flow rate, of 2.3 s, to which needs to be added the additional time-lag due to the lower flow in the sub-sampling lines.
FIG. 5 Example time series of the vertical wind components (w) and the NO3 − concentration derived from m/z 30 for 16 June 2003, 12:30 PM. Shown are the raw 10 Hz data, and also a 1 s running mean for the concentration data (black line on the bottom panel).
For each 30-minute data block, a co-ordinate rotation was performed to account for sensor misalignment and to match the streamwise wind component (u) to the direction of the mean flow. Despite the likely influence of the wind structure on the mean wind flow, the dependence of the vertical rotation angle on wind speed shows a well-defined, near sinusoidal structure (not shown), increasing confidence that co-ordinate rotation is effective. However, it cannot be ruled out fully that the quality of the measured fluxes are somewhat reduced by advection errors due to the complex flow pattern and relatively large angles of attack of up to 22°, a common problem faced when measuring urban fluxes.
The time-lag between the measurement of the wind component and the air concentration, induced by the sampling line, was established through calculation of the cross-correlation spectrum (co-variance as a function of time-lag) and selecting the maximum in a specified possible time-lag range window (3 to 6 s).
2.4.1. Data Filtering
A set of general filtering criteria was established. Flux periods were excluded from the data-series if at least one of the following criteria was met:
| • | The wind direction was in the range 200° to 280°, where large buildings closely upwind were expected to influence the flow. During the Boulder measurements, this criterion removed 9.3% of the data. | ||||
| • | The wind speed was lower than 1 m s−1. This criterion removed 5.2% of the data during the Boulder campaign. | ||||
| • | There was the potential for drift in the Q-AMS m/z calibration during the 29 min averaging period. This was assessed by evaluating the time evolution of the ratio of the ion strength at the expected peak position with the actual peak, saved every 1 min (). Although changes in the m/z calibration did not appear to be a significant problem at this site (2.7% of data removed), this filter criterion could become important if the Q-AMS is operated in environments with strongly fluctuating instrument temperature. | ||||
2.4.2. Quantitative Flux Determination
As described above, in the MS Mode fully quantitative concentrations are usually derived from all m/z fragments. By contrast, fluxes can only be derived from a small number of selected m/z. Hence a mechanism is required to convert the signal on individual m/z into meaningful submicron aerosol mass fluxes. Some fragments may have interferences that are difficult to quantify, and the number of monitored m/z is very restricted. In this case m/z values are selected, for which the contribution to the parent and the lack of major interferences is well characterized from previous studies. For other compounds, information on the mass spectra from the concentration mode is combined with the co-variances between w and the raw ion signals at the different m/z to quantify fluxes.
Laboratory studies have identified the fragmentation patterns of the inorganic components. For example, for NH4NO3, CitationHogrefe et al. (2004) derived for their Q-AMS instrument a fragmentation pattern of 56% (i.e., f = 0.56) at m/z 30 (NO+) and 42% at m/z 46 (NO2 +) (i.e., f = 0.42). Hence, to derive quantitative fluxes of SO4 2− or NO3 − Equation (Equation6) would suffice to derive mass fluxes from each of the major fragments (m/z 30, 46, 48, and 64). For NH4 +, the major peaks at m/z 15 (NH+), 16 (NH2 +), 17 (NH3 +) suffer interferences from methyl ions from fragments from organic compounds (m/z 15), oxygen (O+) ions (m/z 16) from O2 in air, and OH+ ions from water (m/z 17). Since the concentration of O2 in air is constant, the ion intensity at m/z 16 can be corrected for the contribution from O+, which is derived either as the ion intensity detected when the Q-AMS is operated with an inlet filter (to remove any NH4 + from the sample stream) or as the intercept of the graph of m/z 16 versus the sum of NO3 − and SO4 2−.
In the urban environment, HOA is expected to be emitted, while OOA (composed of OOA-I and OOA-II) is likely to be both formed and deposited (CitationVolkamer et al. 2006; CitationZhang et al. 2005a). While for the calculation of organic components (such as HOA or OOA) by MCA or PMF the whole mass spectrum can be taken into account, only few m/z can be monitored in flux mode with the Q-AMS. The analysis of the mass spectrum of the aerosol indicates that m/z 43, 44, and 57 make major contributions to OOA-II, OOA-I, and HOA, respectively (see below). Fluxes of HOA, OOA-I, and OOA-II from the deconvolution of the fluxes measured at these m/z.
| 1. | Firstly, raw fluxes (F j raw) were calculated for each mass j using an eddy-covariance flux routine written in LabView. The fluxes were then normalized by the raw concentration (χ j raw) derived by the same program, to calculate exchange velocities (V ej = F j raw/χ j raw). Raw concentrations and fluxes do not take into account fragmentation patterns, collection efficiencies, long-term changes in multiplier performance, etc. These factors affect flux and concentration in the same way and they are expected to remain constant over a 30 min averaging period. As a result, they cancel in the calculation of V e. | ||||
| 2. | Values of V e were then corrected for density fluctuations due to changes in water vapor mixing ratio (CitationWebb et al. 1980), while temperature fluctuations were estimated to be smoothed out in the inlet. | ||||
| 3. | This approach is sufficient to calculate V e from “pure” mass peaks, where an m/zj can be uniquely attributed to a component i. Contamination from other compounds, including gas phase components, affects concentrations and fluxes in different ways. For example, the V e derived for aerosol mass m/z 44 was corrected for a contribution from gas-phase CO2 fluxes according to: | ||||
| 4. | For those m/z that predominantly represent one single compound, fluxes of the different components (i) of interest were calculated by multiplying the exchange velocity by the concentration, e.g., of NO3 − and SO4 2−, which were calculated from the mass spectra using the method described above. This method is sufficient to calculate fluxes of nitrate and sulfate. | ||||
| 5. | However, further steps are required to calculate fluxes of the organic aerosol classes. The MCA or PMF derives the average mass spectra of HOA, OOA-I, and OOA-II observed during a measurement campaign (see below), which includes the relative contribution (a ji ) of the m/z 43, 44, and 57 (j) to each organic class (i). From the time-series of HOA, OOA-I, and OOA-II derived by the MCA or PMF, associated “m/z concentrations” (ξ j ) can be derived for the three m/z as: | ||||
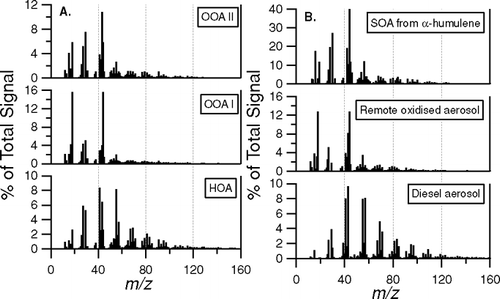
FIG. 10 Comparison of (a) fitted relative mass spectra of HOA, OOA-I and OOA-II with (b) reference spectra representing SOA from humulene + O3 reaction, fully oxidised aerosol from a remote measurement site and diesel exhaust aerosol (FPEAK = −0.2).
lists the m/z monitored during the urban flux measurements in Boulder. Since, with the Q-AMS, fluxes can only be measured for a limited number of m/z, the total organic fluxes has to be derived from a small number of m/z (43, 44, and 57) which, jointly, on average accounted for only 13% of the HOA, 30% of the OOA-I, and 18% of the OOA-II at Boulder.
TABLE 1 Typical m/z chosen for the eddy-covariance JMS mode
2.5. Site Description, Auxiliary Instrumentation for Urban Flux Measurements and Setup of the AMS Eddy Covariance System
For an initial evaluation of the system performance, an urban micrometeorological flux measurement site was established on the roof of the Williams Village Dormitories, overlooking the centre of Boulder, Colorado, USA (105° 15′ 11″ W; 40° 00′ 00″ N; 1625 m asl). For this purpose, a 3 m tall tower was erected on top of a two-storey elevator machine room on the top of Stearns Tower West, a 14-storey, 45 m tall building. This tower supported the ultrasonic anemometer (Model 81000, R.M. Young Company, Traverse City, MI, USA), and a 2.5 μ m cut-off cyclone, which was mounted 0.4 m below the center of the anemometer. A 20 m long copper tube (3/8 inch OD) led into the machine room and was split into three sample streams using a flow splitter (Model 3708, TSI Instruments), leading to an infrared gas analyzer (IRGA) for CO2 and H2O (Model 6262, LiCor Inc.), a condensation particle counter (CPC 3010, TSI Instruments Inc.), a probe for relative humidity (RH) and temperature (T) (Humitter 50Y probe, Vaisala) and the Q-AMS. At an overall flowrate in the inlet line of 15 lpm, the flow was turbulent, which minimizes flux losses, but increases wall losses of particles < 50 nm. The anemometer was not only recorded by the PC of the Q-AMS, but also by a second PC which also recorded T, RH, CO2, and H2O.
Measurements were made over a two-week period (6–20 June 2003) when the dormitories were unoccupied. This measurement period was characterized by some afternoon thunderstorms and highly variable wind directions. Being situated on the boundary between the Foothills of the Rocky Mountains to the west and the Great Plains to the east, the general flow pattern in Boulder is characterized by W down-sloping winds (clean) during the day and E up-sloping winds (more polluted and affected by the Denver conurbation) during the night. The period also included NE winds, which brought to the site more odorous air masses from the agricultural area around Greeley, CO. The flux footprint of the tower comprises residential areas with significant tree cover to the NE and SW, city center region to the NW, a car park and sports facilities to the SE and individual large commuter roads to the S and W (). While the Stearns Tower enjoys uninterrupted fetches to the N, NW, E, and S, other dormitory towers disturb the footprint to the W and SW and these wind directions were therefore excluded from the flux analysis.
FIG. 6 Aerial photograph of the area around the Williams Village measurement site. The white circle at the center of the figure marks the position of the measurement tower.

3. RESULTS
3.1. Aerosol Concentrations
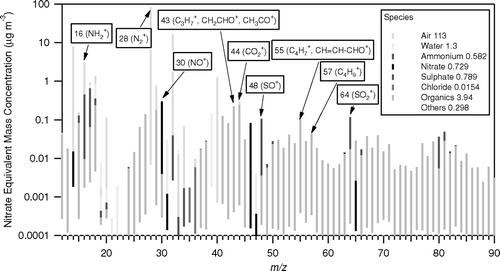
shows the averaged MS for the measurement period. Peaks with identifiable contributions from several parent compounds were de-convolved following the procedure of CitationAllan et al. (2004). Some of the largest organic peaks can be seen at m/z 41, 43, 44, 55, and 57. While m/z 18, 27, and 29 are also major organic peaks, they have major contributions from inorganic and gaseous compounds and it is therefore more difficult to derive organic mass fluxes from these peaks.
FIG. 7 Overall MS spectrum of the measurement period, indicating the m/z monitored for fluxes. The numbers in the legend reflect the average concentrations in μg m−3. Please note the logarithmic scale.
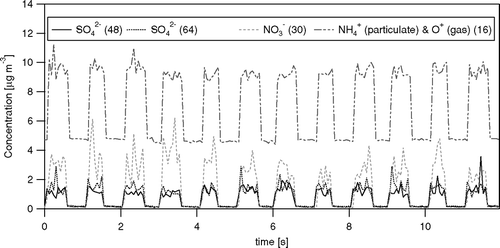
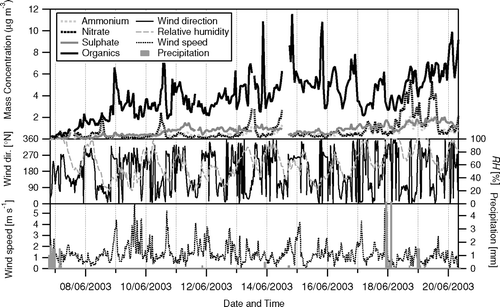
Time-series of the concentrations of the major ions and total organics as measured by the Q-AMS are shown in in relation to meteorological variables. The organics and SO4 2−increased steadily over the measurement period, following the precipitation at the beginning of (and prior to) the measurements, which was more regional than storm related rain events later during the measurement period. NO3 − shows distinct peaks, the largest of which occur at midday and are related to NE wind directions advecting NH3-enriched agricultural air masses from livestock farms near Greeley, 65 km to the NE. Smaller enhancements in NO3 − can be identified during certain night-time periods, likely due to enhanced partitioning with lower temperature and higher RH (CitationNemitz et al. 2004; CitationZhang et al. 2005b). Some sharp rises in the organics appear to be related to rises in wind speed, independent of the time-of-day, suggesting that gusts may ventilate street canyons below the measurement point. However, these gusts are also associated with changes in the wind direction, and thus a change in the air masses. On average, there was a close charge balance between NH4 + cations and the sum of the anions NO3 − and SO4 2−, indicating that the SO4 2− was fully neutralized (not shown).
FIG. 8 Time-series of the 1 h average concentrations of non-refractory ammonium, nitrate, sulfate and total organics in NR-PM1 aerosol measured during the campaign in relation to meteorological variables (RH and precipitation are taken from the Foothills weather station, http://www.atd.ucar.edu/weather/weather_fl/station.html, operated by NCAR/ATD).
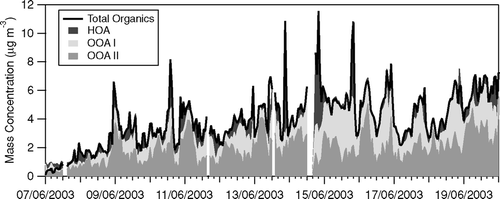
Two different algorithms were applied to decompose the total organic aerosol loading into the dominating aerosol classes, including the MCA of CitationZhang et al. (2007) and PMF (CitationLanz et al. 2007; CitationUlbrich et al. 2008). Both approaches produced similar results and identified three main components, representing HOA and two distinct classes of OOA. The PMF result showed some sensitivity to the value of FPEAK chosen for the fit (CitationUlbrich et al. 2008), a parameter that determines the rotational state in the solution of PMF. This value could be optimized if the solution could be further constrained, for example, with a time series of carbon monoxide or elemental carbon, which may be expected to co-vary with HOA. In the absence of such independent data, we here explore the sensitivity of the fluxes to this solution, by contrasting our main solution (FPEAK = –0.2) to alternative solutions (FPEAK = –0.1 and –0.3) (P. Paatero, private communication). Solutions beyond this range of FPEAK result in unrealistic mass spectra, such as an OOA-II mass spectrum that completely lacks m/z 44, which is unrealistic based on both source studies and results at other locations (CitationAlfarra et al. 2006; CitationBahreini et al. 2005; CitationUlbrich et al. 2008). The time-series of the breakdown of OM according to PMF is shown in . Hourly average concentration ranged from 0.00 to 11.60 μ g m−3 for HOA, from 0.00 to 6.15 μ g m−3 for OOA-I and from 0.00 to 9.11 μ g m−3 for OOA-II. The increase of total organic aerosol over the measurement period was due to an increase in regional OOA-I and II, while HOA concentrations remained relatively constant. HOA displays a “spikier” time series as typical of a more local source, while the OOAs have slower variations indicating a more regional character.
FIG. 9 Breakdown of hourly total organic aerosol loading in HOA, OOA-I and OOA-II using a three-component solution from PMF (FPEAK = −0.2).
compares the PMF results obtained with different FPEAK values. According to the solution for FPEAK = −0.2, HOA, OOA-I, and OOA-II contributed on average 18, 33, and 49% to the organic mass loading, respectively, with a slightly elevated HOA/OOA ratio during daytime (10:00–18:00 hrs), when local traffic activity was largest. It should be noted that the measurements were taken well above the city, while higher HOA/OOA ratios would be expected at street level.
TABLE 2 Summary of the average split of the monitored m/z representing organic aerosol into hydrocarbon like organic aerosol (HOA) and two classes of oxygenated organic aerosol (OOA) as calculated with PMF using different FPEAK values. Also shown is the resulting average concentration of the three different organic aerosol classes and their relative contribution to the total organic aerosol mass (OM), as well as the averaging fluxes derived from the different PMF solutions
The relative mass spectra for the HOA and OOA-I components identified by the analysis routine are similar to mass spectra of fresh diesel exhaust aerosol (CitationCanagaratna et al. 2004) and highly aged rural aerosol (CitationAlfarra et al. 2004), which are generally regarded as proxies for a freshly generated combustion aerosol and a highly oxidized aerosol/regional SOA, respectively (). OOA-II is a less oxidized component which is often associated with fresh/less aged SOA (CitationBahreini et al. 2005; CitationLanz et al. 2007; CitationUlbrich et al. 2008), and its mass spectrum resembles several SOAs produced in smog chambers such as, e.g., that of α -humulene + O3 oxidation products (CitationBahreini et al. 2005; Ulbrich, I. M., and Jimenez, J. L.: AMS Spectral Database. URL:http://cires.colorado.edu/jimenez-group/AMSsd/). From this analysis it becomes evident that m/z's 55 and 57 are dominantly associated with HOA, m/z 44 with OOA-I, and m/z 43 with OOA-II, although the exact contribution is somewhat sensitive to the FPEAK value chosen ().
Average size-distributions of NH4 +, NO3 −, SO4 2−, and organics, derived in the PToF part of the GENALT mode every second half hour, are shown in . The mass median diameters (d va ) are 0.38 μ m for NH4 +, 0.44 μ m for NO3 −, 0.37 μ m for SO4 2−, 0.12 μ m for HOA, 0.40 μ m for OOA-I, and 0.32 μ m for OOA-II. Here the size-distributions for HOA, OOA-I, and OOA-II were derived from the size distributions measured at m/z 57, 43, and 44, by solving the linear system that relates the three components to the three m/z's, according to the contributions from the averaged mass spectra, similar to the method described above for the flux calculation of HOA, OOA-I, and OOA-II. This procedure attributed all of the accumulation mode contribution of m/z 57 to OOA, and indicates that HOA is almost entirely found in the size range 40 to 400 nm, centered around 120 nm. By contrast, OOA-I is dominated by an accumulation mode centered around 300 nm, but shows still some contribution of the smaller mode, while OOA-II shows a broader size distribution, with the main mode centered around 450 nm. Consistent with other urban AMS measurements, the relative importance of the smaller sizes is smallest for SO4 2− and largest for HOA (CitationZhang et al. 2005b; CitationZhang et al. 2005c). However, during this campaign there is clear evidence of an Aitken mode contribution to NO3 − and OOA, which is not always observed. OOA-II shows a larger presence in the smaller sizes when compared with OOA-I, which is also consistent with the fresher character of the first and the more aged character of the second.
3.2. Performance for Flux Measurements
The m/z selected for the flux measurements above Boulder allow two separate estimates of the SO4 2− flux to be inferred from m/z 48 and m/z 64. Since the QMS monitors only one single m/z for each particle, the two flux estimates are derived from independent particle populations, providing a powerful test for the error introduced by sensor noise and statistical constraints. shows scatter plots for the concentration, fluxes and deposition velocities (V d = −F χ/χ). Many previous AMS measurements have shown a close relationship between the SO4 2− concentrations derived from m/z 48 and 64 (e.g., CitationJimenez et al. 2003). There were clear correlations between the fluxes (R2 = 0.78; P < 0.05; N = 258) and deposition velocities (R2 = 0.63; P < 0.05; N = 258) derived from the two m/z. Given the small magnitude of the bi-directional fluxes of typically −10 to 10 ng m−2 s−1 (cf. also and ), these results are very encouraging.
FIG. 12 Comparison of (a) concentrations, (b) fluxes and (c) deposition velocities of SO4 2− derived from two different fragments, i.e., at m/z 48 (SO+) and m/z 64 (SO2 +). For fluxes and Vd, only data with SO4 2− concentrations > 1 μg m−3 are included. Linear regression lines are shown together with 95% confidence intervals.
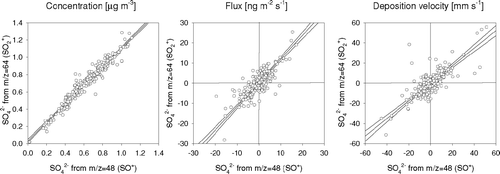
FIG. 14 Example time series of meteorological parameters, concentrations and fluxes observed over a 4 day period. Fluxes of aerosol components (FAMS) are presented as 3 h running means.
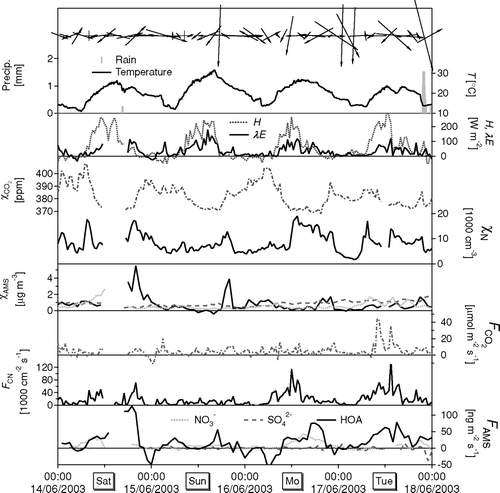
FIG. 16 Averaged diurnal cycles of chemically resolved particle concentrations and fluxes (FPEAK = −0.02).
Power-spectral analysis is often used as a quality control procedure for eddy covariance flux measurements of trace gases. Power spectral analysis has also been successfully applied to particle fluxes measured with CPCs, which detect large numbers of particles (CitationBuzorius et al. 1998; CitationDorsey et al. 2002; CitationMartensson et al. 2002; CitationNemitz et al. 2002a). By contrast, statistical limitations has so far compromised power spectral analysis of fluxes of particles in the Aitken and accumulation mode, measured with optical particle spectrometers (e.g., CitationGallagher et al. 1997; CitationNemitz et al. 2002a). shows normalized co-spectral density functions of the co-variance of w with T, u, nitrate, and HOA. The spectra based on the Q-AMS measurements show good agreement with those of momentum and sensible heat as well as with the f −4/3 response expected for the inertial subrange. There is some indication of slight attenuation for frequencies > 0.3 Hz. At the large measurement height during this study, the associated loss is small (< 5%), given that the contribution of these frequencies is already several decades smaller than the maximum. It is likely that most of the damping occurs in the cut-off cyclone and the short laminar flow region directly in front of the Q-AMS inlet, and could be further reduced when measuring over short vegetation. The relative large contribution of low frequencies suggests that a longer averaging time than the 29 minutes used here would have derived somewhat larger fluxes. Indeed, analysis suggests that up to 10% of the flux may have been lost at the low-frequency end. There is a trade-off, however, between longer averaging time and the need for stationarity over the averaging period.
FIG. 13 Normalised co-spectral density functions of the fluxes of sensible heat, momentum, nitrate (from m/z 46) and HOA (from m/z 57) for an example period (17 June 2003; 16:40), compared with the slope of f −4/3 expected in the inertial subrange. Empty symbols indicate negative contributions which were negated for plotting on the logarithmic scale.
To quantify the limit of detection (LOD) of the Q-AMS flux system, the flux system was operated with an inlet filter, preventing particles from entering the system. For these periods raw fluxes were calculated as usual. However, since concentrations average, by definition, to zero a mass flux could not be calculated. Instead, a LOD was defined as 3× the RMS of these fluxes rescaled for the average contribution of the individual m/z to the associated compound of interest. The LODs thus derived range from 0.24 ng m−2 s−1 for NO3 − to 15.3 ng m−2 s−1 for HOA (). Larger LODs for the organic components is due to the larger instrumental background in the AMS for these species.
TABLE 3 Detection limits of the flux measurements (based on 30-minute averaging), compared with average fluxes measured during the campaign
Errors in particle fluxes are primarily related to the relatively small number of particles detected per averaging time (compared with the much larger number of molecules detected during measurements of gas fluxes). In addition, large particles can carry a significant amount of material, e.g., a study in the Boston area found that 2% of the submicron particles carried 50% of the mass (CitationJimenez et al. 2003). This effect results in spikes in the data series (cf. and ), which cannot be removed by despiking routines but need to be taken into account in the flux calculation. For particle number fluxes a statistical error can be calculated based on the number of particles that were detected and entered the flux calculation (CitationFairall 1984), but this information is not available for the AMS flux measurements. Instead, for SO4 2−, which could be derived from two different m/z, the error in the measurement due to statistical variation in the aerosol populations was estimated as the standard deviation of the differences between the two estimates as 6.2 ng m−2 s−1. This error estimate does not, however, reflect the effect of the aerosol loading, nor does it account for errors in the wind measurement.
3.3. Magnitude of Urban Fluxes
In general, the measured fluxes were highly variable, reflecting the variability in wind direction and wind speed during the measurement period, combined with the heterogeneity of urban matrices in the footprint at different wind directions. Fluxes during a 4-day period are shown in , together with concentrations and meteorological conditions. This example time series demonstrates that fluxes of HOA and (to a lesser extent) NO3 − temporarily follow the total particle number flux, likely due to the influence of primary traffic emissions on both. Reduced emissions of all particle compounds were observed on the Sunday.
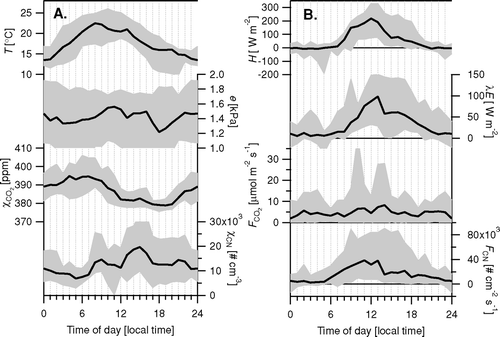
Averaged diurnal cycles of the concentrations and fluxes of CO2 and particle number are shown in , together with the cycle in the fluxes of sensible heat (H) and latent heat (λ E). Temperature peaked before midday, probably due to the effect of the afternoon thunderstorms and the down-sloping winds from the mountains in the afternoon. The cycle in the CO2 concentration is the combined effect of the cycle in the emissions and the boundary layer height and is consistent with earlier measurements in Edinburgh, Scotland (CitationNemitz et al. 2002b). The heat fluxes show the expected diurnal patterns. The sum of λ E + H is only just negative at night, indicating that the urban matrix acts as only a small sink of heat during night. It is likely that in June air conditioning units contributed to the heat output from the city, partly off-setting the natural negative radiation balance. It should be noted, however, that radiative heating of the roof of the building supporting the measure mast may have modified the local sensible heat flux which thus may not have been fully representative for the overall footprint. Fluxes of CO2 were highly variable, with only a weak diurnal cycle and an average emission of 4.6 μ mol m−2 s−1. Fluxes of CN (number flux of particles > 11 nm) showed a more distinct diurnal cycle, with a larger relative range and emissions close to zero during night. A likely cause for this difference is the contribution of vegetation and soils to the net exchange. At night, vegetation and soils provide a source of CO2 (respiration), while, during the day, CO2 is assimilated by the plants. Therefore the biogenic flux would be expected to be anti-correlated with the anthropogenic CO2 emission and dampen the diurnal cycle in the anthropogenic emission. The impact is expected to differ between wind directions from busy roads and residential areas, causing some of the variability. Vegetation was found to significantly affect CO2 exchange over U.S. residential areas in other summer measurements and can result in net deposition fluxes during the day (CitationOfferle et al. 2001). Unfortunately, the present dataset is too limited for a detailed analysis of the dependence of fluxes on time of day and wind direction.
FIG. 15 (a) Averaged diurnal cycles of temperature (T), water pressure (e) and concentrations of CO2 (χCO2) and small particles (χCN). (b) Associated fluxes of sensible heat (H), latent heat (λE), CO2 (F CO2) and small particles (FCN). Shown are medians (bold lines), together with range between 5th and 95th percentiles.
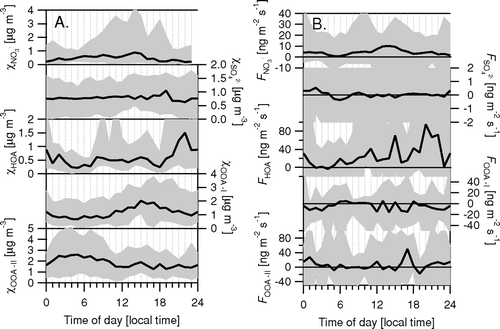
shows the averaged results of the speciated aerosol flux measurements. As with the example 4-day periods of , there was a clear diurnal cycle in the NO3 − concentration and flux with significant emissions during the day. The magnitude of this emission was variable with average daytime peaks of 5 and maximum emissions of 20 ng m−2 s−1. Sulfate fluxes were small and not significantly different from 0, indicating that the city of Boulder is not a significant source of SO4 2− aerosol.
Fluxes of HOA, OOA-I, and OOA-II averaged 31.8, –7.3, and 4.8 ng m−2 s−1, respectively, with median fluxes of 18.2, –3.5, and 3.0 ng m−2 s−1. These averages change somewhat with the choice of FPEAK in PMF introducing an uncertainty of typically +/−20% (). The flux of HOA showed a very clear diurnal pattern of emission, presumably related to traffic activities ( and ). OOA-II is largest at night, possibly due to semivolatility of this component, as observed in previous studies in Zurich and Pittsburgh (CitationLanz et al. 2007; CitationUlbrich et al. 2008), or to correlation of wind direction with time of the day.
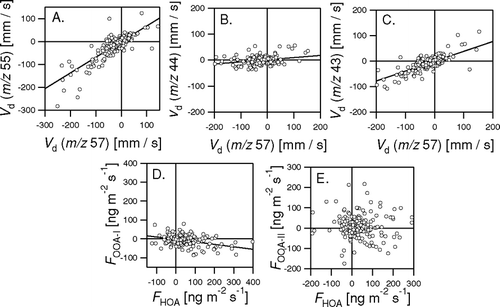
shows the correlations between the raw V d derived for the different m/z representing organic aerosol and between the derived fluxes after de-convolution. As expected, deposition velocities derived from the two m/z that predominantly represent HOA (i.e., 55 and 57) were highly correlated (), with V d(55) some 40% lower, partially possibly due to the higher contribution of OOA at this m/z. Even V d of the m/z that dominantly represent OOA (i.e., 43 and 44) were still significantly correlated with the V d at 57 (, ). After deconvolution, the flux of OOA-I was significantly anticorrelated with the flux of HOA (), while no significant correlation was found between the fluxes of HOA and OOA-II. This lack of correlation may be an effect of the deconvolution routine or it may reflect confounding effects such as wind direction/source areas, transport time or radiation which would influence the fraction of SOA precursors oxidized below the measurement height. Nevertheless, the fact that the average OOA-II flux reflects 15% of the HOA emission suggests that significant amounts of OOA-II are either locally emitted or formed through chemical processing in the urban air space (CitationVolkamer et al. 2006).
FIG. 17 Correlation between the fluxes and deposition velocities derived from different m/z representing different components of the organic aerosol. Regression results: V d(55) = 0.57 V d(57)−4.25 mm s−1 (R2 = 0.85, N = 270); V d(44) = 0.083 V d(57) + 1.31 mm s−1 (R2 = 0.44, N = 270); V d(43) = 0.39 V d(57) − 0.55 mm s−1 (R2 = 0.85, N = 270); F OOA-I = −0.13 F HOA− 3.16 ng m−2 s−1 (R2 = 0.39, N = 270).
4. DISCUSSION
4.1. Particle Emissions and Dynamics in the Urban Environment
4.1.1. Fluxes of CO 2 and CN
Very few studies so far have attempted to measure urban fluxes of pollutants with micrometeorological techniques. Most CO2 flux measurements over city centers have shown emissions throughout the 24 hr diurnal cycle, that follow the pattern of traffic densities (CitationGrimmond et al. 2002; CitationNemitz et al. 2002b; CitationSoegaard and Jansen 2004; CitationVelasco et al. 2005; CitationVogt et al. 2003). By contrast, CO2 flux measurements over U.S. suburban residential areas have derived small fluxes and substantial periods of deposition, dominated by the assimilation of plants in the footprint (e.g., CitationGrimmond et al. 2002; CitationOfferle et al. 2001). During the measurements presented here, the average CO2 flux was small, but positive throughout the diurnal cycle, ranging from 3μ mol m−2 s−1 during night to 7 μ mol m−2 s−1 during the day. This indicates that daytime assimilation by vegetation in the footprint was exceeded by emissions from fossil fuel combustion. By comparison a range of 10 to 40 μ mol m−2 s−1 was observed over Edinburgh city center, U.K., (CitationNemitz et al. 2002b), while monthly averages in the range of 2 to 8 μ mol m−2 s−1 were reported for Copenhagen, DK (CitationSoegaard and Jansen 2004).
Particle number fluxes (> 10 nm) ranged between 5,000 cm−2 s−1 in the early morning hours and 40,000 cm−2 s−1 during the day. By comparison, ranges of 10,000–110,000 cm−2 s−1 have been reported for Stockholm, Sweden (CitationMartensson et al. 2002) and 5,000 to 90,000 cm−2 s−1 for Edinburgh (CitationDorsey et al. 2002).
4.1.2. Concentrations, Size Distributions and Fluxes of Fine Aerosol
Flux measurements of speciated aerosol components above an urban environment have so far only been reported for a scoping study using an REA system (CitationNemitz et al. 2000b). By contrast, the database of concentration measurements by AMS is continuously growing (CitationZhang et al. 2007). Aerosol concentrations in Boulder were moderate during the measurement period, and the fine aerosol fraction was clearly dominated by secondary compounds in the accumulation mode, normally associated with regional accumulation and long-range transport (OOA-I, SO4 2−, NO3 −, and NH4 +), with a smaller contribution from HOA, which is usually attributed to primary emissions (CitationZhang et al. 2005a; CitationZhang et al. 2005c). HOA has the widest size distribution with a significant contribution from the finer mode, which in mass terms is centered around 120 nm (d va ). This is consistent with the large CN number fluxes measured above the city, which are likely to be dominated by small, traffic related particles containing HOA and soot (CitationCanagaratna et al. 2004). From the correlation between CN fluxes and CO2 fluxes (R2 = 0.29; P < 0.01) an average particle number emission factor (d p > 11 nm) of 4.4 × 1014 particles (kg CO2)−1 is derived.
Local secondary aerosol formation by condensation takes place in those sizes that provide most of the surface area. Hence, the size distribution of secondary aerosol components contains some indication whether they formed in the urban environment, where the Aitken mode makes an important contribution to the surface area distribution. By contrast, formation in the rural environment (dominated by the accumulation mode) as well as aging processes, such as coagulation, further condensational uptake of other compounds and size-dependent deposition processes favor secondary compounds to be found in the accumulation mode. Following this reasoning, the Aitken mode contributions in the size distributions () already suggest that some NO3 − and some of the OOA-II are formed in the urban environment, while SO4 2− (and to a lesser extent OOA-I) is purely found in the accumulation mode and is thus produced over more regional scales.
This is directly confirmed by the eddy-covariance flux measurements, which show regular patterns of HOA at an average emission of 31.8 ng m−2 s−1. Although the correlation with the CO2 flux fails to be significant at P = 0.05, the comparison of average fluxes of HOA and CO2 would imply an emission factor of 0.15 g HOA (kg CO2)−1. Using an average stoichiometric H/C ratio for gasoline of 1.85, this emission factor equates to 0.5 g HOA (kg fuel)−1. For comparison, CitationKirchstetter et al. (1999) report emission factors of 0.5 and 0.053 g OC (kg fuel)−1 for heavy duty diesels and light-duty gasoline vehicles, respectively, while CitationCanagaratna et al. (2004) report emissions factor of organic species in the range 0.1 to 0.9 g OC (kg fuel)−1 for diesel vehicles. Traffic in and around the site represents a mixed fleet expected to be dominated by gasoline vehicles, and thus these first flux-derived HOA emission factors may be an upper limit of the range of previous studies. This analysis averages over all sources contributing to the emissions of the two compounds and neglects the effect of terrestrial sources and sinks on the net CO2 exchange. Since, on average, vegetation surrounding the measurement site is likely to represent a net sink of CO2, its emission factor is likely to be a lower estimate.
There is a possible explanation why flux measurements may derive larger HOA emission factors than has been derived from concentration measurements. Here surface emissions are derived from direct flux measurements some 45 m above street level. Depending on turbulent conditions, the time-scale for turbulent transport to the measurement height is of the order of 0.3 to 2.6 min (CitationBrost et al. 1988). By contrast, in many previous studies emission factors were derived from the concentrations that had accumulated across the city, often over hours. Hence, it is likely that these concentration-derived HOA emission factors are more strongly affected by HOA sinks such as deposition, evaporation and oxidation to OOA, and thus underestimate the HOA at the point of emission. With increasing evidence that primary aerosol can evaporate rapidly as the emitted aerosol/gas mixes are diluted (CitationRobinson et al. 2007), it is equally possible that the flux measurements at 45 m underestimate primary emissions. The emission factors derived here need to be confirmed with longer measurement time series.
At 29.9 mm s−1 the average V e of HOA was somewhat larger than the average V e of CN, which, at 16.6 mm s−1, was within the range of previous measurements (CitationDorsey et al. 2002). While the number flux is dominated by particles < 50 nm, the HOA mass flux is dominated by larger particles and lies in the range of size-segregated particle number fluxes above a city (CitationNemitz et al. 2007).
Fluxes of the less oxidized OOA class (OOA-II) are on average upwards, but not significantly correlated with the HOA fluxes. This OOA-II is either emitted or, more likely, chemically formed or partitioned due to semivolatility below the measurement height, which is consistent with the analysis of the size distribution above. An attempt was made to correlate the flux ratio of OOA-II/HOA with the turbulent time scale or temperature, but no significant correlation could be identified and more data are needed to derive chemical time scales for the OOA formation. However, the observation of OOA-II emission fluxes at about 15% of the HOA flux confirms that the urban environment is a net source of primary or secondary weakly oxidized aerosol. By contrast, average fluxes of the more oxidized OOA class (OOA-I) are on average downward.
Fluxes of NO3 − showed a clear diurnal cycle which was more pronounced than that of HOA, but on average of similar magnitude, implying relative emission factors of 0.027 g NO3 − (kg CO2)−1 or 0.09 g NO3 − (kg fuel)−1. Emissions of NO x were not measured during this campaign. However, assuming an average emission ratio of 2.6 g NO x (kg CO2)−1 for a fleet dominated by gasoline vehicles (CitationJimenez et al. 1999; CitationJimenez et al. 2000; US-EPA 1995), the fluxes imply that an average 0.50% of the NO x emitted are fully oxidized to NO3 − below the measurement height. Assuming that the OH + NO2 reaction is the limiting step in the oxidation of NO to aerosol NO3 −, a theoretical NO3 − formation rate may be estimated from the tabulated reaction rate constant of 1.0 (+/−0.1) × 10−11 cm3 molecule−1 s−1 (CitationOkumura et al. 2005). Using an estimated OH concentration of 2 × 106 molecule cm−3, the reaction rate with respect to NO2 is of the order of 0.12% min−1 for the atmospheric conditions during the Boulder campaign. Using the transport time-scale estimated above, this relates to a conversion of 0.03 to 0.30% below the measurement height, which is somewhat smaller than the flux measurements. Again, the flux-derived estimate of the conversion factor may be somewhat overestimated here, as it does not account for terrestrial sources and sinks of CO2. The magnitude of the NO3 − emissions differed greatly between days, more than the emission of CN and CO2. This is consistent with (a) the observation of Aitken mode NO3 − during some days but not during others, often made in cities, and (b) more recent AMS flux measurements above Gothenburg, Sweden, which will be published in more detail elsewhere (CitationThomas et al. 2007b). This day-to-day variability cannot be explained by variability in the equilibrium constant of NH4NO3 in response to changes to temperature and relative humidity alone. However, without supporting measurements of HNO3, NH3, and other N compounds such as N2O5, it is unclear whether the sole controlling mechanism is the gas/aerosol equilibrium of NH4NO3, or whether heterogeneous nitrate formation or the production of organic nitrates may play a role. More measurements are needed to investigate which conditions favor NO3 − formation in the urban environment and this scientific question will certainly form one of the foci for future urban flux work with the Q-AMS.
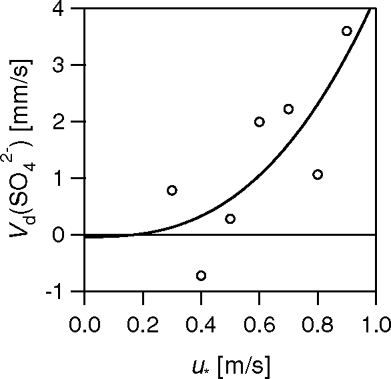
There is almost no information on deposition velocities to urban surfaces. Particle number fluxes derive the net aerosol flux which, in the urban environment, is dominated by emissions. Chemically resolved fluxes offer the opportunity to measure deposition rates of particles, if a chemical compound can be found for which there are no sources in the urban environment. Sulfate fluxes above Boulder were very small, indicating that SO4 2− may be a suitable compound. shows the dependence of the average V d(SO4 2−) on friction velocity (u *), which can be described by the equation:
FIG. 18 Values of the deposition velocities of SO4 2−, classified into bands of u* of width 0.1 m s−1. Each point constitutes an average of 6 to 64 data points.
4.2. System Performance
The flux measurement of individual aerosol chemical components has long been a challenge and so far been mainly confined to gradient measurements. However, even slow continuous analyzers for aerosols struggle to provide the precision to resolve vertical gradients of a few percent and often suffer artefacts due to gas-particle partitioning. In addition, they would not have been applicable to measure fluxes over such heterogeneous terrain such as cities. We here present one of the first operational systems for eddy-covariance flux measurements of aerosol components using a Q-AMS, with emphasis on SO4 2−, NO3 −, HOA, and OOA.
Fluxes above Boulder were highly variable. This was also reflected in the fluxes of CO2 and CN, supporting that this variability was real and probably reflects the nature of the measurement site and the differences in activity mixes in the different upwind directions. However, the high quality of the flux measurements achieved with the AMS is evident in the encouraging agreement between the fluxes derived from different m/z (for SO4 2− and HOA). Since these fluxes are derived from different particle populations, the good agreement implies that the quality of the fluxes is not limited by counting statistics. Our estimates of the LODs, in the region of 1 to 15 ng m−2, depending on compound compare favorably with automated, continuous flux gradient approaches (CitationThomas et al. 2007a). One of the objectives for developing the AMS flux system is to measure dry deposition inputs to vegetation and to quantify deposition velocities (V d). The LODs for fluxes can be used to estimate LODs for V d (for a given aerosol concentration and a size-distribution similar to that observed here). For example, at SO4 2− and NO3 − concentrations of 5 μ g m−3, the AMS ought to be able to resolve a V d of as low as 0.6 mm s−1 and 0.3 mm s−1, respectively.
There are some limitations to the approach. First, the AMS flux system can only measure fluxes of the components detected by the AMS, i.e., non-refractory aerosol in the size-range 35 to 1500 nm. In most environments, this includes virtually all of the SO4 2− and organic material, but only the fine fraction of NO3 − that represents mostly NH4NO3. By contrast, the AMS cannot measure coarse NO3 − which is usually thought to represent NaNO3 and which can make an important contribution to NO3 − in coastal regions. While this restriction makes it easy to study processes controlling NH4NO3, it means that the full NO3 − flux cannot currently be determined. Similarly, while the Q-AMS can measure concentrations and size-distributions of NH4 +, these measurements are noisier, due to interference from other compounds such as CH3 + (on NH+ at m/z 15), O+ (on NH2 + at m/z 16) and OH+ (on NH3 + at m/z 17). Hence fluxes of NH4 + have not yet successfully been quantified. While the AMS captures most of the accumulation mode some clipping at the larger size may occur (as it would with any PM1 measurement) and this also appears to be the case in Boulder (). With the Q-AMS, organic aerosol fluxes have to be derived for a limited number of m/z, and the deconvolution routines used here make the assumption that this contribution is the same for the fluxes as it is for the concentrations. The PMF used to split the organic mass spectra into HOA, OOA-I, and OOA-II shows some sensitivity to the FPEAK rotational parameter, which is estimated to introduce an uncertainty of +/–20% into the flux measurements of the organic classes. Furthermore, while the Q-AMS is field deployable in the sense that it can be removed from the laboratory, it nevertheless needs a water proof shelter with reasonable temperature control, necessitating the use of relative long inlets. In order to minimize flux losses, the flow needs to be turbulent, enhancing diffusive losses to the walls. This mainly affects particles < 50 nm, but also some of the smaller particles detected by the AMS.
Urban areas present a challenge for the application of micrometeorological flux measurement approaches, which nevertheless have produced very reasonable results in an increasing number of studies if they are made above the blending height, where non-stationarities have been shown to be a minor problem (CitationNemitz et al. 2002b). However, the errors due to non-stationarity, advection, and storage below the measurement height could not quantified during this study explicitly, and thus there may be some differences between the measured flux and the surface flux. However, average fluxes should be robust as most errors tend to zero if integrated over the day. In addition, fluxes of different components originating from the same sources should be affected in similar ways so that emission ratios, e.g., of HOA/CO2, should also be robust.
As a future development, it is planned (a) to incorporate into the JMS mode a peak height analysis routine, which will be able to classify particles that contribute to the JMS mode signal according to the amount of the monitored compound contained in these particles and (b) to link these measurements to the signal of a light-scatter probe to obtain fluxes as a function of particle size in addition to composition. In addition, the AMS has recently been coupled to a time-of-flight mass spectrometer (ToF-AMS; CitationDrewnick et al. 2005), which may, in the future, provide the potential to measure fluxes at all m/z simultaneously, and to a high-resolution ToFMS (HR-ToF-AMS; DeCarlo et al. 2006) which may overcome the interferences for the NH4 + fluxes, allow more direct quantification of the organic fluxes, and provide additional information on the nature of the organic species.
Acknowledgments
Funding is gratefully acknowledged through grant NER/T/S/2000/ 01084 of the Core Strategic Measurements for Atmospheric Science (COSMAS) research program and standard grant NE/B504865/1 (CityFlux) of the UK Natural Environment Research Council (NERC), through NASA grant NNG04GA67G, US EPA grant RD-83216101-0, and US NSF CAREER grant ATM-0449815, and through a NASA fellowship to J.A. Huffman (NGT5-30516). Neither funding agency has reviewed this report and thus no endorsement should be inferred. NCAR is sponsored by the US National Science Foundation (NSF).
Related Research Data
REFERENCES
- Aiken , A. C. , DeCarlo , P. F. and Jimenez , J. L. 2007 . Elemental Analysis of Organic Species with Electron Ionization High-Resolution Mass Spectrometry . Anal. Chem. , 79 : 8350 – 8358 . doi:10.1021/ac071150w
- Aiken , A. C. , DeCarlo , P. F. , Kroll , J. H. , Worsnop , D. R. , Huffman , J. A. , Docherty , K. , Ulbrich , I. M. , Mohr , C. , Kimmel , J. R. , Sueper , D. , Zhang , Q. , Sun , Y. , Trimborn , A. , Northway , M. , Ziemann , P. J. , Canagaratna , M. R. , Onasch , T. B. , Alfarra , R. , Prevot , A. S. H. , Dommen , J. , Duplissy , J. , Metzger , A. , Baltensperger , U. and Jimenez , J. L. 2008 . O/C and OM/OC Ratios of Primary, Secondary, and Ambient Organic Aerosols with High Resolution Time-of-Flight Aerosol Mass Spectrometry . Environ. Sci. Technol. , 42 : 4478 – 4485 . doi: 10.1021/es703009q
- Alfarra , M. R. , Coe , H. , Allan , J. D. , Bower , K. N. , Boudries , H. , Canagaratna , M. R. , Jimenez , J. L. , Jayne , J. T. , Garforth , A. , Li , S. M. and Worsnop , D. R. 2004 . Characterization of Urban and Regional Organic Aerosols in the Lower Fraser Valley using Two Aerodyne Aerosol Mass Spectrometers . Atmos. Environ. Part a—General Topics , 38 : 5745 – 5758 .
- Alfarra , M. R. , Paulsen , D. , Gysel , M. , Garforth , A. A. , Dommen , J. , Prevot , A. S. H. , Worsnop , D. R. , Baltensperger , U. and Coe , H. 2006 . A Mass Spectrometric Study of Secondary Organic Aerosols Formed from the Photooxidation of Anthropogenic and Biogenic Precursors in a Reaction Chamber . Atmos. Chem. Phys. , 6 : 5279 – 5293 .
- Allan , J. D. A. E., D. , Coe , H. , Bower , K. N. , Alfarra , M. R. , Jimenez , J. L. , Middlebrook , A. M. , Drewnick , F. , Onasch , T. B. , Canagaratna , M. R. , Jayne , J. T. and Worsnop , D. R. 2004 . Technical Note: A Generalised Method for the Extraction of Chemically Resolved Mass Spectra from Aerodyne Aerosol Mass Spectrometer Data . J. Aerosol Sci. , 35 : 909 – 922 .
- Aubinet , M. , Grelle , A. , Ibrom , A. , Rannik , U. , Moncrieff , J. , Foken , T. , Kowalski , A. S. , Martin , P. H. , Berbigier , P. , Bernhofer , C. , Clement , R. , Elbers , J. , Granier , A. , Grunwald , T. , Morgenstern , K. , Pilegaard , K. , Rebmann , C. , Snijders , W. , Valentini , R. and Vesala , T. 2000 . Estimates of the Annual Net Carbon and Water Exchange of Forests: The EUROFLUX Methodology . Advances in Ecological Research , 30 : 113 – 175 .
- Bahreini , R. , Jimenez , J. L. , Wang , J. , Flagan , R. C. , Seinfeld , J. H. , Jayne , J. T. and Worsnop , D. R. 2003 . Aircraft-Based Aerosol Size and Composition Measurements During ACE–Asia Using an Aerodyne Aerosol Mass Spectrometer . J. Geophys. Res.—Atmospheres , 108 art. no.–8645
- Bahreini , R. , Keywood , M. D. , Ng , N. L. , Varutbangkul , V. , Gao , S. , Flagan , R. C. , Seinfeld , J. H. , Worsnop , D. R. and Jimenez , J. L. 2005 . Measurements of Secondary Organic Aerosol (SOA) from Oxidation of Cycloalkenes, Terpenes, and M-xylene Using an Aerodyne Aerosol Mass Spectromter . Environ. Sci. Technol. , 39 : 5674 – 5688 .
- Brost , R. A. , Delany , A. C. and Huebert , B. J. 1988 . Numerical Modeling of Concentrations and Fluxes of HNO3, NH3, and NH4NO3 Near the Surface . J. Geophys. Res.–Atmospheres , 93 : 7137 – 7152 .
- Buzorius , G. , Rannik , U. , Makela , J. M. , Vesala , T. and Kulmala , M. 1998 . Vertical Aerosol Particle Fluxes Measured by Eddy Covariance Technique Using Condensational Particle Counter . J. Aerosol Sci. , 29 : 157 – 171 .
- Buzorius , G. , Rannik , U. , Nilsson , D. and Kulmala , M. 2001 . Vertical Fluxes and Micrometeorology During Aerosol Particle Formation Events . Tellus Series B–Chemical and Physical Meteorology , 53 : 394 – 405 .
- Canagaratna , M. J. , Jayne , J. T. , Ghertner , D. A. , Herndon , S. , Shi , Q. , Jimenez , J. L. , Silva , P. J. , Williams , P. I. , Lanni , T. , Drewnick , F. , Demerjian , K. L. , Kolb , C. E. and Worsnop , D. R. 2004 . Chase Studies of Particulate Emissions from in–use New York City Vehicles . Aerosol Sci. Technol. , 38 : 555 – 573 .
- Canagaratna , M. R. , Jayne , J. T. , Jimenez , J. L. , Allan , J. D. , Alfarra , M. R. , Zhang , Q. , Onasch , T. B. , Drewnick , F. , Coe , H. , Middlebrook , A. , Delia , A. , Williams , L. R. , Trimborn , A. M. , Northway , M. J. , DeCarlo , P. F. , Kolb , C. E. , Davidovits , P. and Worsnop , D. R. 2007 . Chemical and Microphysical Characterisation of Ambient Aerosols with the Aerodyne Aerosol Mass Spectrometer . Mass Spectrom. Rev. , 26 : 185 – 222 .
- Charlson , R. J. , Schwartz , S. E. , Hales , J. M. , Cess , R. D. , Coakley , J. A. , Hansen , J. E. and Hofmann , D. J. 1992 . Climate Forcing by Anthropogenic Aerosols . Science , 255 : 423 – 430 .
- Crosier , J. , Jimenez , J. L. , Allan , J. D. , Bower , K. N. , Williams , P. I. , Alfarra , M. R. , Canagaratna , M. R. , Jayne , J. T. , Worsnop , D. R. and Coe , H. 2007 . Technical Note: Description and use of the New Jump Mass Spectrum Mode of Operation for the Aerodyne Quadrupole Aerosol Mass Spectrometer (Q-AMS) . Aerosol Sci. Technol. , 41 : 865 – 872 .
- DeCarlo , P. , Slowik , J. G. , Worsnop , D. R. , Davidovits , P. and Jimenez , J. L. 2004 . Particle Morphology and Density Characterization by Combined Mobility and Aerodynamic Diameter Measurements. Part 1: Theory . Aerosol Sci. Technol. , 38 : 1185 – 1205 .
- DeCarlo , P. F. , Kimmel , J. R. , Trimborn , A. , Northway , M. J. , Jayne , J. T. , Aiken , A. C. , Gonin , M. , Fuhrer , K. , Horvath , T. , Docherty , K. , Worsnop , D. R. and Jimenez , J. L. 2006 . Field-Heployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer . Anal. Chem. , 78 : 8281 – 8289 .
- Dockery , D. W. , Pope , C. A. , Xu , X. , Spengler , J. D. , Ware , J. H. , Fay , M. E. , Ferris , B. G. and Speizer , F. E. 1993 . Mortality Risks of Air Pollution: A Prospective Cohort Study . New Eng. J. Med. , 329 : 1753 – 1759 .
- Dorsey , J. R. , Nemitz , E. , Gallagher , M. W. , Fowler , D. , Williams , P. I. , Bower , K. N. and Beswick , K. M. 2002 . Direct Measurements and Parameterisation of Aerosol Flux, Concentration and Emission Velocity Above a City . Atmos. Environ. , 36 : 791 – 800 .
- Drewnick , F. , Hings , S. S. , DeCarlo , P. F. , Jayne , J. T. , Gonin , M. , Fuhrer , K. , Weimer , S. , Jimenez , J. L. , Demerjian , K. L. , Borrmann , S. and Worsnop , D. R. 2005 . A New Time-of-Flight Aerosol Mass Spectrometer (TOF-AMS)—Instrument Description and First Field Deployment . Aerosol Sci. Technol. , 39 : 637 – 658 .
- Duyzer , J. 1994 . Dry Deposition of Ammonia and Ammonium Aerosols over Heathland . J. Geophys. Res.-Atmospheres , 99 : 18757 – 18763 .
- Fairall , C. W. 1984 . Interpretation of Eddy–Correlation Measurements of Particulate Deposition and Aerosol Flux . Atmos. Environ. , 18 : 1329 – 1337 .
- Gallagher , M. W. , Beswick , K. M. , Duyzer , J. , Westrate , H. , Choularton , T. W. and Hummelshoj , P. 1997 . Measurements of Aerosol Fluxes to Speulder Forest Using a Micrometeorological Technique . Atmos. Environ. , 31 : 359 – 373 .
- Gallagher , M. W. , Nemitz , E. , Dorsey , J. R. , Fowler , D. , Sutton , M. A. , Flynn , M. and Duyzer , J. 2002 . Measurements and Parameterizations of Small Aerosol Deposition Velocities to Grassland, Arable Crops and Forest: Influence of Surface Roughness Length on Deposition . J. Geophys. Res.-Atmospheres , 107 : 4154 doi:4110.1029/2001JD000817
- Gaman , A. , Rannik , U. , Aalto , P. , Pohja , T. , Siivola , E. , Kulmala , M. and Vesala , T. 2004 . Relaxed Eddy Accumulation System for Size-Resolved Aerosol Particle Flux Measurements . J. Atmos. Oceanic Technol. , 21 : 933 – 943 .
- Gonzales , D. A. , Allen , J. O. , Delia , A. E. , Jimenez , J. L. , Smith , K. A. , Jayne , J. T. , Canagaratna , M. R. and Worsnop , D. R. Speciated Fine Particle Deposition Velocities Over a Forest Canopy Measured by Eddy-Correlation Mass Spectrometry . 22nd Annual AAAR Conference, October 20–24, 2003 . Anaheim, California : American Association for Aerosol Research . Poster 2PC11, p. 58
- Grimmond , C. S. B. , King , T. S. , Cropley , F. D. , Nowak , D. J. and Souch , C. 2002 . Local-Scale Fluxes of Carbon Dioxide in Urban Environments: Methodological Challenges and Results from Chicago . Environ. Pollut. , 116 : S243 – S254 .
- Grönholm , T. , Aalto , P. P. , Hiltunen , V. , Rannik , U. , Rinne , J. , Laakso , L. , Hyvonen , S. , Vesala , T. and Kulmala , M. 2007 . Measurement of Aerosol Particle Dry Deposition Velocities Using Relaxed Eddy Accumulation Technique . Tellus , 59B : 381 – 386 .
- Held , A. , Hinz , K.-P. , Trimborn , A. , Spengler , B. and Klemm , O. 2003 . Towards Direct Measurement of Trubulent Vertical Fluxes of Compounds in Atmospheric Aerosol Particles . Geophys. Res. Lett. , 30 Article No. 2016, doi: 10.1029/2003GL017854
- Hogrefe , O. , Drewnick , F. , Lala , G. G. , Schwab , J. J. and Demerjian , K. L. 2004 . Development, Operation and Applications of an Aerosol Generation, Calibration and Research Facility . Aerosol Sci. Technol. , 38 ( S1 ) : 196 – 214 .
- Huffman , J. A. , Jayne , J. T. , Drewnick , F. , Aiken , A. C. , Onasch , T. B. , Worsnop , D. R. and Jimenez , J. L. 2005 . Design, Modeling, Optimization and Experimental Tests of a Particle Beam Width Probe for the Aerodyne Aerosol Mass Spectrometer . Aerosol Sci. Technol. , 39 : 1143 – 1163 .
- Jayne , J. T. , Leard , D. C. , Zhang , X. , Davidovits , P. , Smith , K. A. , Kolb , C. E. and Worsnop , D. R. 2000 . Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles . Aerosol Sci. Technol. , 33 : 49 – 70 .
- Jimenez , J. L. , Nelson , D. D. , Zahniser , M. S. , Koplow , M. D. and Schmidt , S. E. 1999 . Characterization of On-Road Vehicle NO Emissions by a TILDAS Remote Sensor . J. Air and Waste Manag. Assoc. , 49 : 463 – 470 .
- Jimenez , J. L. , McRae , G. J. , Nelson , D. D. , Zahniser , M. S. and Kolb , C. E. 2000 . Remote Sensing of Heavy Duty Diesel Truck NO and NO2 Emissions Using Tunable Diode Lasers . Environ. Sci. Technol. , 32 : 2380 – 2387 .
- Jimenez , J. L. , Jayne , J. T. , Shi , Q. , Kolb , C. E. , Worsnop , D. R. , Yourshaw , I. , Seinfeld , J. H. , Flagan , R. C. , Zhang , X. F. , Smith , K. A. , Morris , J. W. and Davidovits , P. 2003 . Ambient Aerosol Sampling Using the Aerodyne Aerosol Mass Spectrometer . J. Geophys. Res.–Atmospheres , 108 : 8425 doi:8410.1029/2001JD001213
- Kirchstetter , T. W. , Harley , R. A. , Kreisberg , N. M. , Stolzenburg , M. R. and Hering , S. V. 1999 . On–Road Measurements of the Particle and Nitrogen Oxide Emissions from Light- and Heavy-Duty Vehicles . Atmos. Environ. Part a–General Topics , 33 : 2955 – 2968 .
- Kowalski , A. S. 2001 . Deliquescence Induces Eddy Covariance and Estimable Dry Deposition Errors . Atmos. Environ. , 35 : 4843 – 4851 .
- Lanz , V. A. , Alfarra , M. R. , Baltensperger , U. , Buchmann , B. , Hueglin , C. and Prevot , A. S. H. 2007 . Source Apportionment of Submicron Organ Aerosols at an Urban Site by Factor Analytical Modelling of Aersosol Mass Spectra . Atmos. Chem. Phys. , 7 : 1503 – 1522 .
- Martensson , E. M. , Nilsson , E. D. and Johansson , C. Emissions of Aerosol Particles in the Urban Environment Measured by Eddy Orrelation . 6th International Aerosol Conference . September 9–13 2002 , Taiwan.
- Mohr , C. , Huffman , J. A. , Cubison , M. J. , Aiken , A. C. , Docherty , K. S. , Kimmel , J. R. , Ulbrich , I. M. , Hannigan , M. , Garcia , J. and Jimenez , J. L. 2008 . Characterization of Primary Organic Aerosol Emissions from Meat Cooking, Trash Burning, and Motor Vehicles with High-Resolution Aerosol Mass Spectrometry and Comparison with Ambient and Chamber Observations . Environ. Sci. Technol. , submitted
- Nemitz , E. , Sutton , M. A. , Wyers , G. P. , Otjes , R. P. , Schjoerring , J. K. , Gallagher , M. W. , Parrington , J. , Fowler , D. and Choularton , T. W. 2000a . Surface/Atmosphere Exchange and Chemical Interaction of Gases and Aerosols Over Oilseed Rape . Agricult. Forest Meteorol. , 105 : 427 – 445 .
- Nemitz , E. , Williams , P. I. , Theobald , M. R. , McDonald , A. G. , Fowler , D. and Gallagher , M. W. 2000b . “ Application of Two Micrometeorological Techniques to Derive Fluxes of Aerosol Components Above a City ” . In EUROTRAC–2 Symposium 2000 , Edited by: Midgley , P. M. , Reuther , M. and Williams , M. Berlin, Garmisch-Partenkirchen, , Germany : Springer Verlag .
- Nemitz , E. , Gallagher , M. W. , Duyzer , J. H. and Fowler , D. 2002a . Micrometeorological Measurements of Particle Deposition Velocities to Moorland Vegetation . Quart. J. Royal Meteorol. Soc. , 128 : 2281 – 2300 .
- Nemitz , E. , Hargreaves , K. J. , McDonald , A. G. , Dorsey , J. R. and Fowler , D. 2002b . Micrometeorological Measurements of the Urban Heat Budget and CO2 Emissions on a City Scale . Environ. Sci. Technol. , 36 : 3139 – 3146 .
- Nemitz , E. and Sutton , M. A. 2004 . Gas–Particle Interactions Above a Dutch Heathland: III. Modelling the Influence of the NH3-HNO3-NH4NO3 Equilibrium on Size–Segregated Particle Fluxes . Atmos. Chem. Phys. , 4 : 1025 – 1045 .
- Nemitz , E. , Sutton , M. A. , Wyers , G. P. , Otjes , R. P. , Mennen , M. G. , van Putten , E. , Hellemond , J. and Gallagher , M. W. 2004 . Gas–Particle Interactions Above a Dutch Heathland: II. Concentrations and Surface Exchange Fluxes of Atmospheric Particles . Atmos. Chem. Phys. , 4 : 1007 – 1024 .
- Nemitz , E. , Theobald , M. R. , McDonald , A. G. , Dorsey , J. R. , Gallagher , M. W. and Fowler , D. 2007 . Size–Segregated Particle Emissions Above a City Derived by a Micrometeorological Technique . Environ. Sci. Technol. , [in preparation]
- Offerle , B. , Grimmond , S. , Oke , T. , Fortuniak , K. , Hom , J. , Salmond , J. , Golub , D. and Walsh , C. 2001 . Energy and CO2 Fluxes from Contrasting Urban Environments (Marseille, France; Lodz, Poland; Baltimore, USA; and Vancouver, B.C.) . American Geophysical Union , San Francisco, December 2001
- Okumura , M. , Mollner , A. K. , Fry , J. L. , Feng , L. , Sander , S. P. and Sivakumaran , V. 2005 . Gas-Phase Formation Rates of Nitric Acid and its Isomers Under Urban Conditions, Final Report CARB Contract No. 03–333., prepared for: State of California Air Resources Board, Sacramento, CA
- Ramanathan , V. , Crutzen , P. J. , Kiehl , J. T. and Rosenfeld , D. 2001 . Atmosphere-Aerosols, Climate, and the Hydrological Cycle . Science , 294 : 2119 – 2124 .
- Robinson , A. L. , Donahue , N. M. , Shrivastava , M. K. , Weitkamp , E. A. , Sage , A. M. , Grieshop , A. P. , Lane , T. E. , Pierce , J. R. and Pandis , S. N. 2007 . Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging . Science , 2 : 1259 – 1262 .
- Salcedo Onac , D. , Canagaratna , M. R. , Dzepina , K. , Huffman , J. A. , Jayne , J. T. , Worsnop , D. R. , Kolb , C. E. , Weimer , S. , Drewnick , F. , Allan , J. D. , Delia , A. E. and Jimenez , J. L. 2007 . Technical note: Use of a Beam Width Probe in an Aerosol Mass Spectrometer to Monitor Particle Collection Efficiency in the Field . Atmos. Chem. Phys. , 7 : 549 – 556 .
- Schery , S. D. , Wasiolek , P. T. , Nemetz , B. M. and Yarger , F. D. 1998 . Relaxed Eddy Accumulation for Flux Measurements of Nanometer–Size Particles . Aerosol Sci. Technol. , 28 : 159 – 172 .
- Slinn , W. G. N. 1982 . Prediction for Particle Deposition to Vegetative Canopies . Atmos. Environ. , 16 : 1785 – 1794 .
- Soegaard , H. and Jansen , L. M. 2004 . “ Towards a Spatial CO2 Budget of Metropolitan Regions Based on Textural Image Classification and Flux Measurements ” . In Remote Sensing and the Environment Article 5925
- Sutton , M. A. 1990 . “ The Surface/Atmosphere Exchange of Ammonia ” . In Institute of Terrestrial Ecology and Resource Mangement , 194 Edinburgh : University of Edinburgh .
- Thomas , R. , Trebs , I. , Otjes , R. , Jongejan , P. A. C. , ten Brink , H. , Phillips , G. , Korner , M. , Meixner , F. X. and Nemitz , E. 2007a . An Automated Analyser to Measure Surface–Atmosphere Exchange Fluxes of Water Soluble Inorganic Aerosol Compounds and Reactive Trace Gases . Environ. Sci. Technol. , [under review]
- Thomas , R. M. , Hallquist , M. , Janhill , S. , House , E. , Coe , H. and Nemitz , E. 2007b . Fluxes of Submicron Aerosol Components Above an Urban Canopy: Measurements During the GOTE–2005 Campaign . Atmos. Chem. Phys. , [in preparation]
- Ulbrich , I. M. , Canagaratna , M. R. , Zhang , Q. , Worsnop , D. R. and Jimenez , J. L. 2008 . Interpretation of Organic Components from Positive Matrix Factorization of Aerosol Mass Spectrometric Data . Atmos. Chem. Phys. Discuss. , 8 : 6729 – 6791 .
- US–EPA . 1995 . Compilation of Air Pollutant Emission Factors, AP–42 , Research Triangle Park, NC : U.S. Environmental Protection Agency .
- Velasco , E. , Lamb , B. , Pressley , S. , Allwine , E. , Westberg , H. , Jobson , B. T. , Alexander , M. , Prazeller , P. , Molina , L. and Molina , M. 2005 . Flux Measurements of Volatile Organic Compounds from an Urban Landscape . Geophysical Research Letters , : 32
- Vogt , R. , Christen , A. , Rotach , M. W. , Roth , M. and Satyanarayana , A. N. V. Fluxes and Profiles of CO2 in the Urban Roughness Sublayer . Fifth International Conference on Urban Climate . September 1–5 2003 , Lodz, Poland. pp. 4
- Volkamer , R. , Jimenez , J. L. , San Martini , F. , Dzepina , K. , Zhang , Q. , Salcedo , D. , Molina , L. T. , Worsnop , D. R. and Molina , M. J. 2006 . Secondary Organic Aerosol Formation from Anthropogenic Air Pollution: Rapid and Higher than Expected . Geophysical Research Letters , 33 doi:10.1029/ 2006GL026899 Article No. L017811
- Watson , J. G. 2002 . Visibility: Science and Regulation . J. Air & Waste Manag. Assoc. , 52 : 628 – 713 .
- Webb , E. K. , Pearman , G. I. and Leuning , R. 1980 . Correction of Flux Measurements for Density Effects Due to Heat and Water Vapour Transfer . Quart. J. Royal Meteorol. Soc. , 106 : 85 – 100 .
- Wesely , M. L. , Cook , D. R. , Hart , R. L. and Speer , R. E. 1985 . Measurements and Parameterization of Particulate Sulfur Dry Deposition Over Grass . J. Geophys. Res.–Atmospheres , 90 : 2131 – 2143 .
- Wyers , G. P. and Duyzer , J. H. 1997 . Micrometeorological Measurement of the Dry Deposition Flux of Sulphate and Nitrate Aerosols to Coniferous Forest . Atmos. Environ. , 31 : 333 – 343 .
- Zhang , Q. , Alfarra , R. , Worsnop , D. , Allan , J. , Coe , H. , Canagaratna , M. and Jimenez , J. 2005a . Deconvolution and Quantification of Hydrocarbon–Like and Oxygenated Organic Aerosols Based on Aerosol Mass Spectrometry . Environ. Sci. Technol. , 39 : 4938 – 4952 .
- Zhang , Q. , Canagaratna , M. R. , Jayne , J. T. , Worsnop , D. R. and Jimenez , J. L. 2005b . Time and Size-Resolved Chemical Composition of Submicron Particles in Pittsburgh-Implications for Aerosol Sources and Processes . J. Geophys. Res.–Atmospheres , 110 D07S09
- Zhang , Q. , Worsnop , D. R. , Canagaratna , M. R. and Jimenez , J. L. 2005c . Hydrocarbon–Like and Oxygenated Organic Aerosols in Pittsburgh: Insights into Sources and Processes of Organic Aerosols . Atmos. Chem. Phys. , 5 : 3289 – 3311 .
- Zhang , Q. , Jimenez , J. L. , Canagaratna , M. J. , Allan , J. D. , Coe , H. , Ulbrich , I. , Alfarra , M. R. , Takami , A. , Middlebrook , A. M. , Sun , Y. L. , Dzepina , K. , Dunlea , E. , Docherty , K. , DeCarlo , P. F. , Salcedo , D. , Onasch , T. , Jayne , J. T. , Miyoshi , T. , Shimono , A. , Hatakeyama , S. , Takegawa , N. , Kondo , Y. , Schneider , J. , Drewnick , F. , Weimer , S. , Demerjian , K. , Williams , P. , Bower , K. , Bahreini , R. , Cottrell , L. , Griffin , R. J. , Rautiainen , J. and Worsnop , D. R. 2007 . Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenically Influenced Northern Hemisphere Mid-Latitudes . Geophys. Res. Lett. , 34 doi:10.1029/2007GL029979 Article No. L013801