Abstract
The Thermal desorption Aerosol Gas chromatograph (TAG) is a recently developed instrument for the in-situ, hourly measurement of speciated organic compounds in atmospheric aerosols. This paper presents a method for the in-field calibration of this instrument, with the objective of providing quantitative concentrations for a large suite of low polarity organic compounds. A new collection and thermal desorption cell was developed that incorporates an injection port for in-situ calibrations with liquid standard mixtures. Two classes of injection standards, instrument tracking and auxiliary, provide the means to calibrate the instrument in the field for a wide range of compounds. A routinely injected tracking standard suite of compounds generates a time-dependent correction of detector drift through the course of a measurement study that accounts for the bulk of the change in response of the TAG instrument. Injection response data for the tracking standard is also used to measure instrument precision and limits of quantitation. Auxiliary standards extend the range of compounds calibrated through use of relative response factors. The accuracy of this in-situ calibration approach is assessed through comparisons of TAG analyzed reference filter punches to published NIST assay values. A subset of compound classes, alkanes and PAHs, are used to illustrate the method and provide a means of reducing an 11-day period of data collected in Riverside, CA during the fall of 2005.
INTRODUCTION
For many years, the composition of atmospheric particulate organic matter has been characterized through GC/MS analyses of filter samples. Even though only a fraction of the total organic mass can be identified at the molecular level, those identified provide important insights regarding the origins of carbonaceous aerosols (CitationMazurek 2002). Yet filter-based methods are constrained by generally poor time resolution and limited continuity. In the last decade, several types of particle beam mass spectrometry instruments such as the AMS (CitationJayne et al. 2000; CitationJimenez et al. 2003) and ATOFMS (CitationGard et al. 1997) have been developed. These offer high time resolution not available from filter methods, as well as size selection and single particle composition. The continuous, detailed characterization provided by these instruments has led to their widespread use in atmospheric research studies. However, because of the complexity of organic aerosols, these instruments do not provide the molecular level composition data needed to both identify sources and to better understand the atmospheric formation and transformation pathways for organic particulate matter (CitationMolina et al. 2004).
Recently, CitationWilliams et al. (2006) introduced a new method to provide hourly, in-situ speciation of organic compounds in atmospheric aerosols. By coupling traditional gas chromatography–mass spectrometry (GC/MS) with in-situ aerosol collection and thermal desorption, this method aims to bridge this gap between compound specific information obtained from off-line filter analyses and the high-time resolution offered by particle beam mass spectrometry methods. Called the Thermal desorption Aerosol Gas chromatograph, or TAG, this automated system has been used to follow the relative, hourly concentrations of dozens to hundreds of ambient organic compounds over periods of several weeks at a time. Thermal desorption of filter based aerosol samples is gaining wider acceptance because of the reduced sample size and handling requirements and has been shown to provide equivalent results to standard liquid extraction techniques for many compound classes (e.g., CitationHays and Lavrich 2007, and citations therein). Like all GC based methods, the TAG instrument is currently restricted to non- to low-polar compounds that elute without derivatization.
The TAG system was initially deployed at a remote site in Nova Scotia (CitationWilliams et al. 2007), and more recently at an urban site in Riverside, California (CitationSOAR 2005). In Nova Scotia, TAG tracked relative, hourly concentrations of 37 organic compounds over a period of three weeks. Through factor analysis, six different source profiles were identified including anthropogenic and biogenic components. In addition, hourly resolution for oxygenated compounds associated with secondary aerosol formation provided insights into atmospheric processes important to their formation. In Riverside, approximately 300 compounds were tracked during two four-week periods in the summer and fall of 2005 (Williams et al., in preparation). More recently a TAG system has been deployed by others for smog chamber and downtown Pittsburgh, PA measurements (CitationLambe et al. 2007) and we have explored increasing compound resolution using 2-D chromatography (CitationGoldstein et al. 2008).
This work focuses on the extension of TAG measurements into the quantitative realm. During the Riverside field deployment, the TAG system was systematically calibrated with various organic standards mixtures introduced directly into the aerosol collection cell. We address here the protocol used for these calibrations, and how the calibration data are applied to ambient TAG measurements. Our objective in these calibrations is to provide the data necessary to correct for slow drifts in instrument response and to assess concentration-dependent response factors for a variety of compounds. While our calibration procedure borrows from established methods used for off-line filter analyses, calibrating the TAG instrument presents a unique set of challenges. As with any on-line system, time taken for calibrating subtracts from time for ambient measurements. Moreover, each TAG calibration run consumes exactly the same amount of time as an ambient sample run. Further, a useful calibration must include the entire TAG analysis system including the collection and thermal desorption cell, the thermal transfer lines to the column as well as the GC/MS itself. Another challenge is the inability currently to introduce an internal standard with each individual ambient sample.
Presented is a protocol using mixtures of liquid standards injected directly into the aerosol collection cell. In applying these standards to ambient data analysis, the following specific questions are addressed:
| • | Are liquid-based injection standards appropriate for calibrating instrument response to samples in a matrix of ambient particulates? | ||||
| • | How stable is the TAG system over the period of a multi-week study? | ||||
| • | What is the precision of measurements with injection based calibrations? | ||||
| • | How can a wide range of compounds be quantified with limited standards? | ||||
The analysis presented here focuses on n-alkanes and polycyclic aromatic hydrocarbons, two classes of compounds that are regularly generated by primary emission sources (CitationSchauer et al. 1996). PAHs are primarily linked to incomplete vehicular combustion and are of inherent interest because of their suspected mutagenicity and carcinogenicity (CitationEPA 2002).The aliphatic alkane series is ubiquitous in the atmosphere with both anthropogenic and biogenic sources and the biogenic contribution level is often estimated through the observed odd-carbon number preference (CitationSimoneit and Mazurek 1982). Because of their relatively low polarity, these classes of compounds pose less difficulty for reproducible chromatography than many other classes of observed compounds, and therefore allow simpler data treatments.
EXPERIMENTAL METHODS
System Description
TAG consists of an aerosol collection system interfaced to a gas chromatograph with parallel mass spectrometry and flame ionization detection (GC/MS-FID). Under normal operation, samples are collected for 30 min, immediately followed by thermal desorption into the GC/MS-FID, with subsequent in-situ analysis. The cycle time is 1-hour in which collection and analysis occur in parallel. Details on the instrument design, laboratory evaluation and initial field deployment are given by Williams et al. (Citation2006; Citation2007).
Aerosol is sampled through a 2 μm cyclone precut and a multi-tube Nafion™ based humidifier (Perma-Pure), and deposited directly into the collection thermal desorption cell (CTD) by inertial impaction. The CTD uses a 9-jet impactor with a 50% collection cut-point of 0.09 μm. The humidifier applies a thin coating of water to reduce solid particle bounce during impaction. The sample flow rate is approximately 9 L/min. During sample collection, the CTD cell is held at a constant temperature of 30°C. The cell interior surfaces along with all sample transfer lines are chemically passivated (Inertium™, AMCX) to reduce wall effects during sampling and optimize compound release from the cell. A heated six-port valve (Valco) isolates the CTD from the GC.
After collection, samples are transferred to the head of the GC column by means of thermal desorption into He carrier gas. This step begins with a 5 min He gas purge at 50°C to vent adsorbed water, which can interfere with chromatographic retention times. The most volatile compounds equivalent to ∼C12 alkane and smaller also desorb at this temperature. The 6 port valve is then switched so that He carrier gas flows from the cell onto the GC column. The cell temperature is ramped to 300°C at ∼30°C/min while the column is held at 45°C, thereby re-focusing desorbed compounds onto the head of the capillary column. The sample is analyzed using an Agilent 6890 GC with column effluent split between an Agilent 5973 MSD with electron impact ionization and an Agilent FID. Chromatographic separation is achieved with an Rtx-5MS column (30 m × 0.25 mm × 0.25 μm, Restek) operated under constant pressure conditions. The analysis protocol uses an 8.6°C/min ramp from 45 to 310°C followed by an 8-min hold. Peak integrations were performed with Chemstation software (Agilent).
As part of the automated sampling sequence, two types of dynamic blanks are collected: filtered ambient and zero-air. Particle free ambient dynamic blanks are measured by switching in-line a Teflon filter upstream of the CTD, between the cyclone and humidifier. Samples free of both particles and gas phase organics are obtained by sampling the output of a zero-air generator (Aadco). The introduction of calibration standards is done manually, as described below.
CTD Cell with Injection Port
To facilitate in-situ field calibrations of TAG, a collection and thermal desorption cell (CTD) was designed, as shown in . This cell differs from that of the original TAG system by the incorporation of an injection port. An Agilent blunt-end microliter syringe is used to deliver liquid standards through a small ∼3 mm OD septum in one arm of a 1/8″ compression fitting tee. Injected standards are deposited near the impaction region of the CTD on the same kind of passivated surface on which aerosol sample collection occurs. A fixed volume of 5 μl is used for all injections in order to ensure constant aliquot surface coverage inside the cell and reproducible droplet release from the syringe needle (see inset detail of ). This direct introduction of standards at the point of ambient sample collection is designed to mimic the complete analysis process including losses during purging of volatiles, surface release, transport through the transfer lines and heated 6-port valve, as well as chromatographic elution characteristics.
FIG. 1 Current design of the collection and thermal desorption (CTD) cell provides in-situ calibrations of TAG via an integrated injection port. Fixed-volume injections of varying concentrations of authentic standards in solution are deposited near the impaction region of the collection cell on the same passivated surface as used for aerosol collection.
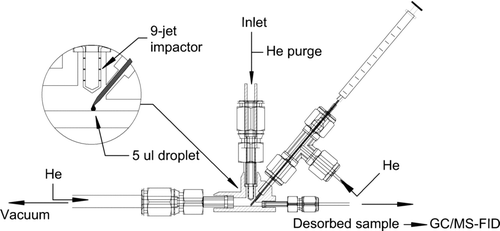
Cell injections are performed during the initial vented purge phase of an otherwise normal thermal desorption cycle. This minimizes solvent exposure of the chromatographic column while exactly following the protocol used for ambient sample analysis. During purging, He gas is passively split between three passages leading to the CTD (see ), including the side-arm of the injection port tee. Approximately half of the flow is introduced upstream of the impactor jet to insure a steady stream across an ambient sample while the remainder of the purge flow is nearly equally split between the downstream and injection ports of the cell. Typically, He flow is 50–100 cc/min during the purge before switching to the 2 cc/min carrier gas delivered to the GC column during the thermal desorption phase. System flow path and valve sequencing details can be found in CitationWilliams et al. (2006).
Details of the CTD design not shown include a 100 W cartridge heater and Type K thermocouple probe both inserted into the CTD body and used with PID electronic control of the thermal desorption cycle. Additionally, a finned heat sink attached to the base of the cell provides rapid post-desorption cooling back down to 30°C prior to resumption of ambient sampling. For a typical hour long sampling cycle, 30 min are used for sampling and roughly 15 min each for heating and cooling.
Authentic Standards
During the Riverside study, 11 distinct sets of authentic standard mixtures were employed, including more than 200 compounds providing measurable responses. In this paper we discuss three sets of these standards, as listed in . These include (1) a tracking standard consisting of a custom multi-class mixture of compounds obtained from the Wisconsin State Laboratory of Hygiene, (2) an n-alkane windowing standard (Alkane) covering the carbon number range of C8-C40 obtained from Accustandard (#DRH-008S-R1), and (3) a 116-compound semi-volatile aromatic mix (PAH) obtained from Cerilliant (EPA Method 8270c, #ERS-026). Standards were stored at <0°C in original sealed ampoules until injection samples were prepared by dilution and transferred to amber mini-vials with hard caps for storage or septa caps for injection use. Injection vials were removed from the freezer approximately 30 min prior to each injection to allow thermal equilibration.
TABLE 1 Subset of authentic standards used for TAG calibration during summer and fall Riverside field studies. The tracking standard was injected regularly throughout both study periods while the auxiliary standards were infrequently injected and primarily serve to confirm MS compound identification through retention times
Reagent and HPLC grade solvents were obtained from VWR, Sigma-Aldrich, and Fisher Scientific at 99.5% to 99.9% purity levels. Solvent blends were chosen for each standard's dilution to match the solubility of each standards solvent base but minimize toxicity for field use. Toluene was substituted for benzene in the field preparations for the custom tracking standard and toluene plus acetone replaced methylene chloride plus benzene in the EPA standard. The alkane standard was diluted with chloroform, the original solvent base for this standard. Dilution ratios ranged from a low of 25:1 for an organic acid mix and a high of 6400:1 for the tracking standard during limits of detection measurements. On column mass levels per compound ranged from tens of nanograms to tens of picograms, respectively.
For the summer (fall) field study a total of 58 (64) distinct standard injections were performed. Of these, approximately one-half were of the tracking standard and the remainder spread across the other standard sets. The tracking standard injections were done on a nearly daily basis. The majority (80% in summer, 90% in fall) of these were done at consistent level to gauge changes in instrument response. Multi-point calibrations with the tracking standard were done on 2 days in the summer and 3 days in the fall. Three point calibrations were done with the EPA Method 8270c standard, once in the summer and twice in the fall. The alkane windowing standard was run at two levels in the summer and fall.
Assessment of Accuracy
The accuracy of our liquid standard based calibration was tested through the direct analysis of NIST reference material RM8785 (CitationSchantz et al. 2006) using the TAG system. RM8785 is a PM2.5 sample on 47 mm quartz filters consisting of re-suspended baghouse samples of airborne urban particulate matter (CitationKlouda et al. 1996). Punched sections of these NIST reference filters were inserted into the TAG collection cell and analyzed according to standard protocol. Liquid-based calibrated TAG responses of the reference material were compared for eight polycyclic aromatic hydrocarbons to the published assay levels in the NIST filters.
Filter sections were obtained with an oblong arch punch with width 0.28 cm and length 1.3 cm and area of 0.41 cm2. Each 47 mm filter has approximately 1 mg total particulate gravimetric mass and a relative loading between 0.1 and 6 ng/mg for individual PAH compounds. Typical per punch mass loadings per co-eluting compound group analyzed ranged from 0.1–0.7 ng, which are reasonable values for atmospheric PAH concentrations assuming a 0.25 m3 sample volume.
The comparison experiment consisted of running liquid standards interspersed with one or two RM8785 punches, all analyzed with the same TAG method protocol except for sample introduction to the cell. The liquid standards were injected via the cell's integral injection port using fixed 5 μl aliquots per the instrument's calibration convention. Either one or two filter punches were inserted into the cell using tweezers by temporarily opening the sample flow exhaust port with a 0.5 cm ID. During this procedure He purge gas was allowed to flow out the opening to avoid contamination.
A liquid PAH standard (Cerilliant ERS-009) was injected 6 times at 5 distinct levels spanning in all cases each compound's single and double punch mass level. Non-linear calibrations as outlined below were used to reduce the integrated peak areas of the RM8785 data and convert them to on-column mass levels for comparison with NIST assay values.
Assessment of Precision and Limits of Quantitation
Compound precision was measured through four repeated injections of 5 μl injections of the tracking standard each immediately followed by a normal thermal desorption analysis. The single ion peaks from the MS were integrated and compared across injections. The resulting relative standard deviation (RSD) of these peak areas is taken as a baseline or minimum precision of the instrument response to sample injections for the representative compounds of the tracking standard.
Limits of quantitation (LOQ) for the tracking standard were obtained by injecting successive dilutions to determine at which point single ion peaks could no longer be distinguished from background noise. The specific dilution factors used were 2, 4, 8, 32, and 64 relative to the standard concentration level used for tracking purposes. A single compound LOQ is defined here as the geometric mean mass of the lowest concentration with an easily quantifiable single ion signal and the next lower dilution level without a quantifiable signal. This operational definition is more stringent than the usual 3-sigma definition of limit of detection based on instrument noise.
Field Deployment
The first field deployment of the TAG instrument incorporating the new CTD cell occurred as part of the multi-investigator Study of Organic Aerosols at Riverside (SOAR). This 2005 field campaign was conducted on the University of California Riverside (UCR) campus during two periods, one in the summer from July 17 to August 15 and one in fall from October 27 to November 30. Measurements focused on carbonaceous aerosols. Instrumentation ranged from single-particle mass spectrometers to multi-hour filter samples for GC/MS analysis. A complete list of study participants and their respective measurements may be found elsewhere (CitationSOAR 2005).
The UCR field site was located at the southeastern edge of campus approximately 0.6 km east of interstate highway 215. The TAG instrument was stationed inside a trailer along with instruments for monitoring gas phase organics and meteorological parameters. Aerosol samples were drawn from a dedicated insulated stainless steel inlet line from a height of approximately 5 m above ground on a 3 m tower mounted to the trailer roof.
CALIBRATION METHODOLOGY
The TAG calibration methodology employed sought to calibrate the instrument for a wide range of organic compounds and additionally correct for instrument drift over the multi-week campaign. This calibration protocol utilized two categories of standards (): (1) a single tracking standard consisting of a mixture of several compound groups and (2) multiple auxiliary standards, each of which represents a single family of compounds. Importantly, many of the auxiliary standards contained at least one compound also found in the tracking standard.
The tracking standard (TS) was regularly injected at a fixed concentration level and used for gauging both absolute system response and system drift. On two days in the summer and three days in the fall, the TS was introduced at multiple levels to establish calibration curves and LOQs. Auxiliary standards were used to positively identify compounds by retention times and were injected less frequently. This protocol was designed to maximize the calibration information while minimizing the time TAG was not acquiring ambient air samples.
The quadrupole mass spectrometer was tuned at the outset of each study period using the internal perfluorinated calibrant, PFTBA, and then allowed to run continuously without retuning for the two 4-week field campaigns. Re-tuning during the course of a study would have required more extensive calibrations, and would have severely restricted the amount of time available for ambient sampling.
The repeated (near daily) fixed concentration TS injections were used to quantify the long-term drift in system response during each 4-week study period. For each of the compounds found in the TS, all of the data (standards and ambient) were corrected based on the measured change in system response to the fixed concentration TS injection data. We refer to this system drift correction as “de-trending.”
For each of the two study periods, the de-trended TS data was then used to generate a single, time-independent, multipoint calibration curve for each compound in the TS. These resulting calibration curves were then applied to similarly de-trended raw ambient data. All MS analyses were performed on single ion peak areas wherein the selected ion mass was chosen for maximum relative abundance and non-interference with any co-eluting compounds.
For compounds found only in the auxiliary standards, we assessed the response relative to a compound that is common to the auxiliary and tracking standards. This reference or cross-over compound provides a relative response factor that is applied to the calibration curve for the reference compound, extending quantification to all of the compounds common to both standards. Specifically, the ambient data for compounds found in the auxiliary standard not-present in the TS are de-trended in the same manner as for the cross-over TS compound, and then reduced using the calibration curve for the latter multiplied by the relative response factor. This assumes a similar concentration-dependent response for the two compounds. Thus care is taken to use an appropriate concentration level for assessing the relative response factor. Also, we note that the determination of the relative response factors is aided by the fact that all compounds in the auxiliary standard are at the same concentration level. To date we have not fully addressed the quantification of compounds in the auxiliary standards that do not contain a compound common with the tracking standard.
RESULTS
Accuracy
Accuracy of the liquid calibration approach was assessed through laboratory measurements of NIST reference material inserted into the cell. Since RM8785 filters contain a full range of ambient particulates, using this method of sample introduction permitted us to evaluate response within the context of a complex matrix of organic matter found in any ambient aerosol, and to assess whether the simple compound mixes used for the liquid standards provide an appropriate calibration.
and compare the indicated on-column mass concentrations obtained by TAG to that reported for the NIST reference material for 8 different PAHs. Shown are results for 5 single-punch runs and 3 double-punch runs. Repeat desorptions of the filter punches indicate 90–100% recoveries but were hard to quantify. Furthermore, as judged by filter blank results, no intrusion of PAHs was detected as a result of opening the cell to insert the punches. Therefore, no attempt was made to correct for small losses in compound recovery from the filter matrices. We note that this incomplete recovery does not occur with injected standards or ambient samples but only from the analysis of filter punches. The resulting regression slope of 0.92 is shown in is within 1/3 standard error of unity but is also consistent with small recovery losses. The high correlation (R2= 0.95) implies little systematic bias exists for the different PAH compounds.
FIG. 2 Comparison of calibrated TAG masses to NIST assay values for select PAH compounds on one or two RM8785 filter punches.
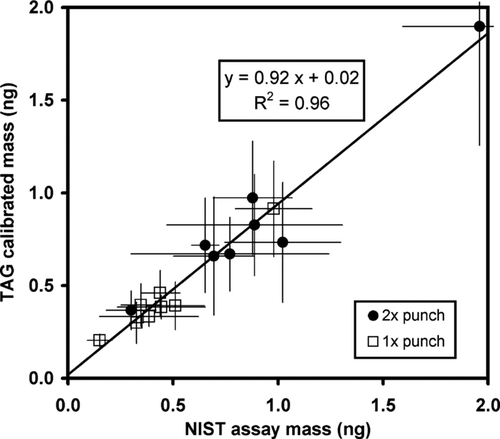
TABLE 2 Comparison of calibrated TAG masses to NIST assay values of select PAH compounds on RM8785 filter punches
The TAG uncertainties, shown by vertical error bars, are ±1 standard deviations from replicate analyses of the NIST filter punches. Horizontal error bars are derived from the inter-laboratory variability associated with analysis of the NIST RM8785 reference material plus an estimated inter-punch variability. Heterogeneity experiments (CitationLippa and Schantz 2007) suggest that the uniformity of the original particulate material can give rise to inter-filter variability on the order of 4%. No published data exists on uniformity of material on the filters themselves but extrapolating the sub-sampling trends of CitationLippa and Schantz (2007) analysis predicts inter-punch variability on the order of 6%. This analysis is based on the average of 10 PAHs that include all of the compounds included in this analysis. Thus, inter-punch variability will contribute to the uncertainty of any comparison between TAG analyzed responses to the published assay levels on the original particulate samples and has been taken into account as a systematic error of 6%. Uncertainties associated with the liquid standard concentrations and punch area were neglected.
Results for individual PAH compounds are summarized in . Ratios of the two population means are within a single propagated standard deviation of unity. The reproducibility of the analysis of the filter punches is the dominant source of uncertainty in this comparison but no significant bias can be detected between the two analytical methods. The data indicate that further effort, such as the incorporation of an internal standard with each sample, is needed to reduce the uncertainty observed among individual samples when employing this method of sample introduction. Yet, within the level of uncertainty present, the liquid based calibration of the TAG system is in agreement with the NIST certified levels, and justifies the use of liquid standards for TAG calibrations.
Precision and LOQ
For the Riverside field study, the system precision was obtained from repeat injections of the tracking standard; these results are presented in . For the non-polar compounds used in this treatment (shown in bold), the relative standard deviations for four injections ranged from 1.1% (cholestane) to 3.8% (hexadecane), thus demonstrating the excellent repeatability achievable with the CTD based injection port. For the two least volatile compounds, cholesterol and hexatriacontane, the relative standard deviations were 8.8% and 15%, respectively, indicating that the reduced sensitivity (higher LOQs) of these compounds affected the reproducibility. Lowest precision (23%) was obtained for decanoic acid, a compound known to have poor reproducibility in GC work without derivatization.
TABLE 3 Compounds contained in the Tracking standard
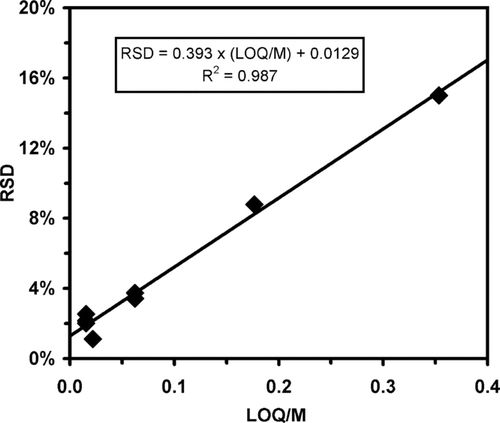
LOQs range from 0.02 ng for cholestane to 7 ng for hexatriacontane (right most column of ). This conservative measure of system response is higher than a limit of detection often defined as 3 times noise level and includes non-linear effects of the thermal desorption sample introduction system (i.e., adsorption site density throughout the heated transfer line pathway). demonstrates a linear correlation exists between the precision of repeat injections and the LOQ of individual compounds. Plotted are the RSD values for the less-volatile set of compounds (i.e., excluding those that elute prior to hexadecane) versus the ratio of LOQ to the injection mass level for that compound. For either smaller LOQs or larger injected mass levels or both, the precision improves and the RSD is observed to decrease. The minimum or baseline precision of the injection based calibrations may be inferred from the intercept of the regression line (R2= 0.99) to be 1.3%. Conversely, to maintain injection precision below 15% towards calibrating a collected analyte requires a sample size greater than 2.9 × LOQ for that analyte based on the regression. For example, calibrating octacosane (LOQ = 0.15 ng) at a baseline precision level of 15% would require an injection mass level of 0.44 ng on column. For a 30 minute sample collection period, this mass corresponds to an ambient concentration of 1.7 ng/m3.
FIG. 3 Relationship of injection repeatability (RSD) versus the ratio of LOQ to the individual compound injection mass level M. Compounds that elute before hexadecane (i.e., those listed above the shaded region of ) where excluded. For these less-volatile species, the baseline precision for calibration standards can be taken as the limit of LOQ/M∼0, which is the intercept of the regression line = 1.3% RSD.
System Stability and Drift Corrections
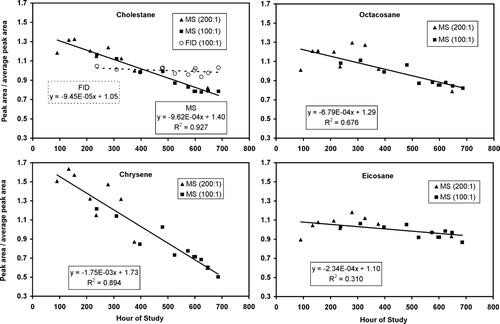
Temporal trends in TAG response were obtained during the fall study by examining 19 separate injections of the tracking standard that spanned 22 out of the 30 days in the fall period. shows the relative system response for the four TS compounds, cholestane, eicosane, octacosane, and chrysene as a function of time for the fall dataset. For purposes of inter-comparison, each compound's single ion peak areas have been normalized by the study average response for that compound. The distinct symbols are results from three different calibration solutions that were used during the course of the fall study.
FIG. 4 Fall instrument response trends are shown for four selected tracking standard compounds used in this analysis. Plotted is the detector peak area divided by each compound's average response versus hour of study. Mean single ion areas are 6.7 × 105 (cholestane), 4.0 × 106 (chrysene), 2.2 × 106 (eicosane), and 4.2 × 106 (octacosane). These compounds demonstrate a consistent, downward trend in MS response as a function of time. For a uniformly distributed subset of the tracking standard data, the cholestane FID data show no significant trend. Different filled symbols indicate distinct standard solutions. Drift regressions shown exclude the first set of standard solutions (triangles) due to uncertainty in that standard's concentrations.
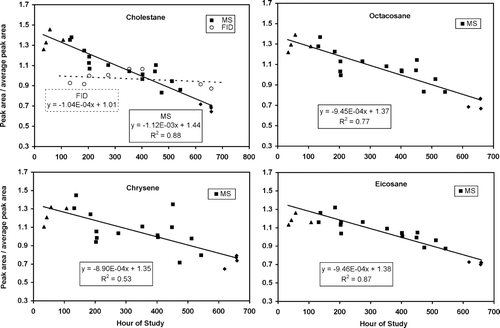
For all four compounds, the MS data show a steady decline in response over the study with total relative response changes ranging from −43% (chrysene) to −54% (cholestane). Notably, eicosane and octacosane exhibited essentially the same study-wide decline of −45% and −46%, respectively, as might be expected given that the same ion (57 m/z) was used for both integrations. Some of the compounds, e.g., chrysene, exhibit much greater variability on a time scale of days that is evidently superimposed on the downward trend observed in all compounds.
In contrast to the MS data, the FID data for cholestane shows no trend across the study, with a regression slope less than 1/10 that of the MS, and not significantly different from zero (R2= 0.079). The lack of a similar trend with the FID establishes that the main source of change in response for TAG stems from the MS detector itself. Co-elution interferences prevented similar analyses of FID response on the other compounds.
The MS drift is assessed by linear fits to the data of . Even though systematic changes across all compounds are evident for the individual injections, the density in the time series data permits only a linear regression for the trend analysis. The first set of data (triangles) used a relatively older standards solution with less reliable concentrations and so was excluded from the trend analysis. Results of the linear fits are shown on graphs. Coefficients of determination range from 0.53 (chrysene) to 0.88 (cholestane). These regression lines are used to correct the raw data for the detector drift. This correction or “de-trending” is applied to both the calibration and ambient data, in accordance with the relation:
where A and B are the slope and intercept values, respectively, shown in except for a normalization factor applied to the coefficients for presentation purposes. For a given time t, the value of Gs varies with compound with a range relative to each compound mean from near 1.4 at the beginning of each study period to near 0.7 towards the end.
Similar trend results were observed for the summer SOAR data, where 20 TS injections were made over a period of 25 out of 28 days total for that campaign. shows the resulting trends where symbols represent different dilution levels used with the tracking standard (either 200:1 or 100:1 from stock). The 200:1 data during the first week were corrected to a 100:1 level by application of a preliminary calibration curve to the concentration ratio of two (i.e., scalar factor = 2 b s , where bs is the exponent of the power law, see following section for details). A similar downward trend to that observed with the fall data is detected with the MS data from the SOAR data, but with a larger range of maximum changes in relative response of −15% (eicosane) to −69% (chrysene). As with the fall dataset, the FID signal for cholestane shows an insignificant decrease in response over the majority of the study period.
FIG. 5 Summer instrument response trends are shown for four selected tracking standard compounds used in this analysis. Plotted is the detector peak area divided by each compound's average response versus hour of study. Mean single ion areas are 8.4 × 105 (cholestane), 6.3 × 106 (chrysene), 2.6 × 106 (eicosane), and 5.6 × 106 (octacosane). These compounds demonstrate a consistent, downward trend in MS response as a function of time. For a uniformly distributed subset of the tracking standard data, the cholestane FID data shows no significant trend. Different filled symbols are for two standard concentration levels with the indicated stock dilution ratio. The more dilute response data was scaled up to the more concentrated level by application of a preliminary calibration using the more concentrated subset of data.
One notable difference between the two study periods is in the consistency in alkane trends for the fall relative to the alkane trends in the summer. While the fall dataset possess trend slopes for eicosane and octacosane that are equal to each other, the same trends in the summer differ by nearly a factor of three. This difference may be partly due to the greater scatter in the data and the use of a mixture of two concentration levels with the necessary re-scaling of the more dilute data (triangles). An examination of the more volatile compound hexadecane for the summer showed no downward trend within limits of uncertainty (slope = −5.1 × 10−5 ± 4.4 × 10−4 with 90% confidence intervals). Therefore, other unidentified factors affecting the overall instrument response beyond the MS drift were affecting the more volatile alkanes. Treating these compounds on an individual basis is necessary for the summer dataset, whereas the alkanes C16 to C28 in the fall dataset possess trend slopes that differ at most by 11% and could be corrected similarly.
Time-Independent, Multi-Point Calibrations
Since TAG is operated near the detection limits for many individual compounds, the use of a non-linear calibration form is required. The functional form of the field calibration F was chosen to be a power law:
Time-independent calibrations are formed from the de-trended responses to the selected TS compounds injected at multiple concentrations, as illustrated with the summer SOAR data in . Here are shown both linear (dotted line) and power law (solid line) fits of the de-trended calibration data to demonstrate that the non-linear form is much better at tracking the data for lower concentrations. The figure insets show with expanded axes that extrapolations of the linear fits always miss the lowest level mass calibration data. In all cases, the R2 values of the power laws are equal to or greater than the linear fit results. For two compounds, eicosane and chrysene, the linear fits are observed to be slightly closer to the data at the highest injection level but since ambient levels are at or below the 1 ng per sample level, or 4 ng/m3 equivalently, this distinction between the two functional forms at high mass loadings is not important.
FIG. 6 Time-independent calibration of TAG for four of the tracking standard compounds for the Summer SOAR study. Non-linear fits (solid lines) are shown to be superior to linear fits (broken lines) for all four compounds, especially near the limits of detection of the instrument (insets). Vertical spread in calibration points indicates residual variation of data after de-trending.
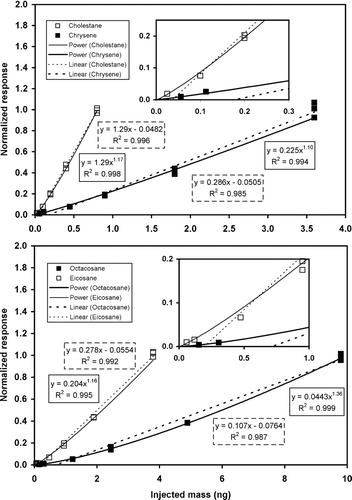
The summer TS calibration is comprised of a 3-point set on 7/27, a 5-point set during the LOQ injections on 8/12 and finally a 3-point set on 8/13. The number of datum from the LOQ sequence of injections contributed to a compound's calibration set varies by the individual LOQs. All four of the compounds under discussion were detected at five dilution levels except for cholestane which was only observed at four levels.
The vertical spread in the normalized response data indicate the degree to which de-trending successfully removes systematic changes from detector drift. The highest level injection was made on all three calibration days and shows that points with the lowest response with a 16 day drift correction are comparable to the variation seen in the pair of datum taken 1 day apart. A small systematic under-correction in the de-trended data is detectable at this highest level of mass injection. The 7/27 data differed relative to the average of the data from 8/12 and 8/13 by +0.2%, −11%, −4.6%, and −3.8% for cholestane, chrysene, octacosane, and eicosane, respectively.
Relative Response Factors for n-Alkanes and PAHs
Calibrations for compounds not present in the tracking standard were obtained from less frequently applied auxiliary standards (). Using compounds common to both the tracking standard and the auxiliary standards allows relating the response of all of the auxiliary compounds to that of the over-lapping TS compound. A scaling relationship was constructed between the response of the cross-over TS compound within the auxiliary standard, ys , and the response to the remaining compounds, yi , as the simple ratio
This response factor was constructed to apply to the n-alkanes in the range of C15-C32 () using the reference compound C20. All single ion peak areas were computed for the alkanes using mass fragment m/z = 57. For the PAHs in the range of 3–5 fused aromatic rings (e.g., phenanthrene to benz(a)pyrene, ) the response factor was generated using chrysene (m/z = 228) as the reference compound. In these two standards all compounds were present in equal masses, and therefore it was not necessary to correct for variations in concentrations among compounds within the auxiliary standards.
FIG. 7 Relative response factors for n-alkanes defined as the ratio of the single ion peak areas (57 m/z) for compound Cn to the reference compound eicosane (C20) for summer and fall periods. Sensitivity to mass level is indicated at both the low and high volatility ends of the carbon number spectrum. Response factors from the alkane auxiliary standard (triangles, squares) provide the complete range of response for TAG and are in general agreement with similar ratios obtained from the multipoint calibration data using C16, C20, and C28 in the tracking standard (circles), shown with 1-σ error bars, over the indicated mass injection levels.
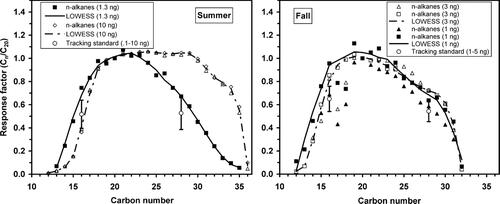
TABLE 4 Relative response factors for PAHs in the EPA auxiliary standard relative to the tracking standard compound chrysene, as defined by Equation (Equation4). R i, chy is the response at the 1 ng injection level, and R i, chy is the average response over a range of injection levels
Alkane Response Factors
shows relative response factors for the n-alkane windowing standard for the summer and fall studies for several concentration levels referenced to eicosane (C20) as a function of carbon number. A similar result would be obtained if boiling point or vapor pressure had been used instead of carbon number. Evident is a similar volatility window observed by TAG for these two periods with a slightly better response for the largest alkanes in the summer. Solid lines are locally weighted least-squares smoothed curves to be used for R i,s .
The fall data show response factors at 1 ng per compound and at 3 ng per compound. Little difference in response factor is seen for these two injection masses. In the summer, two more-disparate levels were used: 1.3 and 10 ng. These data show a strong mass dependence at high carbon numbers, and a lesser dependence on the low carbon number side, with an overlap for carbon numbers near 20. The 10 ng level standards were done twice, with consistent results, while the 1.3 ng injection was performed only once and was co-injected with another standard at similarly low levels. However, the 1 ng level is closer to the range of concentrations measured in the ambient sample, and co-injection of standards that otherwise do not over-load the column is not expected to change the instrument's response. Also, the summer data are closest to the 13 ng data from the fall. For these reasons we employ the response factor from the 1 ng level for both study periods.
The presence of over-lapping compounds between the TS alkanes and the auxiliary alkanes, allows for an independent measurement of two of the response factors, C16 and C28, shown in by grey circles. The range bars represent ±1 standard deviation of the TS data from the multi-point injection subset for the indicated average mass levels. A consistent discrepancy between these two approaches is observed, with the combined results from both factors from the two studies showing TS values on average smaller relative to the auxiliary standard by 24% ± 5%. This discrepancy is unresolved, and must be included as an uncertainty in the ambient concentrations derived there from.
PAH Response Factors
Response factors for a collection of PAHs present in the EPA method 8270c auxiliary standard were obtained during both study periods, as collected in . For the normalization, the average of benz(a)anthracene and chrysene was chosen because these compounds elute from the column without sufficient separation to allow consistent independent peak integrations to be performed. Similarly, the response factors for the two isomers benz(b)fluoranthene and benz(k)fluoranthene were combined. Response factors for 1 ng/compound injections are shown in columns three and five with the average responses for the entire range of injections levels given in columns four and six. The ranges of injection levels differed between the two study periods, with 1–8 ng and 1–3 ng employed in the summer and fall, respectively. The uncertainty for the 1 ng level response factors can conservatively be taken to be equal to the standard deviations obtained for the average responses over the stated ranges. As with the alkane response factors, independent information such as expected ambient concentration levels needs to be used to justify using the 1-ng results.
The m/z value for the chrysene analysis ion, 228, falls in the middle of the range of m/z values for the other PAHs listed in . The chrysene trend with time was used to correct these other compounds because it was observed that the drift dependence on ion mass is smaller than the dependence on compound class. This distinction can be seen by the example compounds shown in – and the trends for other compounds not shown.
Application of the TAG Calibration
Application of the calibration results to the ambient aerosol data involves (1) identifying the compound through mass spectrum and retention time matching, (2) pairing it with a TS reference compound, (3) integrating the single-ion peak area, (4) correcting for the time-dependent MS drift during the study period according to the de-trending results of the chosen TS compound, (5) applying, if needed, the mass dependent relative response factor selected from the auxiliary standard, and (6) using the calibration for the matched TS compound to obtain the absolute sample mass for the desired compound. Here we summarize this procedure in mathematical terms.
With an assumed linear drift in the MS data, we correct the temporal variation of TS compound s by the linear regression de-trending function of time Gs (t) as described above. The de-trended calibration data, given by Equation (Equation1), is used to construct a time-independent function of system response and time given by Equation (Equation3) which can be inverted to obtain mass as a function of system response. If the compound is a member of the auxiliary standards, the relative response factor R i,s given by Equation (Equation4), is used to convert the expected response to compound i from that of the TS standard s.
To apply the resulting calibration relationship, raw TAG responses for compound i, yi (t), are first de-trended just as with the calibration data to form y' i (t). Then the calibrated TAG response for compound i, is obtained from the inverse function F −1 s given by Equation (Equation3) in conjunction with the scaling response factor R i,s to obtain:
When calibrating a compound present in the tracking standard, s≡i and R i,s =1 so that the calibration simplifies to:
Dividing these quantities, mi (t) or ms (t), by the sample volume (typically 0.25 m3 for a 30 min sample) gives the resulting ambient mass concentration.
Application to Fall Riverside Ambient Measurements
Example data for the eleven day period of November 4–15, 2005 from the SOAR field study are shown in . The octacosane and chrysene data were reduced by direct application of the TS calibrations using Equation (Equation6) since these compounds are in the tracking standard. The two identified hopanes summed in the time series, 17βH)-28-norhopane and 17αH), 21βH)-hopane, were calibrated using the closest TS compound available, cholestane, with a relative response correction of 1.25 and 0.79, respectively, based on subsequent co-injections of an authentic standard with these hopanes plus 5α-cholestane.
FIG. 8 Calibrated TAG response for octacosane, chrysene, and the sum of two hopanes, during the fall study. The calibration for the tracking standard cholestane was applied to each of the hopane responses with a relative response factor prior to summing. Gaps greater than 2 h are shown with line breaks indicating when calibrations or other interruptions of normal operation occurred.
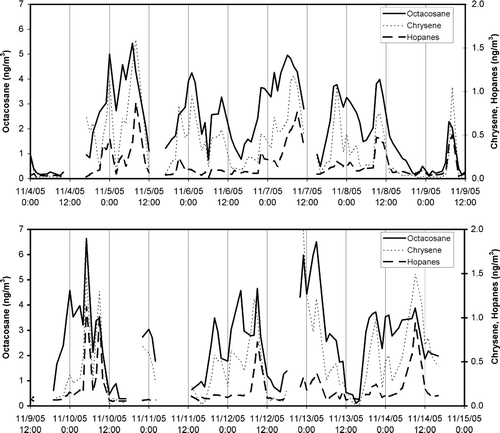
The data of illustrate the hourly variability in the observed atmospheric concentrations. Diurnal patterns are apparent, especially the morning and evening peaks in all of these primary emission compounds. Note that this variability is on a timescale that would be missed entirely with a 24 h integrated sample and greatly obscured with even 6 h samples. The day to day variability, moreover, is much smaller than the diurnal variability so that the higher time resolution afforded by TAG should lead to improved source apportionment analyses.
We plan to compare calibrated TAG responses to other aerosol measurements taken during these studies when data sets are finalized. During the summer, 4 and 6 h filter samples were collected for analysis by traditional GCMS methods and these could provide a valuable comparison. We note that differences in sampling methods such as impaction versus filtration, 30 min. versus multi-hour collection and differing approaches for blank corrections may complicate these comparisons, but could also provide insights with regard to sampling methodologies for semi-volatile constituents.
SUMMARY
This article summarizes the approach used to obtain and apply calibration data for individual organic compounds measured by the TAG system. Since TAG is an on-line instrument, the allotted time used for the analysis of standards must be carefully balanced with the time for ambient measurements. A protocol was therefore devised utilizing a single tracking standard in combination with several auxiliary standards. The tracking standard contains a range of compounds, including several alkanes, aromatic hydrocarbons, a sterane, and several organic acids. This standard was injected at fixed concentration levels on a nearly daily basis, and was used to correct MS and system drift. Additionally, multipoint calibrations with the tracking standard and the most common auxiliary standards were done on one to three days of each one-month study period. Other auxiliary standards were applied less frequently, and at only one or two concentration levels. Analysis of ambient data for compounds in the auxiliary standards was done by evaluating the relative response to a “reference” compound present in both the tracking standard and the auxiliary standard.
Analysis of the tracking standard data show a consistent drift in the system response throughout each study period, that was well-represented by a simple linear fit over time. Comparison of the MS and FID data for cholestane showed that this drift was attributable to the mass spectrometer. After correcting this linear drift, we find that for each compound a single multipoint calibration curve could be used to represent the system response over the entire study period. Near the detection limit, this response was best represented by a power law. Analysis of an n-alkane windowing standard showed a systematic carbon number dependence for the response relative to C20. Ambient data reduced using these calibrations yield concentrations for alkanes, PAHs and hopanes that exhibit temporal variations on the scale of hours that would be difficult to catch via filter-based methods.
Laboratory tests were performed to assess the accuracy of liquid based injection standards through the analysis of NIST reference filters. Analyzed punches inserted into the TAG cell showed within the uncertainty little bias relative to NIST assay values for 8 PAH compounds common to the authentic standard used to calibrate the instrument and the reference material. The slightly lower response (92%) measured by the TAG system relative to NIST is consistent with incomplete thermal desorption from the filter punches relative to aerosol or injected samples. Furthermore, the presence of a large background of ambient particulates on the RM8785 filters demonstrates that at least for these analyzed compounds a liquid standard based calibration is applicable to aerosols in an urban matrix.
The data presented here represent the first systematic calibrations applied to the TAG instrument in the field. The current effort provides an internally consistent data set, and is a significant step towards quantification of TAG measurements. The removal of instrument drift, an integral part of the calibration procedure, represents a useful advance in itself when compound timelines are to be used in correlation studies (e.g., factor analysis). Future refinements to the calibration protocol described here may include the use of customized standard mixtures to optimize compound selection and levels, thereby reducing the number of auxiliary standards required and extending the range of calibration applicability.
Acknowledgments
Thanks to Prof. James Schauer for providing the tracking standard, Prof. Paul Ziemann for his gracious hospitality, and Prof. Jose Jimenez and Dr. Ken Docherty for their efforts at organizing the SOAR campaign. Funding was provided by the Department of Energy Small Business Innovative Research program (Grant #DE-FG02-02ER83825), the California Air Resources Department (Grant #03-324) and DOE's Global Change Education Program (graduate research fellowship for BJW).
Notes
a (1) Custom standard from Wisconsin State Laboratory of Hygiene, (2) Accustandard, catalogue number DRH-008S-R1, (3) Cerilliant, catalogue number ERS-026.
1NIST RM8785 intercomparison mean and standard deviation of mass per punch (CitationSchantz et al. 2006).
2TAG calibrated mean and standard deviation of mass per one or two punches.
3Ratio of TAG mean to NIST mean and propagated standard deviation.
a Bold entries are most relevant to this article.
b M = on-column mass injection level.
c m/z = single ion mass to charge ratio used for integration analysis.
d RSD = relative standard deviation of repeated injections at level M.
e LOQ = limit of quantitation defined here as the geometric mean of the mass injection level at which a peak integration is well defined and the next more dilute level where the peak is poorly defined but still above the level of noise.
a Average and standard deviation in the response factor for standard injections of 1 to 8 ng.
b Range of injections is 1 to 3 ng.
c Combined response for co-eluting compounds.
Related Research Data
REFERENCES
- EPA . 2002 . Health Assessment Document for Diesel Engine Exhaust , Washington, DC : U.S. Environmental Protection Agency . EPA/600/8–9/057F
- Gard , E. , Mayer , J. E. , Morrical , B. D. , Dienes , T. , Fergenson , D. P. and Prather , K. A. 1997 . Real-Time Analysis of Individual Atmospheric Aerosol Particles: Design and Performance of a Portable ATOFMS . Anal. Chem. , 69 : 4083 – 4091 .
- Goldstein , A. H. , Worton , D. R. , Williams , B. J. , Hering , S. V. , Kreisberg , N. M. , Panić , O. and Górecki , T. 2008 . Thermal Desorption Aerosol Comprehensive Two-Dimensional Gas Chromatographic Resolution for In-Situ Measurements of Organic Aerosols . J. Chrom. A , : 340 – 347 .
- Hays , M. D. and Lavrich , R. J. 2007 . Developments in Direct Thermal Extraction Gas Chromatography-Mass Spectrometry of Fine Aerosols . TrAC Trends Analyt. Chem. , 26 ( 2 ) : 88 – 102 .
- Jayne , J. T. , Leard , D. C. , Zhang , X. , Davidovits , P. , Smith , K. A. , Kolb , C. E. and Worsnop , D. R. 2000 . Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles . Aerosol Sci. & Technol. , 33 : 49 – 70 .
- Jimenez , J. L. , Jayne , J. T. , Shi , Q. , Kolb , C. E. , Worsnop , D. R. , Yourshaw , I. , Seinfeld , J. H. , Flagan , R. C. , Zhang , X. and Smith , K. A. 2003 . Ambient Aerosol Sampling Using the Aerodyne Aerosol Mass Spectrometer . J. Geophys. Res. , 108 : 8425
- Klouda , G. A. , Klinedinst , D. B. , Steel , E. B. , Benner , B. A. and Parish , H. J. 1996 . Exploring a Method to Produce an Urban Dust Particle Filter Standard . Aerosol Sci. & Technol. , 27 : 351 – 352 .
- Lambe , A. T. , Logue , J. M. , Robinson , A. L. , Donahue , N. M. , Worton , D. R. , Williams , B. J. , Goldstein , A. H. , Kreisberg , N. M. , Gauthier , A. and Hering , S. V. September 24–28 2007 . Highly Time-Resolved Ambient Measurements of Organic Molecular Markers and Air Toxics in Pittsburgh Using Thermal Desorption Aerosol GC-MS (TAG) , September 24–28 , Reno, NV : American Association of Aerosol Research .
- Lippa , K. A. and Schantz , M. M. 2007 . Micro heterogeneity evaluation of polycyclic aromatic hydrocarbons in particulate standard reference materials . Anal. Bioanal. Chem. , 387 ( 7 ) : 2389 – 2399 .
- Mazurek , M. A. 2002 . Molecular Identification of Organic Compounds in Atmospheric Complex Mixtures and Relationship to Atmospheric Chemistry and Sources . Environ. Health Perspect. , 110 : 995 – 1003 .
- Molina , M. J. , Ivanov , A. V. , Trakhtenberg , S. and Molina , L. T. 2004 . Atmospheric Evolution of Organic Aerosol . Geophys. Res. Lett. , 31 : L22104 doi:10.1029/2004GL020910
- Schantz , M. M. , Poster , D. L. , Kucklick , J. R. , Wise , S. A. , McDow , S. and Lewtas , J. 2006 . “ Intercomparison Program for Organic Speciation in PM2.5 Air Particulate Matter: Description and Results for Trial III ” . 7303 NISTIR .
- Schauer , J. J. , Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. , Cass , G. R. and Simoneit , B. R. T. 1996 . Source Apportionment of Airborne Particulate Matter Using Organic Compounds As Tracers . Atmos. Environ. , 30 : 3837 – 3855 .
- Simoneit , B. R. T. and Mazurek , M. A. 1982 . Organic Matter of the Troposphere. II—Natural Background of Biogenic Lipid Matter in Aerosols Over the Rural Western United States . Atmos. Environ. , 16 : 2139 – 2159 .
- SOAR . 2005 . http://cires.colorado.edu/jimenez-group/Field/Riverside05/
- Williams , B. J. , Goldstein , A. H. , Millet , D. B. , Holzinger , R. , Kreisberg , N. M. , Hering , S. V. , White , A. B. , Worsnop , D. R. , Allan , J. D. and Jimenez , J. L. 2007 . Chemical Speciation of Organic Aerosol During the International Consortium for Atmospheric Research on Transport and Transformation 2004: Results from in Situ Measurements . J. Geophys. Res. , 112 : D10S26 doi:10.1029/2006JD007601
- Williams , B. J. , Goldstein , A. H. , Kreisberg , N. M. and Hering , S. V. 2006 . An in-situ instrument for speciated organic composition of atmospheric aerosols: Thermal desorption aerosol GC/MSFID (TAG) . Aerosol Sci. & Technol. , 40 : 627 – 638 .