Abstract
An Aerodyne quadrupole aerosol mass spectrometer (AMS) was deployed in China in the summer of 2006. The measurements were made in the Pearl River Delta region in July 2006 (PRD campaign) and also in Beijing in August–September 2006 (CAREBEIJING campaign). The AMS successfully measured size-resolved chemical composition of submicron non-refractory aerosol (vaporized at 600°C in vacuum) with a time resolution of 10 min, although some quantification issues have been identified. We observed extremely large signals at m/z 39 ( 39 K + ) and 41 (41K + ), which significantly exceeded m/z 28 (N + 2 ) signals. We also found large signals of m/z 85 ( 85 Rb + ), 87 (87Rb + ), and 133 (Cs + ). Laboratory experiments suggest that the large enhancement of K + could have been due to the presence of K-containing particles in ambient air. The interferences of alkali metals at m/z 41, 85, 87, and 133 were significant and need to be corrected for better quantification of organic aerosol. The AMS measurements are compared with other, collocated measurements: a particle-into-liquid sampler combined with an ion chromatograph (PILS-IC), a Sunset Laboratory semi-continuous carbonaceous aerosol analyzer, and a Berner impactor sampler followed by off-line ion chromatography analysis (for major inorganic ions). We have found good agreement between the AMS and the other instruments when we assume an AMS particle collection efficiency (CE) of 0.5 for the PRD data and CE = 1.0 for the CAREBEIJING data. These results suggest that the AMS CE could be significantly different in different locations. Possible factors affecting the variability in the CE values are discussed.
1. INTRODUCTION
Recent advances in on-line aerosol instruments have improved our understanding of the emission and formation processes of aerosol particles both from anthropogenic and natural sources. The Aerodyne aerosol mass spectrometer (AMS) is one of the most advanced on-line aerosol instruments, as it can quantitatively measure size-resolved chemical composition of submicron non-refractory aerosol (vaporized at 600°C in vacuum) with a time resolution of the order of seconds to minutes (CitationJayne et al. 2000; CitationJimenez et al. 2003; CitationAllan et al. 2003a). The AMS has been widely used at various locations in the world, as summarized by CitationZhang et al. (2007). Previous AMS measurements were conducted mostly in the United States, Europe, and Japan, while intensive measurements in Asia, especially China, Southeast Asia, and India, are still very limited. Although CitationZhang et al. (2007) have presented some AMS data obtained in Beijing, no detailed description of the AMS measurements in China has been published.
There have been a substantial number of off-line aerosol measurements at urban and suburban sites in China (e.g., CitationDuan et al. 2004; CitationZheng et al. 2005; CitationGuinot et al. 2007). These studies have provided useful insights into the diurnal and seasonal average chemical composition of fine particles. However, the origins of aerosol and its precursors in this region, especially organic aerosol, are highly complex and uncertain because many types of emission sources, including vehicles, power plants, domestic coal/biofuel burning, and open biomass burning, are often collocated (CitationStreets et al. 2003). In fact, CitationDuan et al. (2004) showed that biomass burning can be a significant source of organic aerosol even in the urban area of Beijing. The performance of the AMS (mass spectrum patterns, particle collection efficiency, etc.) has been tested mostly in urban areas in the United States, Europe, and Japan, where the types of emission sources are generally well defined (e.g., CitationAllan et al. 2003b; CitationDrewnick et al. 2003; CitationJimenez et al. 2003; CitationTakegawa et al. 2005), but it has not been fully tested in China or other environments with complex mixtures of various types of emission sources, the contributions of which are highly uncertain.
We have conducted on-line measurements of aerosol using a quadrupole AMS (Q-AMS) in China. The Q-AMS is hereafter referred to simply as the AMS. The measurements were made at a rural site (Backgarden site) in the Pearl River Delta (PRD) region in July 2006 and also at a rural site (Yufa site) in the Beijing region in August–September 2006. The former is referred to as the PRD 2006 intensive campaign (PRD campaign) and the latter as the Campaigns of Air Quality Research in Beijing 2006 (CAREBEIJING campaign). Both campaigns were organized by Peking University (PKU) and involved many scientists from China, Germany, Korea, and Japan. Detailed descriptions of the measurement site and interpretation of the temporal variations of aerosol will be presented elsewhere. In this paper we focus on the performance of the AMS during those intensive campaigns based on intercomparison with other, collocated instruments.
2. MEASUREMENTS
The Backgarden site was a 3 story building (23°30'N, 113°02'E) located in a rural area at ∼50 km northwest of the center of the Guangzhou city, which is the capital city of the Guangdong Province. The Yufa site was a 4-story building (39°31'N, 116°18'E) located in a rural area at ∼50 km south of the center of the Beijing city.
In this study we use data obtained by the AMS, a particle-into-liquid sampler combined with an ion chromatograph (PILS-IC), a Sunset Laboratory semi-continuous carbonaceous aerosol analyzer (Sunset EC/OC), and a five-stage Berner cascade impactor sampler (Hauke, Austria) followed by laboratory analysis. The AMS was operated from July 12 to 31 during the PRD campaign and from August 16 to September 9 during the CAREBEIJING campaign. The PILS-IC was operated only from July 4 to 13 during the PRD campaign, and the intercomparison was made only for a 32-h period from July 12 to 13. Nevertheless, these data are useful to characterize the performance of the AMS during the PRD campaign. The Sunset EC/OC analyzer was operated from July 3 to 31 during the PRD campaign and from August 11 to September 9 during the CAREBEIJING campaign. The AMS, PILS-IC, and EC/OC analyzer were installed in the same room of the observation building but used different sampling lines. The Berner impactor was installed on the rooftop of the observation building. Impactor samples were collected from July 1 to 26 during the PRD campaign and from August 15 to September 2 during the CAREBEIJING campaign. However, the Berner impactor data obtained during the PRD campaign are not included in the intercomparison because they are not available at the present stage. The mass concentrations of aerosol species reported in this paper are in units of μg m−3 at 25°C and 1 atm. Brief descriptions of the individual instruments are given below.
2.1. Aerodyne Aerosol Mass Spectrometer (AMS)
Size-resolved concentrations of major inorganic (sulfate (SO2− 4), nitrate (NO− 3), chloride (Cl−), and ammonium (NH+ 4)) and organic matters (OM) were measured with a time resolution of 10 min. Details of the AMS have been described in previous publications (CitationJayne et al. 2000; CitationJimenez et al. 2003; CitationAllan et al. 2003a, Citation2004a, Citation2004b). The inlet temperature was controlled at a constant value (40 or 42°C, depending on ambient dew point) to dry particles in the sample air, as we did for our Tokyo measurements (CitationTakegawa et al. 2005). CitationTakegawa et al. (2005) assumed a particle collection efficiency (CE) of 0.5 for inorganic and organic compounds under dry conditions and found consistent results with PILS-IC and Sunset OC measurements. However, we assume different CE values in this study. This assumption is based on intercomparison with other, collocated instruments, as described in section 3.2. The sample air for the AMS was aspirated from the rooftop of the building using a stainless steel tube (inner diameter (ID): 10 mm). The length of the inlet tube was ∼6 m for the PRD campaign and ∼9 m for the CAREBEIJING campaign. Because of the relatively long sampling line and small sample flow of the AMS (∼84 cc min−1), an additional pump (operated at a flow rate of ∼10 l min−1) was used to reduce the residence time of air in the sampling line.
We should note that mass peaks at higher m/z (>100) were slightly broader than unit mass resolution most of the time during the measurement campaigns. This is because the radio frequency (RF) power supply for the QMS did not generate RF of sufficient amplitude at higher voltages (i.e., higher m/z). The fragment patterns of SO2− 4 and NO− 3 showed that the quality of mass peaks at lower m/z (<100) was normal. The resolution problem was relatively minor in terms of mass concentration because almost all m/z peaks for major inorganic compounds and the majority of organic peaks are included in the lower m/z range (<100). The effect of the broadening of an m/z peak on its peak height (transmission efficiency) and on neighboring peaks (leak) was estimated to be ∼30% at m/z 149. The contributions of the sum of organic signals at m/z> 100 to the total OM mass loadings were <15%. Based on these estimates, the total OM mass could have been overestimated by <5% due to the resolution problem. We do not make a correction for this possible overestimation.
The AMS standard analysis procedure does not include the m/z 28 signal originating from organic compounds (CO+) because the observed m/z 28 signal is dominated by nitrogen (N+ 2) in air. It has been shown that m/z 28 could be an important peak for the quantification of organic compounds (CitationZhang et al. 2005; CitationTakegawa et al. 2007). CitationTakegawa et al. (2007) estimated the average ratio of m/z 28 to m/z 44 to be 1.0 for Tokyo data during the summer of 2003 using particle time-of-flight data (P-ToF), in which m/z 28 signals from N+ 2 and organics can be separated. Here we estimated the m/z 28 signal from organics using the same method as CitationTakegawa et al. (2007). We selected time periods with moderately or very high loadings of OM (11 cases for PRD and 15 cases for CAREBEIJING) and calculated the m/z 28 and 44 signals from organics using the P-ToF data. The average ratio of m/z 28 to m/z 44 was estimated as the linear regression slope of the P-ToF m/z 28 versus 44 signals to be 0.85 for PRD and 1.55 for CAREBEIJING. The difference in the ratio between PRD and CAREBEIJING may be indicative of some difference in the chemical composition of organics. Conversely, the ratio of m/z 28 to m/z 44 signals originating from organics can provide additional information of organic aerosol if m/z 28 signals from organics can be well quantified.
The accuracy for major inorganic compounds (SO2− 4 and NO− 3) was estimated based on routine calibrations. The limit of detection (LOD) was estimated by placing a particle filter in the sampling line, as was done by CitationTakegawa et al. (2005). As for the PRD campaign, the accuracy of the measurements was estimated to be 14%. The LODs (10 min) were 0.09, 0.03, 0.03, 0.5, and 0.4 μg m–3 for SO2− 4, NO− 3, Cl−, NH+ 4, and OM, respectively. As for the CAREBEIJING campaign, the accuracy was estimated to be 18%, and the LODs were 0.1, 0.04, 0.02, 0.4, and 0.4 μg m−3 for SO2− 4, NO− 3, Cl−, NH+ 4, and OM, respectively.
2.2 Other, Collocated Instruments
The PILS-IC provides real-time measurements of water-soluble inorganic ions such as SO2− 4, NO− 3, Cl–, NH+ 4, sodium (Na+), potassium (K+), calcium (Ca2+), and other ions. The configuration of the PILS-IC used in this study is basically the same as that described by CitationOrsini et al. (2003) and CitationTakegawa et al. (2005). The PILS produces supersaturated water vapor by adding steam to the sample air. The particles in the sample air grow into droplets (diameter > 1 μm) large enough to be collected by an impactor. The collected droplets are introduced into an IC by adding a continuous liquid water flow. The sampling interval employed for this study was 15 min. The sample line for the PILS-IC was a stainless steel tube with ID of 10 mm and the length of ∼6 m. A PM1 cyclone (URG Corp., USA) was used for the PILS-IC inlet. As with the major inorganic ions used in this study, the LOD (15 min) was estimated to be 0.001, 0.002, 0.001, 0.02, and 0.07 μg m–3 for SO2– 4, NO– 3, Cl–, NH+ 4, and K+, respectively.
The Sunset EC/OC analyzer can measure mass concentrations of EC and OC with a time resolution of 1 h. Ambient particles were collected on a quartz filter and then analyzed based on the thermal-optical-transmittance method. The temperature protocol is based on that proposed by the National Institute for Occupational Safety and Health (NIOSH) (Birch and Cary 1996) with slight modification to fit our measurement conditions. A carbon denuder provided by the Sunset Laboratory was used to reduce the effects of volatile organic compounds (VOCs) on the OC measurements. The artifacts of VOCs, which were likely due to adsorption of VOCs on the quartz filter, were measured by placing a particle filter in the sample line. The OC concentrations were corrected by subtracting the VOC artifact from the measured OC concentrations (2.0 μgC m–3 for PRD and 1.9 μgC m–3 for CAREBEIJING). Without enough data points to estimate the LOD of OC (i.e., variability of the VOC artifact), we assume that the LOD was better than 2.0 μgC m–3 during the two campaigns. The sample line for the EC/OC analyzer was a stainless steel tube with ID of 10 mm. The length of the inlet tube was ∼6 m for the PRD campaign and ∼9 m for the CAREBEIJING campaign. A PM1 cyclone (URG Corp., USA) was used for the EC/OC inlet from July 17 to 31 during the PRD campaign and the entire period during the CAREBEIJING campaign. Because the sample flow rate of the EC/OC analyzer was 8 l min–1, an additional pump was used to match the flow rate required for the cyclone (16.7 l min–1).
The Berner impactor had cut points of 0.05, 0.14, 0.42, 1.2, 3.5, and 10 μm in the continuum/transition-regime aerodynamic diameter (see appendix) at ambient relative humidity. The sample flow rate was 75 l min–1 and the integration time ranged from 2.5 to 10 h during the CAREBEIJING campaign. Pre-heated (2 hours at 350°C) aluminum foils were used as sampling substrates. The aerosol samples were stored at–20°C and sent to Germany for chemical analysis. The analyzed parameters/species include total mass, major inorganic ions (SO2– 4, NO– 3, Cl–, NH+ 4, Na+, K+, and Ca2+), EC, OC, and others. Aliquots of the impactor foils were cut into small pieces and extracted in 1.5 ml deionized water (Nanopure, Barnstead, USA) by shaking and ultrasonication. After filtration through 0.45-μm pore size syringe filters (Acrodisc 13, Pall, USA), the extract was analyzed by a variety of techniques. Analysis of inorganic ions was carried out by ion chromatography (IC690, Metrohm, Switzerland). Organic and elemental carbon (OC/EC) was determined by a two-step thermal method (without optical correction for the separation of EC and OC) using a commercial carbon analyzer (C-mat 5500, Ströhlein, Germany). Analytical uncertainties are estimated to be ∼10% for major ions and ∼5% for OC. We use the sum of 0.05–0.14, 0.14–0.42, and 0.42–1.2 μm stages for intercomparison with the AMS.
3. RESULTS
3.1. Interference in AMS Mass Spectra
3.1.1. Enhanced Signals of Alkali Metals in AMS Mass Spectra
Time series of ion count rates at m/z 23 (23Na+), 39 (39K+ and C3H+ 3), 41 (41K+ and C3H+ 5), 43 (C3H+ 7 and C2H3O+), and 85 (85Rb+, C6H+ 13, etc.) during the PRD and CAREBEIJING campaigns are shown in and , respectively. “Beam open” indicates that the chopper position was off the particle beam so that all particles introduced into the vacuum chamber could reach the vaporizer. “Beam blocked” indicates that the chopper was completely blocking the particle beam so that no particles reached the vaporizer. The difference between the beam open and blocked modes, which is hereafter referred to simply as “signal,” indicates contributions from ambient particles. The dwell time for each mode was 4 s. Note that the ion rates at m/z 39 in the beam open position were sometimes saturated because they reached the upper limit of the analog-to-digital converter of the data acquisition system. Except for such saturation cases, the observed ratio of the m/z 41 to 39 signals shows excellent agreement with the isotopic ratio of 41K to 39K, indicating that these signals mainly originated from K+. The m/z 39 and 41 signals were much larger than the m/z 28 signal (air beam: AB) of ∼2.5 × 106 Hz, especially for the CAREBEIJING data.
FIG. 1 Time series of ion count rates at m/z 23 (23Na+), 39 (39K+ and C3H+ 3), 41 (41K+ and C3H+ 5), 43 (C3H+ 7 and C2H3O+), and 85 (85Rb+, C6H+ 13, etc.) during the PRD campaign. The solid and shaded lines represent the ion count rates in the beam open and blocked modes, respectively. The AMS-PILS intercomparison period (∼32 h) is shown as a horizontal bar. “Filter” indicates the time period (∼30 min) when ambient particles were removed using a particle filter.
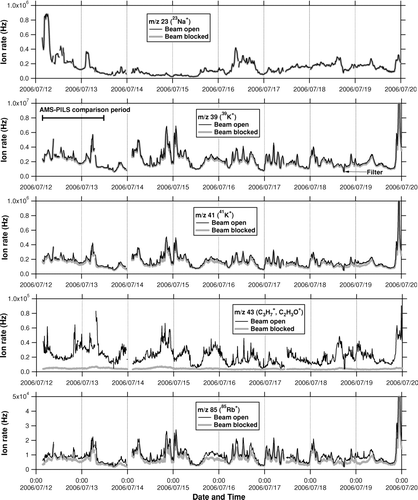
FIG. 2 Same as , but for the CAREBEIJING campaign. “Zero air” indicates the time period when particle-free purified air (zero air) was introduced in the AMS.
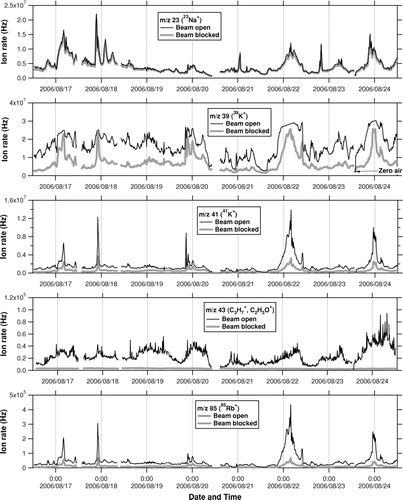
As for the m/z 85 and 87 signals, these signals likely originated mainly from Rb because the observed ratio of the m/z 87 to m/z 85 signals showed excellent agreement with the isotopic ratio of 87Rb to 85Rb. It is also likely that the m/z 133 signal mainly originated from Cs because it was highly correlated with the m/z 85 signal (r 2 = 0.97), with ∼8 times lower intensity (not shown). The m/z 85 and 133 signals were also significant because they were comparable to the m/z 43 signal, which is one of the major peaks of organic compounds. While the AMS standard analysis procedure assumes that m/z 85, 87, and 133 signals originate entirely from organic compounds, we have found large contributions from Rb+ and Cs+. This phenomenon has not been observed in our previous studies in Tokyo, and it has never been reported by other investigators to the best of our knowledge.
3.1.2. Possible Origins of Alkali Metal Signals
It is known that alkali metals (Na, K, Rb, and Cs) often coexist in naturally occurring rocks and sometimes as contaminates in metal products. In previous AMS experiments, it has been shown that large amounts of Na+ and K+ ions are emitted from the AMS vaporizer (porous tungsten) by surface ionization (e.g., CitationAllan et al. 2003b). In the present case, however, it is unlikely that the observed alkali metal ions originated entirely from the vaporizer because these ions were significantly reduced when ambient particles were removed using a particle filter; particle-free purified air (zero air) was introduced in the AMS; and routine calibrations were performed using pure ammonium nitrate (NH4NO3) particles during the campaigns. It should be noted that the Na+ and K+ ion rates could be dependent on the vaporizer position relative to the ionizer and also on the electrical potential of the vaporizer relative to the ionizer (so-called heater bias voltage), which was kept at a constant value unless the AMS was vented to the atmosphere. The fact that the alkali metal ions were significantly reduced during the filter/zero-air measurements and NH4NO3 calibrations indicates that this phenomenon was not due to inappropriate adjustment of the heater bias voltage but to some influence from ambient particles. Significant reduction of alkali metal ions occurred not only in the beam open mode but also in the beam blocked mode during the filter/zero-air measurements ( and ). In other words, the background levels of alkali metal ions were largely affected by the presence of ambient particles, which could have resulted in an underestimation of alkali metal signals from ambient particles (i.e., beam open minus blocked). A possible explanation for the enhanced background levels is that some fraction of alkali metal compounds bounced from the vaporizer, were adsorbed around the ionizer region, and vaporized more slowly than the dwell time (4 s) of the beam open mode. As with potassium (m/z 41), the average ratio of the beam blocked to open ion rates was 0.80 for PRD and 0.31 for CAREBEIJING, suggesting that K-containing particles during PRD could have been vaporized more slowly than those in CAREBEIJING. Note that we used m/z 41 here because the ion rates at m/z 39 in the beam open mode were sometimes saturated.
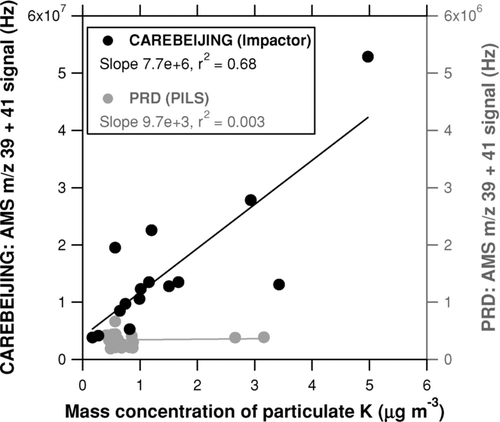
The significance of K in biomass/biofuel burning aerosols has been identified in many studies, as summarized by CitationAndreae and Merlet (2001). The presence of Rb in ambient aerosol has been identified by a single-particle mass spectrometer (CitationMurphy et al. 2006), although the signal intensity was very small. The presence of Cs in aerosol has never been reported to the best of our knowledge. Here we focus on K because measurements of Rb and Cs were not available. depicts the relationship between the sum of the m/z 39 and 41 signals and the mass concentration of particulate K measured by the PILS-IC (for PRD) or Berner impactor (for CAREBEIJING). The Berner impactor data are not available for the PRD campaign, as mentioned earlier, and thus the data for the PRD campaign is limited to July 12–13, 2006 (i.e., overlap of the AMS and PILS-IC operation). The m/z 39 signal was calculated from the m/z 41 signal using the isotopic ratio, because it was sometimes saturated. A positive correlation of the m/z 39 + 41 signal versus mass concentration of particulate K was found for the CAREBEIJING data. The ionization efficiency of K relative to nitrate (RIE K ) is estimated to be ∼8 × 102 for CAREBEIJING, assuming that the potassium compounds were detected by the AMS with the same collection efficiency as nitrate (CE = 1; see section 3.2.2). On the other hand, the m/z 39 + 41 signals during the PRD were much lower than those during the CAREBEIJING campaign and showed no correlation with the mass concentrations of K, although the temporal coverage was short. Note that the m/z 39 + 41 signals from ambient particles could have been underestimated due to possible effects of slow vaporization, especially for PRD. For comparison, CitationDrewnick et al. (2006) estimated an RIE K of 2.9 for aerosol in fireworks plumes using a time-of-flight AMS (ToF-AMS). The RIE K inferred from the CAREBEIJING data was much higher than that by CitationDrewnick et al. (2006).
FIG. 3 Relationship between the sum of the m/z 39 and 41 signals and the mass concentration of particulate K measured by the PILS-IC during the PRD campaign (shaded) and by the Berner impactor during the CAREBEIJING campaign (solid). The m/z 39 signal was calculated from the m/z 41 signal using the isotopic ratio because it was sometimes saturated. The lines represent the regression lines.
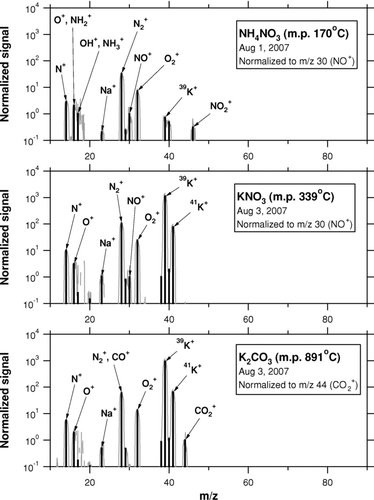
Laboratory experiments were performed after the campaign to investigate the mass spectra of K-containing particles (). Potassium nitrate (KNO3: melting point 339°C, dissociation at 400°C) and potassium carbonate (K2CO3: melting point 891°C) were chosen as examples of K-containing particles with different thermodynamic properties. K2CO3 can be regarded as “more refractory” than KNO3. NH4NO3 (melting point 170°C, dissociation at 210°C) was chosen for comparison. The thermodynamic parameters for these compounds were taken from the Chemical Dictionary Citation(1994). The experimental apparatus was the same as that described by CitationTakegawa et al. (2007). The high voltage applied to the QMS detector (electron multiplier) was properly adjusted so that the m/z 39 signal was not saturated. In , the signals have been normalized to m/z 30 (NO+) for NH4NO3 and KNO3 and normalized to m/z 44 (CO+ 2) for K2CO3. No large enhancements of m/z 39 and 41 were found in the mass spectrum of NH4NO3, while extremely large enhancements of those peaks were found in the mass spectra of KNO3 and K2CO3. The RIE K values for KNO3 and K2CO3 are found to be ∼2 × 103 and ∼1 × 103, respectively. The ratio of beam blocked to open ion rates at m/z 39 was 0.12 for KNO3 and 0.15 for K2CO3. Our laboratory experiments show the plausibility of very high RIE K but do not show significant dependence of RIE K and background levels at m/z 39 on refractoriness, at least for the two compounds tested.
FIG. 4 Mass spectra of ammonium nitrate (NH4NO3), potassium nitrate (KNO3), and potassium carbonate (K2CO3) obtained in the laboratory. The shaded lines represent the raw mass spectra and the sticks are the average signals for unit mass resolution.
In summary, the large m/z 39 and 41 signals observed during the PRD and CAREBEIJING campaigns cannot be explained by the vaporizer artifact alone, and they could potentially be explained by the presence of K-containing particles in ambient air. PRD, CAREBEIJING, and CitationDrewnick et al. (2006) give different AMS responses to K-containing particles, and the cause of the differences has not been fully determined. It should be noted that the AMS employs a two-stage ionization scheme: thermal decomposition and electron impact ionization (EI). Alkali metals tend to leave the surface of the vaporizer in an ionized state because of their low ionization potential and thus bypass the EI process that is necessary for other species to be detected by QMS. The observed difference in the AMS response to K between CitationDrewnick et al. (2006) and our cases may be partially due to some differences in the vaporizer configuration and perhaps in the chemical form of potassium. Because of the uncertainties described above, the relationship between the m/z 39 + 41 signal and K concentration in should be regarded as qualitative.
Finally, effects of alkali metals on the quantification of other species are discussed. The ion rates at m/z 39 were extremely high during the entire period, resulting in significant leak into m/z 38 even though the QMS was operated at unit mass resolution. Although m/z 38 (C3H+ 2) is one of the m/z peaks from organic compounds, we ignore this peak for the quantification of OM because of its minor contribution in most cases. There was another quantification issue regarding the m/z 41 signal. It has been identified in many environments that m/z 41 is one of the major peaks of organic compounds and should be included for calculating the mass concentrations of OM. In the AMS standard analysis procedure, the m/z 41 signal from organics is calculated by subtracting the 41K+signal estimated from 39K+, which cannot be applied in the case of this study because the ion rates at m/z 39 were sometimes saturated. We estimate the m/z 41 signal from organics using m/z 27 and 55 on the basis of a delta analysis: i.e., m/z 41 (organics) = (m/z 27 + m/z 55)/2. This procedure is conceptually the same as those for some organic m/z peaks with significant interference from other compounds (CitationAllan et al. 2004b). As for m/z 85, 87, and 133, we ignore these m/z peaks for the present analysis because the m/z 85, 87, and 133 signals originating from organics are generally minor. The interference from alkali metals discussed here may not be important for recently developed high-resolution ToF-AMS (CitationDeCarlo et al. 2006).
3.2. Intercomparison
In the following sections we show intercomparisons of the AMS with the other, collocated instruments for both the PRD and CAREBEIJING campaigns. Based on the work of CitationAllan et al. (2004a) and CitationTakegawa et al. (2005), the inlet temperature was controlled at a constant value (40 or 42°C) to dry particles in the sample air, and we assumed CE = 0.5 for preliminary intercomparisons. We have found reasonable agreements between the AMS and the other measurements for the PRD data but significant overestimation by the AMS for the CAREBEIJING data. These results suggest that the assumption of CE = 0.5 could be applicable to the PRD data but may not be applicable to the CAREBEIJING data even though the sample air was dried in both two cases.
The assumption of CE = 0.5 has been widely used for many AMS studies. However, it is rather empirical and there is no physical basis to apply it in all cases. As mentioned earlier, the assumption of CE = 0.5 has been tested mostly in urban areas, where emission sources are well defined and often dominated by diesel/gasoline vehicles (e.g., CitationDrewnick et al. 2003, CitationJimenez et al. 2003; CitationTakegawa et al. 2005). On the other hand, CE has not been well investigated in mixtures of urban pollution and other sources such as coal combustion and biomass/biofuel burning. Without an established method to determine the AMS CE values, we assume CE = 0.5 for the PRD data and CE = 1 for the CAREBEIJING data for all species (SO2– 4, NO– 3, Cl–, NH+ 4, and OM). Because CE is smaller than or equal to unity, the mass concentrations reported for the CAREBEIJING campaign should be regarded as lower limits.
In the following sections we use a bivariate regression analysis that considers the errors in both variables (CitationPress et al. 1992). As for the intercomparison of inorganic compounds, the linear regression slope is forced through zero because of the limited number of data points.
3.2.1. PRD Campaign
shows time series of SO2– 4 measured by the AMS and PILS-IC during the PRD campaign. Although the number of data points for this intercomparison is limited, we see that the concentration level and temporal variation of SO2– 4 measured by the AMS agreed well with those by the PILS-IC. shows plots of the AMS versus PILS-IC data for the inorganic compounds. We use 1-h average data for the plots. Because the AMS cannot detect sodium chloride (NaCl) under normal operating conditions (600°C vaporizer), we estimated non-sea salt (nss) Cl– for the PILS-IC data assuming that all of the observed Na+ originated from sodium chloride (NaCl) in sea salt. The nss-Cl– concentration estimated above is a lower limit because Cl– from sea salt could be partially/fully depleted by reaction between NaCl and nitric acid (HNO3).
FIG. 5 Time series of SO2– 4 measured by the AMS (solid) and PILS-IC (shaded) during the PRD campaign.
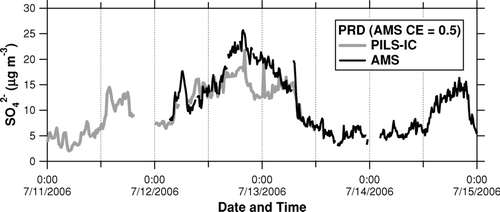
FIG. 6 Plots of AMS versus PILS-IC data for inorganic compounds during the PRD campaign. We use 1-h average data for the intercomparison. The solid lines represent the regression lines with forced through origins. The dot-dashed line indicates 1:1 correspondence line. The dashed lines indicate 1.5:1 and 1:1.5 correspondence lines.
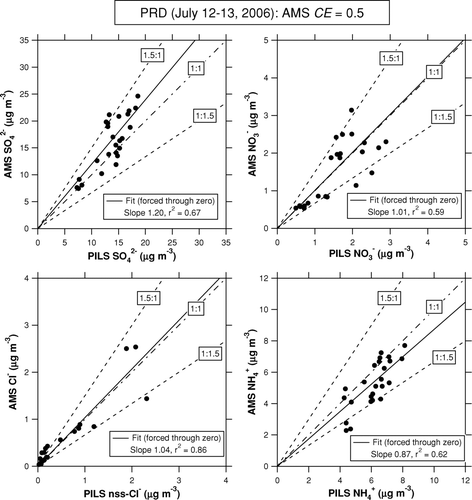
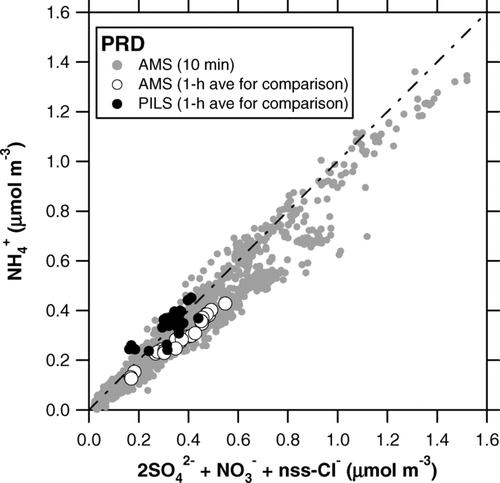
For major inorganic compounds (SO2– 4, NO– 3, and NH+ 4), 75–90% of the data points lie within the 50% range, and the linear regression slopes range from 0.87 to 1.20. This result indicates that agreement between the AMS and PILS-IC during this period was 50% with respect to individual 1-h data points and 20% on average. The agreement between the AMS and PILS-IC is also good for Cl–, although some data points were close to the limit of detection. It should be noted that the AMS tends to overestimate the PILS-IC values for SO2– 4 and underestimate those for NH+ 4. These results may not be consistent if we consider the ion balance. shows a plot of the molar concentration of NH+ 4 versus 2SO2– 4 + NO– 3 + nss-Cl–. The PILS-IC data show that these anions were fully neutralized by NH+ 4 during the intercomparison period. On the other hand, the AMS data show that the anions were not fully neutralized in some cases, including the intercomparison period. The low cation/anion ratios in the AMS data were systematically found under southeasterly conditions (i.e., transport of polluted air from Guangzhou city), suggesting that the deficit in AMS NH+ 4 was not primarily due to variability in RIE NH4, which should be independent of wind direction, but to the possible presence of acidic particles in polluted air from Guangzhou. However, this is not a definitive conclusion at this stage, considering the inconsistency with the PILS-IC measurements during the intercomparison period.
FIG. 7 Plot of the molar concentration of NH+ 4 versus 2SO2– 4+ NO– 3+ nss-Cl– during the PRD campaign. The shaded circles represent the 10-min AMS data during the entire period. The open and solid circles represent the 1-h averaged AMS and PILS data, respectively, for the intercomparison. The dot-dashed line indicates 1:1 correspondence line.
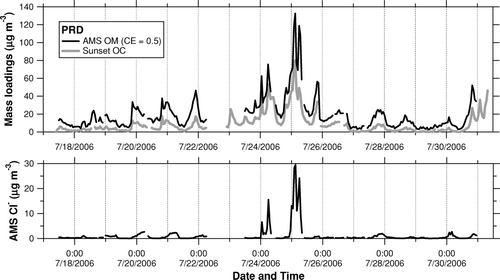
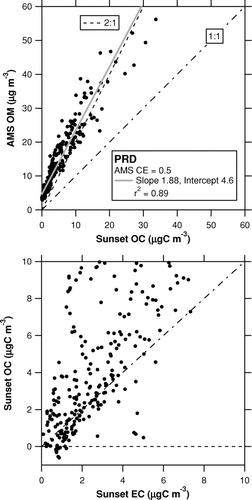
shows time series of AMS OM and Sunset OC and that of AMS Cl–. The temporal variation of the AMS OM tracks very well with that of the Sunset OC. The very high concentrations of OM and OC on July 24–25, which were associated with large enhancements of Cl–, were likely due to influence of local emission sources (Takegawa et al. manuscript in preparation). A scatterplot of AMS OM versus Sunset OC is displayed in . The linear regression slope and intercept of the correlation are calculated to be 1.88 and 4.6 μg m–3, respectively. The data affected by the local emission sources on July 24–25 were excluded for the linear regression analysis. The slope of 1.88 is within the range of the OM/OC ratios of 1.6–2.1 given by CitationTurpin and Lim (2001). There is a non-negligible offset of 4.6 μg m–3 in the regression of AMS OM versus Sunset OC.
FIG. 8 Time series of AMS OM (top: solid), Sunset OC (top: shaded), and AMS Cl− (bottom) during the PRD campaign.
also displays a scatterplot of Sunset OC versus EC concentrations. The OC concentrations sometimes show slightly negative values (as low as–0.5 μgC m–3) at EC concentrations of ∼1 μgC m–3. Considering that OC concentrations are generally higher than EC concentrations in many cases (i.e., OC/EC ratios > 1), the non-negligible offset could be mainly due to the subtraction of VOC artifacts in the Sunset OC data.
FIG. 9 Scatterplot of AMS OM versus Sunset OC (top) and that of Sunset OC versus EC (bottom) during the PRD campaign. The shaded line represents the regression line. The dot-dashed and dashed lines (top) indicate 1:1 and 2:1 correspondence lines, respectively.
3.2.2. CAREBEIJING Campaign
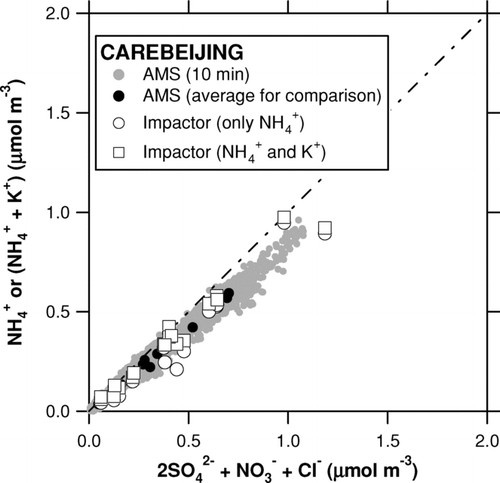
shows time series of SO2– 4 measured by the AMS and Berner impactor. The 10-min AMS data were averaged according to the integration time of the Berner impactor. Despite some missing AMS data, the overlap of the two instruments covers a substantial time period during the CAREBEIJING campaign. shows plots of the AMS versus Berner impactor for the inorganic compounds. We do not apply a correction for nss-Cl– to the Berner impactor data because it yields many data points with negative nss-Cl– concentrations. This would be a reasonable assumption since the measurement site is removed from coastal regions. shows a plot of the molar concentration of NH+ 4 (or NH+ 4+ K+) versus 2SO2– 4+ NO– 3+ Cl–. Both AMS and Berner impactor data show that the measured concentrations of NH+ 4 were slightly lower than those required to fully neutralize the observed anions during the entire period. If we add K+ from the Berner impactor analysis, the data points come closer to the 1:1 correspondence line. This result suggests that SO2– 4, NO– 3, Cl– were mostly neutralized by NH+ 4, and that K+ could have also contributed to the neutralization of SO2– 4, NO– 3, and Cl–.
FIG. 10 Time series of SO2− 4 measured by the AMS (shaded line and solid circles) and Berner impactor (open squares). The shaded line represents the 10 min AMS data and the solid circles represent those averaged according to the integration time of the Berner impactor.
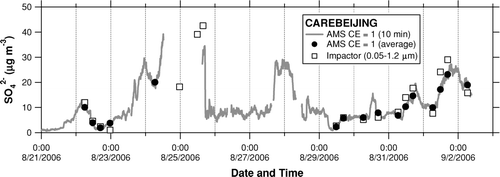
FIG. 11 Plots of the AMS versus Berner impactor data for inorganic compounds during the CAREBEIJING campaign. The solid lines represent the regression lines with forced through origins. The dot-dashed line indicates 1:1 correspondence line. The dashed lines indicate 1.5:1 and 1:1.5 correspondence lines.
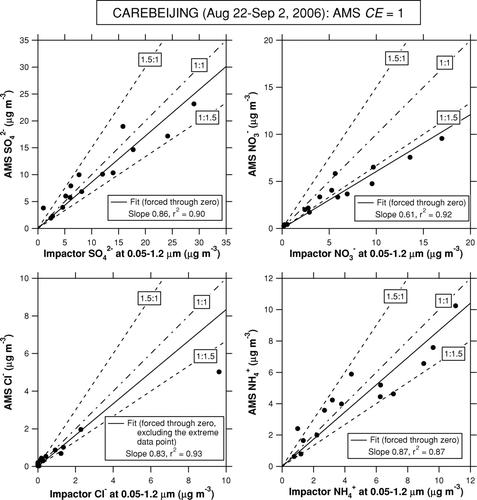
FIG. 12 Plot of the molar concentration of NH+ 4 (or NH+ 4 + K+) versus 2SO2− 4 + NO− 3 + Cl− during the CAREBEIJING campaign. The shaded circles represent the 10 min AMS data and the solid circles represent those averaged according to the integration time of the Berner impactor. The AMS data include only NH+ 4 as cations. The open circles represent the Berner impactor data including only NH+ 4 as cations and the open squares represent those including NH+ 4 and K+. The dot-dashed line indicates 1:1 correspondence line.
As for SO2– 4 and NH+ 4 in , 80–95% of the data points lie within the 50% range, and the linear regression slopes are 0.86–0.87. This result indicates that agreement between the AMS and Berner impactor for SO2– 4 and NH+ 4 was 50% with respect to individual impactor samples and better than 15% on average. On the other hand, AMS NO– 3 was systematically lower than Berner impactor NO– 3. This may be either due to presence of some nitrate compounds with lower CE (i.e., higher vaporization/dissociation temperature) than NH4NO3, to positive artifacts in the Berner impactor data, or to a difference in the size cut between the AMS and Berner impactor. As for the first possibility, sodium nitrate (NaNO3) and calcium nitrate (Ca(NO3)2) would have made minor contributions because of low concentrations of Na+ (<0.6 μg m–3) and Ca2+ (<0.5 μg m–3) identified in the Berner impactor samples at 0.05–1.2 μm.
shows the size distribution of SO2– 4 and NO– 3 measured by the AMS and Berner impactor for two cases (August 30 and September 1). The AMS sizing (free molecular regime aerodynamic diameter (dva ) under dry conditions) is different than the impactor sizing (continuum/transition-regime aerodynamic diameter (da ) at ambient relative humidity), as described in the appendix. Nevertheless, the comparison of the size distribution can provide some insights into the difference in NO– 3 concentrations between the AMS and Berner impactor. The first case (14:30–19:00 on August 30) is a period when the AMS NO– 3 showed good agreement with the impactor NO– 3. The second case (9:30–14:00 on September 1) is a period when the AMS NO– 3 was significantly lower than the impactor NO– 3. shows the ratio of the AMS mass to the impactor mass (M AMS/M 0.05–1.2) as a function of the contribution of the 0.42–1.2-μm size range (M 0.42–1.2/M 0.05–1.2). As for NO– 3, the AMS tends to underestimate the impactor as the contribution of the 0.42–1.2-μm size range value increases. As for SO2– 4, however, there is no such tendency. A possible explanation for this feature is that a significant fraction of NO– 3 was externally mixed with SO2– 4 and distributed around 1 μm, where the transmission efficiency of the AMS aerodynamic lens is largely reduced. Positive artifacts in the Berner impactor data were unlikely the cause of this feature because they should not be size-dependent. From the preceding results, it is suggested that the underestimation of NO– 3 by the AMS compared to the Berner impactor could be due to the difference in the size cut between the AMS and Berner impactor.
FIG. 13 Size distribution of SO2– 4 and NO– 3 measured by the AMS (solid) and Berner impactor (shaded) for two cases during the CAREBEIJING campaign (August 30 and September 1).
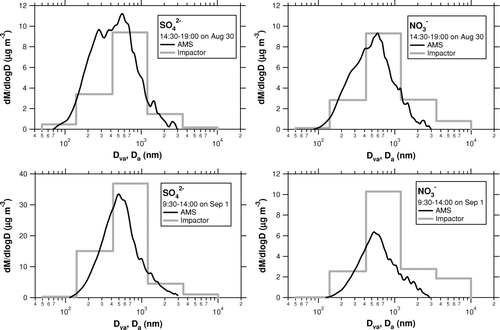
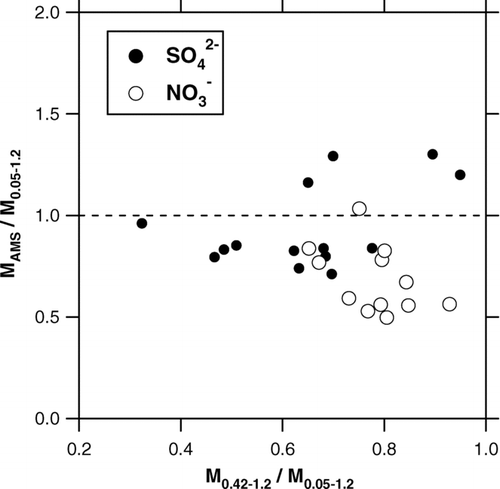
FIG. 14 Ratio of the AMS mass to the Berner impactor mass (MAMS/M 0.05−1.2) as a function of the contribution of the 0.42–1.2-μm size range (M 0.42−1.2/M 0.05−1.2) during the CAREBEIJING campaign. The solid and open circles represent SO2– 4 and NO– 3, respectively.
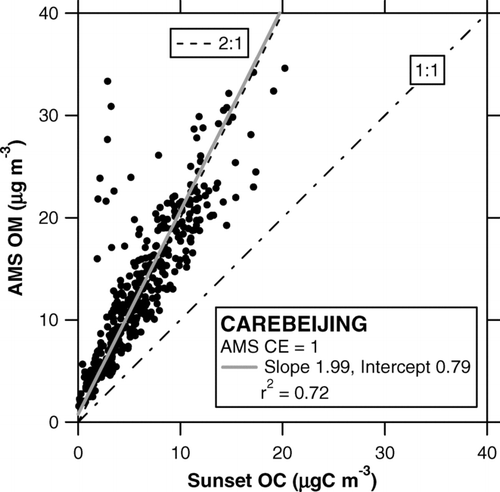
shows time series of AMS OM, Sunset OC, and Berner impactor OC. The OC concentrations measured by the two independent methods agreed reasonably well. Except for the data point at 6:30 on September 2, the linear regression slope and intercept of the Sunset versus impactor OC are calculated to be 1.07 and–0.7 μgC m–3, respectively (r 2 = 0.70). shows a scatterplot of AMS OM versus Sunset OC. The linear regression slope and intercept of the correlation are calculated to be 1.99 and 0.79 μg m–3, respectively. It appears that the offset in the Sunset OC due to the subtraction of VOC artifacts was not significant during the CAREBEIJING campaign as compared to the PRD campaign. The slope of 1.99 is within the range of the OM/OC ratio (1.6–2.1) given by CitationTurpin and Lim (2001).
FIG. 15 Time series of AMS OM (solid line), Sunset OC (shaded line), and Berner impactor OC (open squares) during the CAREBEIJING campaign.
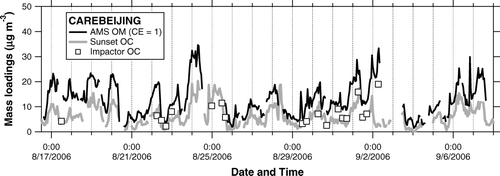
FIG. 16 Scatterplot of AMS OM versus Sunset OC during the CAREBEIJING campaign. The shaded line represents the regression line. The dot-dashed and dashed lines indicate 1:1 and 2:1 correspondence lines, respectively.
4. DISCUSSION
The AMS CE issue has been extensively studied by several investigators. CitationHuffman et al. (2005) introduced an equation to describe CE as a function of particle transmission efficiency through the aerodynamic lens (EL ), particle focusing efficiency onto the vaporizer (ES ), and particle vaporization (or bouncing) efficiency at the vaporizer (Eb ).
Previous studies have proposed that ratios of inorganic compounds (NH+ 4/SO2– 4 or NO– 3/SO2– 4 ratio) could be used for empirically estimating CE for ambient particles (CitationCrosier et al. 2007; CitationKleinman et al. 2007) and for laboratory-generated particles (CitationMatthew et al. 2008). In our cases, however, no systematic dependence of CE (ratio of AMS-SO2– 4 to PILS-/Impactor-SO2– 4) on NH+ 4/SO2– 4 or NO– 3/SO2– 4 ratio has been found in the PRD and CAREBEIJING data (not shown). Considering that inorganic compounds are often internally mixed with a substantial amount of organic compounds (e.g., CitationMurphy et al. 2006), the estimate of CE based on the ratios of inorganic compounds would be applicable only to some specific situations. Without information on thermodynamic properties (phase, vaporization temperature, etc.) and mixing state of organic aerosol in our cases, it may not be very meaningful to consider an empirical formula for estimating CE based on the AMS composition data. In fact, we do not find a systematic dependence of CE on OM/SO2– 4 ratio in the PRD and CAREBEIJING data.
Although it is highly speculative, a possible explanation is that the large K+ signals and CE = 1 for the CAREBEIJING data could both be related to influence from biomass/biofuel burning. CitationLi et al. (2003) have found “tar ball” particles in filter samples obtained in biomass burning smoke in Africa. Such tar ball particles could be very sticky and may not efficiently bounce on the AMS vaporizer. However, this hypothesis assumes that these organic compounds were almost always internally mixed with major inorganic compounds and affected the bouncing efficiencies of particles to a large extent during the entire period, which has not been validated at the present stage. In addition, this hypothesis may not explain the difference between PRD and CAREBEIJING. Further experiments are needed to systematically investigate AMS CE issues in various environments, especially in regions with significant influences from biomass/biofuel burning.
5. CONCLUSIONS
An Aerodyne AMS was deployed in China during the PRD and CAREBEIJING campaigns in the summer of 2006. The performance of the AMS during the two campaigns was evaluated. We found extremely large signals from potassium at m/z 39 and 41 in the AMS mass spectra. This phenomenon cannot be accounted for by a vaporizer artifact alone, and it may be explained by the presence of K-containing particles in ambient air. The interference could be important for the quantification of organic compounds because m/z 41 is one of the most important peaks from organic compounds.
The AMS measurements were compared with other, collocated measurements including a PILS-IC, a Sunset Laboratory OC analyzer, and a Berner impactor sampler. For major inorganic compounds, we found good agreement between the AMS and the other instruments when we assume an AMS CE = 0.5 for the PRD data and CE = 1.0 for the CAREBEIJING data. The agreement for SO2– 4 and NH+ 4 was ∼20% on average for the two campaigns. The AMS underestimated the impactor data by ∼40% for NO– 3 during the CAREBEIJING campaign, which may have been due to the difference in the size cut between the AMS and the impactor. The regression slope of AMS OM versus Sunset OC was 1.88 for PRD and 1.99 for CAREBEIJING, which are within the range of OM/OC ratios given by CitationTurpin and Lim (2001). The two intercomparison results suggest that the AMS CE could be significantly different in different locations. It is possible that the large K+ signals and the variability in the CE values could both be related to influence from biomass/biofuel burning, although this hypothesis is highly speculative and needs further experiments in future studies.
APPENDIX
The definitions of aerodynamic diameters given below are based on the work of CitationDeCarlo et al. (2004). In the following equations, the dynamic shape factors in the continuum, transition, and free-molecular regimes are assumed to be the same. The aerodynamic diameter, da , is given by
The transmission efficiency of the aerodynamic lens (EL ) is one of the most important quantification issues for the AMS. CitationJayne et al. (2000) showed that EL ∼ 1 at dva = 60–500 nm, although recent experiments suggest that EL starts to decrease above ∼300 nm (CitationLiu et al. 2006). NH4NO3 particles were used in these previous studies because they can be well focused (ES ∼ 1) and efficiently vaporized (Eb ∼ 1). We also tested the transmission efficiency of our aerodynamic lens system at a larger dva region, as shown in . The particle generation system used in this study was basically the same as that described by CitationTakegawa et al. (2005). Dry, monodisperse particles were generated using an atomizer (Model 3076, TSI, Inc., USA) and a differential mobility analyzer (DMA: Model 3080, TSI, Inc., USA). Number concentrations of the monodisperse particles measured by the AMS were compared with those measured by a condensation particle counter (CPC: Model 3022A, TSI, Inc., USA), and the AMS CE was derived as the ratio of AMS to CPC counts. Effects of multiply charge particles in the CPC data were not corrected, and thus the derived AMS CE may have been slightly underestimated. We used KNO3 (ρ = 2.10 g cm–3) and sodium nitrate (NaNO3: ρ = 2.30 g cm–3) in addition to NH4NO3 (ρ = 1.72 g cm–3) in order to generate aerodynamically larger particles. The vaporizer temperature was set to 600°C for NH4NO3 and 800°C for the others because of the higher boiling/melting temperatures of KNO3 and NaNO3 than NH4NO3. The three experiments gave a similar dependence of AMS CE on dva , although the CE values for KNO3 and NaNO3 at dva < 600 nm were almost constant at 0.8–0.9. This result is likely due to the lower Eb for KNO3 and NaNO3 even at 800°C. This is not important because we focus on the dependence of AMS CE on dva . The three experiments gave systematically higher CE values at dva > 300 nm than the theoretical curve from a computational fluid dynamics calculation (FLUENT model: D. R. Worsnop, personal communication).
FIG. A1 AMS collection efficiency measured using laboratory-generated particles. The solid, open, and shaded circles are the results for NH4NO3, NaNO3, and KNO3, respectively. The values in parentheses in the legend indicate the vaporizer temperature. The solid line indicates the theoretical curve from a computational fluid dynamics calculation (FLUENT model: D. R. Worsnop, personal communication).
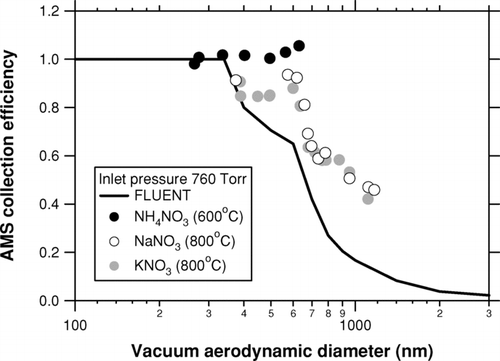
Consequently, our results suggest that EL can be approximated as 1 at dva < 600 nm (at least dva > 260 nm) and that EL is higher than the FLUENT model calculation at dva > 300 nm. We also suggest that AMS size cut can be characterized by 50% cut-off at dva = 1 μm. On the other hand, the classical PM1 size cut is defined as the 50% cut-off at da (not dva ) = 1 μm. The approximation of the AMS size cut to classical PM1 is applicable when ρ/χ of accumulation mode particles is close to unity. Additional information on particle density and shape may reduce uncertainties in comparison of AMS data with other measurements having a PM1 size cut.
Acknowledgments
The authors thank J. D. Allan, J. L. Jimenez, Q. Zhang, and D. R. Worsnop for providing the AMS analysis software. This study was funded by the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Related Research Data
REFERENCES
- Allan , J. D. , Jimenez , J. L. , Williams , P. I. , Alfarra , M. R. , Bower , K. N. , Jayne , J. T. , Coe , H. and Worsnop , D. R. 2003a . Quantitative Sampling using an Aerodyne Aerosol Mass Spectrometer, 1, Techniques of Data Interpretation and Error Analysis . J. Geophys. Res. , 108 : 4090 doi:10.1029/2002JD002358
- Allan , J. D. , Alfarra , M. R. , Bower , K. N. , Williams , P. I. , Gallagher , M. W. , Jimenez , J. L. , McDonald , A. G. , Nemitz , E. , Canagaratna , M. R. , Jayne , J. T. , Coe , H. and Worsnop , D. R. 2003b . Quantitative Sampling using an Aerodyne Aerosol Mass Spectrometer, 2, Measurements of Fine Particulate Chemical Composition in Two U.K. Cities . J. Geophys. Res. , 108 : 4091 doi:10.1029/2002JD002359
- Allan , J. D. , Bower , K. N. , Coe , H. , Boudries , H. , Jayne , J. T. , Canagaratna , M. R. , Millet , D. B. , Goldstein , A. H. , Quinn , P. K. , Weber , R. J. and Worsnop , D. R. 2004a . Submicron Aerosol Composition at Trinidad Head, CA during ITCT 2K2, Its Relationship with Gas Phase Volatile Organic Carbon and Assessment of Instrument Performance . J. Geophys. Res. , 109 : D23S24 doi:10.1029/2003JD004208
- Allan , J. D. , Delia , A. E. , Coe , H. , Bower , K. N. , Alfarra , M. R. , Jimenez , J. L. , Middlebrook , A. M. , Drewnick , F. , Onasch , T. B. , Canagaratna , M. R. , Jayne , J. T. and Worsnop , D. R. 2004b . A Generalised Method for the Extraction of Chemically Resolved Mass Spectra from Aerodyne Aerosol Mass Spectrometer Data (Technical Note) . J. Aerosol Sci. , 35 : 909 – 922 .
- Andreae , M. O. and Marlet , P. 2001 . Emissions of Trace Gases and Aerosols from Biomass Burning . Global Biogeochem. Cycles , 15 : 955 – 966 .
- Chemical Dictionary . 1994 . Edited by: Ooki , M. , Oosawa , T. , Tanaka , M. and Chihara , H. Tokyo, , Japan : Tokyo Kagaku Dozin, Co., Ltd. . (in Japanese)
- Crosier , J. , Allan , J. D. , Coe , H. , Bower , K. N. , Formenti , P. and Williams , P. I. 2007 . Chemical Composition of Summertime Aerosol in the Po Valley (Italy), Northern Adriatic and Black Sea . Q. J. R. Meterol. Soc. , 133 ( S1 ) : 61 – 75 .
- DeCarlo , P. , Slowik , J. G. , Worsnop , D. R. , Davidovits , P. and Jimenez , J. L. 2004 . Particle Morphology and Density Characterization by Combined Mobility and Aerodynamic Diameter Measurements, Part 1: Theory . Aerosol Sci. Technol. , 38 : 1185 – 1205 .
- DeCarlo , P. F. , Kimmel , J. R. , Trimborn , A. , Northway , M. J. , Jayne , J. T. , Aiken , A. C. , Gonin , M. , Fuhrer , K. , Horvath , T. , Docherty , K. S. , Worsnop , D. R. and Jimenez , J. L. 2006 . Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer . Anal. Chem. , Article 10.1021/ac061249n
- Drewnick , F. , Schwab , J. J. , Hogrefe , O. , Peters , S. , Husain , L. , Diamond , D. , Weber , R. and Demerjian , K. L. 2003 . Intercomparison and Evaluation of Four Semi-Continuous PM2.5 Sulfate Instruments . Atmos. Environ. , 37 : 3335 – 3350 .
- Drewnick , F. , Hings , S. S. , Curtius , J. , Eerdekens , G. and Williams , J. 2006 . Measurement of Fine Particulate and Gas-phase Species During the New Year's Fireworks 2005 in Mainz, Germany . Atmos. Environ. , 40 : 4316 – 4327 .
- Duan , F. K. , Liu , X. D. , Yu , T. and Cachier , H. 2004 . Identification and Estimate of Biomass Burning Contribution to the Urban Aerosol Organic Carbon Concentrations in Beijing . Atmos. Environ. , 38 : 1275 – 1282 .
- Guinot , B. , Cachier , H. , Sciare , J. , Tong , Y. , Xin , W. and Jianhua , Y. 2007 . Beijing Aerosol: Atmospheric Interactions and New Trends . J. Geophys. Res. , 112 : D14314 doi:10.1029/2006JD008195
- Huffman , J. A. , Jayne , J. T. , Drewnick , F. , Aiken , A. C. , Onasch , T. , Worsnop , D. R. and Jimenez , J. L. 2005 . Design, Modeling, Optimization, and Experimental Tests of a Particle Beam Width Probe for the Aerodyne Aerosol Mass Spectrometer . Aerosol Sci. Technol. , 39 ( 12 ) : 1143 – 1163 .
- Jayne , J. T. , Leard , D. C. , Zhang , X. F. , Davidovits , P. , Smith , K. A. , Kolb , C. E. and Worsnop , D. R. 2000 . Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles . Aerosol Sci. Technol. , 33 ( 1–2 ) : 49 – 70 .
- Jimenez , J. L. , Jayne , J. T. , Shi , Q. , Kolb , C. E. , Worsnop , D. R. , Yourshaw , I. , Seinfeld , J. H. , Flagan , R. C. , Zhang , X. , Smith , K. A. , Morris , J. W. and Davidovits , P. 2003 . Ambient Aerosol Sampling using the Aerodyne Aerosol Mass Spectrometer . J. Geophys. Res. , 108 : 8425 doi:10.1029/2001JD001213
- Kleinman , L. I. , Daum , P. H. , Lee , Y. N. , Senum , G. I. , Springston , S. R. , Wang , J , Berkowitz , C. , Hubbe , J. , Zaveri , R. A. , Brechtel , F. J. , Jayne , J. T. , Onasch , T. B. and Worsnop , D. R. 2007 . Aircraft Observations of Aerosol Composition and Ageing in New England and Mid-Atlantic States during the Summer 2002 New England Air Quality Study Field Campaign . J. Geophys. Res. , 112 : D09310 doi:10.1029/2006JD007786
- Li , J. , Posfai , M. , Hobbs , P. V. and Buseck , P. R. 2003 . Individual Aerosol Particles from Biomass Burning in Southern Africa: 2. Compositions and Aging of Inorganic Particles . J. Geophys. Res. , 108 ( D13 ) : 8484 doi:10.1029/2002JD002310
- Liu , P. S. K. , Deng , R. , Smith , K. A. , Williams , L. R. , Jayne , J. T. , Canagaratna , M. R. , Moore , K. , Onasch , T. B. , Worsnop , D. R. and Deshler , T. 2006 . Transmission Efficiency of an Aerodynamic Focusing Lens System: Comparison of Model Calculations and Laboratory Measurements for the Aerodyne Aerosol Mass Spectrometer . Aerosol Sci. Technol. , 41 ( 8 ) : 721 – 733 .
- Matthew , B. M. , Middlebrook , A. M. and Onasch , T. B. 2008 . Collection Efficiencies in an Aerodyne Aerosol Mass Spectrometer as a Function of Particle Phase for Laboratory Generated Aerosols . Aerosol Sci. Technol. , 42 : 884 – 898 .
- Murphy , D. M. , Cziczo , D. J. , Froyd , K. D. , Hudson , P. K. , Matthew , B. M. , Middlebrook , A. M. , Peltier , R. E. , Sullivan , A. , Thomson , D. S. and Weber , R. J. 2006 . Single-Particle Mass Spectrometry of Tropospheric Aerosol Particles . J. Geophys. Res. , 111 : D23S32 doi:10.1029/2006JD007340
- Orsini , D. , Ma , Y. , Sullivan , A. , Sierau , B. , Baumann , K. and Weber , R. 2003 . Refinements to the Particle-Into-Liquid Sampler (PILS) for Ground and Airborne Measurements of Water Soluble Aerosol Composition . Atmos. Environ. , 37 : 1243 – 1259 .
- Press , W. H. , Teukolsky , S. A. , Vetterling , W. T. and Flannery , B. P. 1992 . Numerical Recipes in C: The Art of Scientific Computing , Cambridge : Cambridge University Press . New York, Port Chester, Melbourne, Sydney
- Salcedo , D. , Onasch , T. B. , Canagaratna , M. R. , Dzepina , K. , Huffman , J. A. , Jayne , J. T. , Worsnop , D. R. , Kolb , C. E. , Weimer , S. , Drewnick , F. , Allan , J. D. , Delia , A. E. and Jimenez , J. L. 2007 . Technical Note: Use of a Beam Width Probe in an Aerosol Mass Spectrometer to Monitor Particle Collection Efficiency in the Field . Atmos. Chem. Phys. , 7 : 549 – 556 .
- Streets , D. G. , Bond , T. C. , Carmichael , G. R. , Fernandes , S. D. , Fu , Q. , He , D. , Klimont , Z. , Nelson , S. M. , Tsai , N. Y. , Wang , M. Q. , Woo , J.-H. and Yarber , K. F. 2003 . An Inventory of Gaseous and Primary Aerosol Emissions in Asia in the Year 2000 . J. Geophys. Res. , 108 doi:10.1029/2002JD003093
- Takegawa , N. , Miyazaki , Y. , Kondo , Y. , Komazaki , Y. , Miyakawa , T. , Jimenez , J. L. , Jayne , J. T. , Worsnop , D. R. , Allan , J. D. and Weber , R. J. 2005 . Characterization of an Aerodyne Aerosol Mass Spectrometer (AMS): Intercomparison with Other Aerosol Instruments . Aerosol Sci. Technol. , 39 : 760 – 770 .
- Takegawa , N. , Miyakawa , T. , Kawamura , K. and Kondo , Y. 2007 . Contribution of Selected Dicarboxylic and ω-Oxocarboxylic Acids in Ambient Aerosol to the m/z 44 Signal of an Aerodyne Aerosol Mass Spectrometer . Aerosol Sci. Technol. , 41 : 418 – 437 .
- Turpin , B. J. and Lim , H. J. 2001 . Species Contributions to PM2.5 Mass Concentrations: Revisiting Common Assumptions for Estimating Organic Mass . Aerosol Sci. Technol. , 35 : 602 – 610 .
- Zhang , Q. , Alfarra , M. R. , Worsnop , D. R. , Allan , J. D. , Coe , H. , Canagaratna , M. R. and Jimenez , J. L. 2005 . Deconvolution and Quantification of Hydrocarbon-Like and Oxygenated Organic Aerosols Based on Aerosol Mass Spectrometry . Environ. Sci. Technol. , 39 : 4938–4952 doi:10.1021/es048568l
- Zhang , Q. 2007 . Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenically-Influenced Northern Hemisphere Midlatitudes . Geophys. Res. Lett. , 34 ( L13801 ) doi:10.1029/2007GL029979
- Zheng , M. , Salmon , L. G. , Schauer , J. J. , Zeng , L. M. , Kiang , C. S. , Zhang , Y. H. and Cass , G. R. 2005 . Seasonal Trends in PM2.5 Source Contributions in Beijing, China . Atmos. Environ. , 39 : 3967 – 3976 .