Abstract
The development of a thermal desorption chemical ionization ion trap mass spectrometer for the chemical characterization of ultrafine aerosol particles is reported and first experimental results are presented. Atmospheric particles are size-classified and collected using a unipolar charger, a radial differential mobility analyzer and an electrostatic precipitator, and analyzed after thermal desorption and chemical ionization using an ion trap mass spectrometer. Integration of an ion trap mass spectrometer allows for fast scans of the entire mass spectrum every 0.5 s and bears the potential to identify unknown particulate compounds by tandem mass spectrometry. Particle collection efficiencies range from 90–100% for 25 nm particles to about 50% for 40 nm particles. In the current configuration, the absolute sensitivity of the instrument with regard to ammonium is in the range of 10–100 pg NH + 4 . In ambient samples collected in the Colorado Front Range, NH + 4 was the major signal peak in the positive ion spectrum, and additional minor signals and peak patterns of organic compounds including methylamine were found.
1. INTRODUCTION
The application of mass spectrometry to in-situ analysis of atmospheric aerosol particles in real-time has rapidly expanded and has recently led to significant advances in the experimental characterization of particulate matter in the atmosphere. Over the past years, many research groups have developed and adapted mass spectrometric techniques to analyze aerosol particles, including laser desorption/ionization instruments for single particle analysis (e.g., CitationSullivan and Prather 2005; CitationMurphy 2007), and thermal vaporization instruments (e.g., CitationCanagaratna et al. 2007) for the quantitative detection of the chemical composition of size-resolved particles. Among time-of-flight mass spectrometers and quadrupole mass spectrometers, various ion trap-based aerosol mass spectrometers have been developed, e.g., a transportable single-particle laser-ablation ion trap (CitationHarris et al. 2005), a thermal desorption ion trap aerosol mass spectrometer (CitationKürten et al. 2007), and a thermal desorption ion trap-based instrument for semivolatile aerosol components (CitationHarris et al. 2007). Many aerosol mass spectrometers are limited to the analysis of particles with diameters above ∼50–100 nm due to a sharp decrease in the transmission efficiency of smaller particles. Advances in the use of aerodynamic focusing lens systems have led to molecular composition analysis of ∼20 nm particles (e.g., CitationKane et al. 2001; CitationPhares et al. 2002), and atomic composition analysis of even smaller particles (e.g., CitationReents and Ge 2000). More recently, the development of mass spectrometers designed specifically for sub-20 nm particles such as the Thermal Desorption Chemical Ionization Mass Spectrometer (CitationVoisin et al. 2003; CitationSmith et al. 2004) and the NanoAerosol Mass Spectrometer (CitationWang et al. 2006) have added substantially to our knowledge about the chemical composition of ultrafine aerosol particles. The TDCIMS and NAMS instruments extend the size range of aerosol mass spectrometers to the nucleation mode size range (3–20 nm), which is essential for our understanding of the mechanisms of new particle formation and growth, the chemistry of potential cloud condensation nuclei, and atmospheric heterogeneous chemistry in general. Near real-time measurements are crucial to capture the temporal evolution of these highly dynamic processes. In addition, advances in the analytical capabilities to unambiguously identify individual molecular compounds are required to further our knowledge of the chemical reactions involved in particle nucleation and growth.
In this study, we report advances in the development of a thermal desorption chemical ionization ion trap mass spectrometer for the chemical characterization of ultrafine aerosol particles based on the TDCIMS design established by Smith and colleagues (CitationVoisin et al. 2003; CitationSmith et al. 2004; CitationSmith et al. 2005; CitationSmith and Rathbone 2008). The quadrupole ion trap mass spectrometer adds improved in-situ identification capabilities of organic compounds through multidimensional mass spectrometry (MSn) of characteristic molecules, and allows for fast acquisition of full mass spectra to better characterize the rapid changes in the chemical compound mixture evaporated during thermal desorption. In addition, a more compact design of the instrument facilitates its application in future field experiments.
2. INSTRUMENT DEVELOPMENT
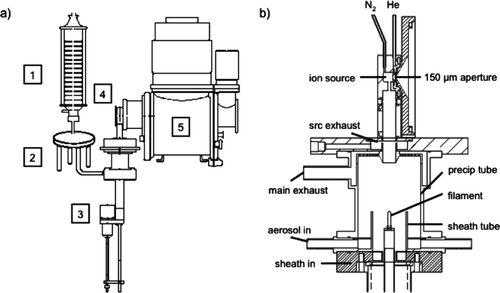
Thermal desorption chemical ionization mass spectrometry (TDCIMS) of ultrafine aerosol particles was introduced by CitationVoisin et al. (2003), and the general ideas and techniques of TDCIMS using a triple-quadrupole mass spectrometer have been described in detail in CitationSmith et al. (2004). Here, the development of a thermal desorption chemical ionization ion trap mass spectrometer is presented. A schematic overview of the instrument is given in . Atmospheric aerosol particles are sampled and introduced into the unipolar charger, where ultrafine particles are efficiently charged. Size-classification in the radial differential mobility analyzer allows for the selection of a monodisperse aerosol sample, which is transferred to an electrostatic precipitator for collection on a metal filament. After a collection period of typically 20–30 min, the sample is thermally desorbed from the filament, and analyzed with an ion trap mass spectrometer after chemical ionization reactions in the ion source region.
FIG. 1 (a) Schematic overview of the thermal desorption chemical ionization ion trap mass spectrometer with (1) unipolar charger, (2) radial differential mobility analyzer, (3) electrostatic precipitator, (4) ion source region, and (5) mass spectrometer. (b) Detail of the electrostatic precipitator and ion source.
2.1. Unipolar Charger (UPC)
Aerosol particles are charged in a high efficiency unipolar charger (UPC) built in-house after a modified design by CitationChen and Pui (1999). The ambient sample is exposed to α particles emitted from Po-210 to create ions. An electric field applied across the ion production region forces ions of the desired polarity downstream, while ions of the opposite polarity are lost to the inlet walls. After attachment of unipolar ions to aerosol particles in the charging region, the charged particles are rapidly carried out of the charger by an electric field to minimize diffusive losses. As described in CitationSmith et al. (2004), there are two major differences to the original design by CitationChen and Pui (1999): No sheath flow is used to reduce wall losses because this would dilute the sample, and larger inner dimensions in combination with a stronger radioactive source are used to allow for higher flow rates through the charger.
2.2. Radial Differential Mobility Analyzer (RDMA)
A quasi-monodisperse particle sample is selected in a radial differential mobility analyzer (RDMA) downstream of the UPC. The compact RDMA design based on a radial inward flow between parallel disks has been developed specifically for the classification of ultrafine aerosol particles (CitationZhang et al. 1995). In this study, the disk separation was b = 1.0 cm, the exit radius r1= 0.24 cm, and the entrance radius r2= 5.04 cm. The RDMA is designed to reduce the aerosol residence time and diffusive losses in the DMA. The transmission efficiency varies from 0.85 to 0.95 in the particle diameter range from 3–100 nm, with a very high resolution for ultrafine particles (CitationZhang and Flagan 1996). During field operation, the RDMA was operated with a sheath flow rate of 3.0 l min−1, and an aerosol flow rate of 1.0 l min−1.
2.3. Electrostatic Precipitator (EP)
The electrostatic precipitator (cf. ) consists of an 8.0 cm long collection tube made of aluminum with an inside diameter (ID) of 6.0 cm, a 3.2 cm ID aluminum sheath tube extending 2.5 cm into the collection tube, and a central hollow shaft made of stainless steel with an exchangeable ceramic top holding a nichrome (90% Ni, 10% Cr) filament for particle collection. A linear actuator drives the filament rod to the optimal collection position in the precipitator, and moves the filament into the ion source region for thermal desorption of the collected particles. The precipitator, while functionally similar to that described previously, has been completely redesigned to reduce diffusive losses as well as to provide more optimal flows for both sample and sheath gas.
The aerosol sample flow contains charged and size-classified particles from two identical pairs of UPCs and RDMAs. The sample flow enters the collection tube through the outer annulus around the sheath tube. The sheath tube itself is flushed with nitrogen to protect the collection filament from contamination by gaseous compounds potentially present in the aerosol sample flow.
The main fraction of the sheath flow and the aerosol sample flow less the particles collected on the filament is drawn out through a pattern of holes positioned at the top of the collection tube to maintain plug flow throughout the precipitator (cf. “main exhaust” in ). A small fraction of the combined precipitator flow, and the ion source excess flow, which is directed opposite to the precipitator flow, is drawn through an additional annular exhaust slit downstream of the main precipitator exhaust (cf. “src exhaust” in ), thus effectively isolating the ion source region from the precipitator flow and reducing potential contamination. During collection, the ion source flow is increased from 0.7 l min–1 to 1 l min–1 and the src exhaust flow correspondingly increased by 0.3 l min–1, to improve this isolation. As explained below, this had an unintended consequence of perturbing the measurement of collected particulate mass, which was however accounted for through our standard background sampling protocol.
For particle collection, the filament is biased to a potential of 4 kV, while the tip of the filament protrudes from the sheath tube. The modular filament holder allows for quick exchange of filaments if different materials or geometries are required for particle collection. The collection efficiency of ultrafine particles on the NiCr filament depends mainly on particle size, the filament position, and the sheath flow rate. In this study, 31 nm particles were collected with a sheath flow rate of 1.0 l min–1 and the filament protruding 0.6 cm from the end of the sheath tube. shows the total particle masses collected during several 20 min collection periods on 13 December 2007 in Marshall, CO, derived from particle collection efficiencies and size distribution transfer functions (–) measured with an additional scanning mobility particle sizer connected to the main precipitator exhaust. Error bars in the calculated mass are derived from the standard deviations of the size distributions, the averages of which are shown in –. The two white bars are particle mass estimates during “background” analyses, i.e., when no voltage was applied to the collection filament. However, the collection procedure is otherwise exactly replicated. As mentioned above, during both collection and background sampling periods we increased ion source and src exhaust flows to improve isolation of the ion source from the precipitator flow. This change in flow coincided exactly with the starting and ending of the collection period, and is believed to be the reason why the particle concentration decreased slightly during background sampling when no other forces exist that would modify particle concentration. The information obtained by these two background sampling periods was used to correct the sampled masses calculated from the collection periods. During the first sampling period, a particle mass of about 250 pg was collected, while particle masses of 500 to 700 pg were collected during the four subsequent sampling periods.
FIG. 2 (a) Estimates of collected particle mass during 20 min collection periods on 13 December 2007 at Marshall, CO. Two “background” periods are indicated by white bars. Error bars are derived from the standard deviations of the particle size distributions. (b–f) Average particle size distributions during collection (grey) and before/after collection (black), and particle collection efficiencies (grey diamonds) during the sampling periods indicated in . The grey shading represents the average collection efficiency of all collection periods ± one standard deviation.
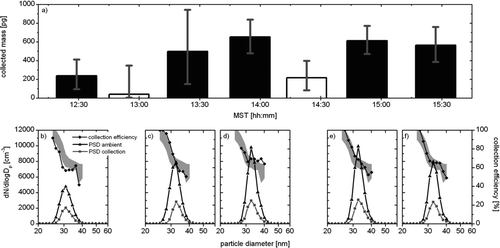
In –, the black particle size distributions represent averages of two 2 min size distribution scans of ambient aerosol taken before and after the particle collection period with no voltage applied to the collection filament. The grey size distributions represent averages of typically ten 2 min size distribution scans taken during the particle collection period when the collection filament was biased to a high voltage. The size-resolved collection efficiency (grey diamonds) is calculated from these measurements as the ratio of the particle size distributions during collection and before/after collection. Clearly, the collection efficiency decreases with increasing particle diameter, from typically 90–100% at particle diameters around 25 nm to about 50% at a particle diameter of 40 nm. Using this measurement setup, as well as deviations in particle concentration obtained by background sampling, the particle collection efficiency can be determined in-situ during field measurements for each collection period individually, thus allowing for an accurate estimation of the particle mass collected in the electrostatic precipitator.
In laboratory experiments with ammonium sulfate and ammonium oxalate, there was no indication of sample evaporation from the collection filament, however, in ambient air it is possible that some semivolatile species will evaporate from the sample during collection. This is a common issue for instruments that collect particles on a substrate for chemical analysis, and as with these other techniques the results obtained with the TDCIMS should be interpreted with due consideration of this.
2.4. Ion Source Region
The ion source region is a 1.0 cm3 half-cylindrical chamber made of stainless steel with an opening slit for the collection filament at the bottom, and a 150 μm aperture on the flat side towards the ion trap vacuum chamber (cf. ). On the aperture side, the chamber is flushed with helium gas from the top, while nitrogen is fed into the ion source region on the opposite side. The curved wall of the chamber accommodates a piece of Am-241 foil (375 μCi) emitting α particles to ionize the reactant gas mixture at atmospheric pressure, and form H3O+, O– 2 and CO– 3 ions and clusters of these ions with water as the primary stable ions.
For thermal desorption, the NiCr filament is moved into the chamber and resistively heated by applying a known current to it. The thermal desorption profiles can be adjusted using current pulses, steps, or ramps. The compounds evaporated from the aerosol sample are finally ionized through reaction with the primary stable ions according to typical CIMS reactions (CitationEisele and Tanner 1993). This study focuses on the evaluation of positive ion spectra to investigate particulate ammonium. Thermal desorption of particulate ammonium will produce NH3, which reacts with H3O+ water clusters to create NH+ 4. Electrostatic fields direct the analyte ions into a collision cell, where the ions are declustered from neutral compounds present in the gas, and directed into the ion trap chamber. In order to avoid re-condensation of the desorbed material on the steel walls, the entire ion source block is heated to 75°C by two thermostatically controlled heating cartridges.
2.5. Ion Trap Mass Spectrometer
Ion trap mass spectrometry is a versatile technique to analyze ions generated from the thermal desorption of aerosol particles. In a first step, ions are trapped in a superposition of oscillating and static electric fields between a ring electrode and two electrically connected end cap electrodes. In a second step, the stored ions are ejected from the trap according to their mass-to-charge ratio by gradually changing the electric fields.
In this work, a commercial quadrupole ion trap (C-1251, R.M. Jordan, Grass Valley, CA, USA) is used to accumulate ions and record mass spectra in the mass-selective instability scan mode. A radiofrequency (RF) field oscillating at 1.1 MHz is applied to the ring electrode. For improved particle ejection, an axial modulation waveform (460 kHz) is applied to the end cap electrodes. A typical mass scan includes a 500 ms trapping period where the low m/z cutoff for trapping was typically set to m/z = 10 amu. This is followed by ramping of the RF voltage with a typical dwell time of 100 μs/amu, and a 50 ms flushing period at the end of the scan to eject any remaining ions from the trap. Thus, using a typical scan range from m/z 10 to 210, the actual scan and analysis period is only 20 ms. This configuration requires a total of 570 ms for the collection of a complete mass spectrum and results in unit mass resolution. Varying the trapping period from 100 ms to 10 s yielded a linear relationship of the total signal intensity and the trapping period throughout this range in laboratory experiments. Thus, the limited number of ions that can be accumulated in the trap is not regarded an issue in the presented mode of operation. Increasing the dwell time to 200 μs/amu slows down the scan period but increases the mass resolution. To achieve the objectives of this work, fast scanning is desirable and unit mass resolution is considered to be sufficient. During analysis and flushing periods, ions are deflected at a gate lens upstream of the ion trap to avoid interference with the detection of ions ejected from the trap. Positive ions are detected using an electron multiplier (Model 14612, ETP, Ringwood, Australia) and additional amplification of the detector signal. For negative ion detection, a conversion dynode is used. Using helium as buffer gas, the ion trap is operated at a pressure of about 1.3 × 10–6 bar. Under these conditions, the ions are constricted to trajectories close to the center of the trap and finally ejected more quickly and efficiently, thus improving mass resolution.
Mass-selective manipulation of trapped ions allows for multidimensional mass spectrometry (MSn) capabilities using an ion trap mass spectrometer. This feature will help to characterize and identify unknown organic compounds in ultrafine aerosol particles. Unwanted ions are ejected from the ion trap through multiple-frequency resonance ejection using a filtered noise field (FNF) applied to the end cap electrodes. The remaining target compound is fragmented by resonance excitation and subsequent analysis of the fragment ions.
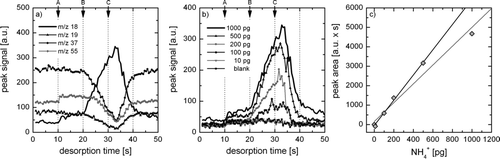
In order to evaluate the sensitivity of the system for NH+ 4, 1 μl of different (NH4)2SO4 solutions were applied directly to the filament and thermally desorbed in the ion source region. The total amount of ammonium in the 1 μl samples ranged from 10 to 1000 pg. The mass spectra were evaluated for the analyte ion NH+ 4 at the mass-to-charge ratio m/z = 18, and the reagent ions H3O+ (m/z 19), H3O+H2O (m/z 37), and H3O+(H2O)2 (m/z 55). shows the temporal evolution of NH+ 4 and the water cluster reagent ions during a 50 s desorption experiment with a total amount of 1 ng NH+ 4 applied to the filament. The displayed curves are averages of five repetitions of this experiment, and vertical dashed lines indicate the times at which operations described below are performed. The filament is transferred to the ion source region 10 s after the start of the experiment (A). Thermal desorption begins after 20 s (B) through application of a 1.3 A current to the filament. In a second desorption step, a current of 2.7 A is applied after 30 s (C). When the filament enters the ion source region, the signals of m/z 18 and 55 are slightly elevated, however, a clear increase of NH+ 4 begins just after the first desorption step (B). At the same time, m/z 19 and 37 clearly decrease, while the decrease of m/z 55 sets in later when most of the m/z 19 and 37 signal has vanished. Briefly after the second desorption step (C), NH+ 4 and the reagent ions are basically depleted, and the NH+ 4 signal decreases to background levels after about 45 s, while the reagent ion signals reappear. Clearly, a 1 ng sample of NH+ 4 depletes the reagent ions almost completely. shows the m/z 18 traces of five different NH+ 4 amounts ranging from 10 to 1000 pg, and a blank sample. Again, all curves are averages of five repetitions of the experiment. The 100 pg sample is still clearly above the blank signal, and shows the same evolution as the experiments with larger amounts of NH+ 4. In contrast, the 10 pg sample is not significantly different from the blank. Thus, we conclude that under the experimental conditions selected for this experiment, the sensitivity of the instrument for ammonium is between 10 and 100 pg NH+ 4. In comparison, the original triple quadrupole TDCIMS exhibits a sensitivity of about 1 pg (CitationVoisin et al. 2003). This difference in sensitivity may be expected due to the reduced ion throughput in the ion trap setup. Given this sensitivity assessment, it may still be advantageous to use the original quadrupole TDCIMS if ten or less individual m/z peaks are to be monitored, or if MS n is not required for ion identification. In this case, shorter collection periods and switching between the m/z peaks of interest are possible due to the higher sensitivity of the quadrupole. On the other hand, the ion trap instrument yields full mass spectra for each collection period and allows for the analytical versatility described above. In , the integrated peak areas of the NH+ 4 signals shown in are displayed after subtraction of the “background” signal. The relationship between the total amount of NH+ 4 and the integrated peak area is linear between 10 and 500 pg NH+ 4, however, the peak area of the 1000 pg experiment is clearly too low. This is due to the depletion of reagent ions as indicated in . Thus, a linear response of the ion trap mass spectrometer can be expected for NH+ 4 samples below 500 pg.
FIG. 3 (a) Temporal evolution of NH+ 4 (m/z 18) and three water cluster reagent ions (m/z 19, 37, 55) during a 50 s desorption experiment of 1 ng NH+ 4. (b) Temporal evolution of the m/z 18 signal during 50 s desorption experiments of 0, 10, 100, 200, 500, and 1000 pg NH+ 4. (c) Total amount of NH+ 4 vs. m/z 18 peak area after “background” subtraction. The black line is the linear regression of all data points except 1000 ng NH+ 4, the grey line is the linear regression of all data points. Further explanations in the text.
2.6. Instrument Control and Data Acquisition
The individual instrument components are controlled and monitored by software developed in-house. This includes the control of the electrostatic field in the unipolar charger, the RDMA flows and electrostatic field, the position, voltage bias and temperature of the collection filament, the mass flow controllers for ion source, sheath, sample and exhaust flows, the turbo pump operation and pressure in the vacuum chamber, the drive of the voltages applied to the ring and end cap electrodes of the ion trap, and the lenses guiding the ions from the source region to the trap. A typical measurement sequence includes cleaning of the filament by pre-heating for 1 min, collection of size-selected aerosol particles for 20 min by electrostatic precipitation, and a 2 min desorption and analysis period using three desorption steps with increasing temperature.
3. FIELD MEASUREMENTS
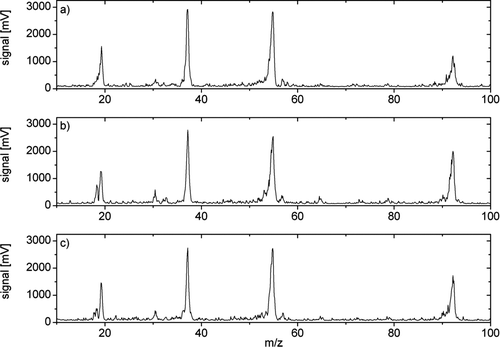
The thermal desorption chemical ionization ion trap mass spectrometer was employed in the field for the first time from 22 November 2007 to 16 December 2007 at the Marshall Field Site of the National Center for Atmospheric Research south of Boulder, CO in the Colorado Front Range (Latitude: 39.949 Longitude: –105.195 Altitude: 1742.2 meters). For most of the measurement period, the particle number concentrations were relatively low, especially under very clean conditions with W winds from the mountains. Because the presence of sub-20 nm particles and indications of new particle formation were scarce, large particles with diameters of ∼30 nm were collected and analyzed on several days. presents a detail of ambient mass spectra from m/z = 10 to 100 of 31 nm aerosol particles collected on 13 December 2007 following a 20 min collection period ending at 12:42 MST. The total particle mass collected during this period is estimated to be about 240 pg (cf. ). is an individual mass spectrum taken before the start of thermal desorption. The dominant signals are the reagent ion peaks at m/z = 19, 37, and 55 (H3O+(H2O)n, n = 1–3), and a peak at m/z = 93. This last peak is identified as (C4H10O)H3O+, a butanol/water cluster not derived from the aerosol sample, but from butanol vapors emitted from the condensation particle counters used in the SMPS system and measuring the ambient particle number concentration. The butanol signature is also evident in a small peak at m/z = 57, the dehydrated ion signal (C4H+ 9). shows a mass spectrum during the first desorption step. The most prominent additional signal is a clear peak at m/z = 18 (NH+ 4). The baseline separation of m/z 18 and 19 allows for an unambiguous attribution of the NH+ 4 and H3O+ signals in order to follow the desorption profile of NH+ 4. Additional minor signals clearly above the background can be found at m/z = 30, 32, 65, and 79. NO+ at m/z 30 indicates the presence of nitrogen-containing compounds, most probably nitrate. The signal at m/z 32 is tentatively assigned to protonated methylamine (CH3NH2H+), which has been found previously in TDCIMS measurements of ambient aerosol particles. The signals at m/z 65 and 79 are consistent with ethanol and propanol water clusters, (C2H6O)H3O+ and (C3H8O)H3O+. After the second desorption step, the mass spectrum () still exhibits a clear signal at m/z = 18, but the other minor signals at m/z = 32, 65 and 79 have decreased to background values comparable with .
FIG. 4 Ambient mass spectra from m/z = 10 to 100 of 31 nm aerosol particles collected from 12.22 to 12.42 MST on 13 December 2007. (a) Before the start of thermal desorption, (b) during the first desorption step, and (c) during second desorption step.
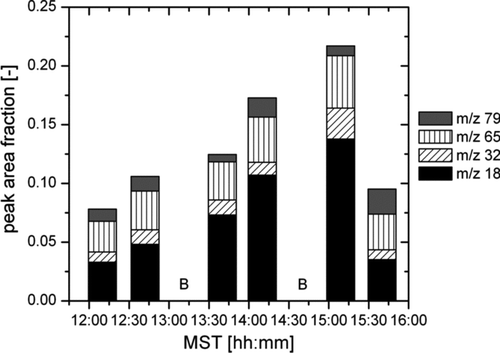
In order to distinguish particle-derived mass spectra and “background” spectra, two 20 min periods were used as background references, in which the collection filament was exposed to the aerosol sample flow without a voltage applied to the wire. The mass spectra acquired from otherwise identical thermal desorption and analysis sequences are considered background signals and should not be attributed to the aerosol sample. They are derived from the contamination of the collection filament by diffusion of ambient air and/or contamination of the ion source over time from the desorbed aerosol species. Under the assumption that the magnitude of these peaks remains constant during the time required to obtain the aerosol collection and the associated background mass spectra, the background peaks can be subtracted from the collection period peaks to yield the particle-derived signal (CitationSmith et al. 2005). For the current study, we have adopted the conservative approach of presenting only those ions that consistently appear in background-subtracted mass spectra over several sampling periods. This is a reasonable approach considering that the lifetime of 30 nm particles is on the order of a day, and few local sources of aerosol exist at the Marshall Field Site. shows the peak area fractions of m/z = 18, 32, 65, and 79 for six collection periods after subtraction of two “background” periods on 13 December 2007. The fraction of these four signals totals between 8 and 22% of the entire mass spectrum during collection periods if the background signal is subtracted. The dominating signal in all collection periods is NH+ 4, which is clearly above background levels, and thus particle derived. From the sensitivity study in , this result indicates 10–100 pg of NH+ 4 in the aerosol sample. Given the estimates of collected particle mass of about 500 pg (cf. ), this corresponds to a NH+ 4 mass fraction of 2–20%. While the signal at m/z = 65 is relatively stable throughout the sampling period, m/z = 79 is strongest in the fourth and the last collection sample, and lowest in the third and fifth sample. In contrast, the signal at m/z = 32 (methylamine) is only slightly above background levels, and strongest in the third and fifth collection period.
FIG. 5 Peak area fractions of m/z = 18, 32, 65, and 79 for six collection periods after “background” subtraction on 13 December 2007. B indicates “background” periods.
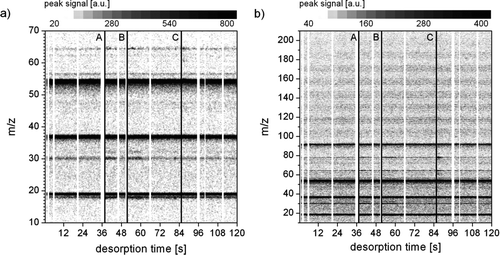
The fast acquisition of complete mass spectra every half second can be used to follow the response of peak signals to several desorption steps. Particulate compounds with a high vapor pressure should be volatilized and detected at lower temperatures, while low vapor pressure compounds may be detected in a later desorption step. This was recently implemented with the TDCIMS for the characterization of organic acids in solid samples (CitationSmith and Rathbone 2008). Here we present the advantages of employing this “temperature programmed thermal desorption” technique with the ion trap mass spectrometer. shows the temporal evolution of mass spectra acquired during a 2 min period using three desorption steps with increasing temperature. focuses on the low m/z range from 10 to 70 given on the ordinate, while presents the entire mass range from m/z = 10 to 210. The shading indicates the strength of the signal at any given m/z ratio and desorption time. Again, the mass spectra are dominated by the signals of the three reagent ions at m/z = 19, 37, and 55. In addition, some minor traces are present throughout the entire desorption period, e.g., at m/z = 57 and m/z = 93. Particle-derived peaks become more intense just after one of the desorption steps indicated by A, B and C. For example in , a slight trace of m/z = 32 appears after step B, the signals of m/z = 18 and 30 increase after steps A and B, and the strength of m/z = 65 increases after step A, B, and C. This is also the case for m/z = 79 in . Another remarkable feature of is the wave pattern of lighter and darker regions in the mass range above m/z = 100. A closer investigation of these signals reveals a distance of 14 amu between the peaks and valleys, thus indicating a pattern of organic compounds varying in their number of CH2 groups. CitationHearn and Smith (2006)made a similar observation in ambient aerosol samples using their aerosol chemical ionization mass spectrometer. In our measurements, most of this spectral pattern is lost when the “background” signal is subtracted, but a non-negligible fraction remains, especially in the period following desorption step B.
FIG. 6 Temporal evolution of mass spectra of 31 nm aerosol particles collected from 13:19 to 13:39 MST on 13 December 2007 in the mass range (a) m/z = 10 to 70 and (b) m/z = 10 to 210. Three desorption steps are indicated by vertical lines A, B, and C.
4. EXTENSION OF TECHNIQUE TO FLUX MEASUREMENT CAPABILITIES
The instrument's sampling procedure by electrostatic precipitation over a period of typically 20 min is inherently compatible with conditional sampling, i.e., atmospheric aerosol particles are introduced into the instrument only when pre-defined sampling conditions are met. This allows for collection of aerosol samples depending on control parameters such as particle number concentration or wind direction. Possible applications of this mechanism include the rejection of contaminated air samples detected by elevated particle concentrations, or conditional sampling in relaxed eddy accumulation, a micrometeorological technique to measure turbulent fluxes of atmospheric constituents (e.g., CitationBusinger and Oncley 1990). A prototype conditional sampling inlet was tested during the field measurements at Marshall. Sampling of aerosol particles was controlled by a fast-responding pinch valve mechanism that allowed either ambient aerosol or filtered, particle-free air to enter the instrument depending on the vertical wind direction. The main sampling and the filtered air lines were united after passage of a two-way pinch valve (Series 384, Asco Scientific, Florham Park, NJ, USA) and connected to the unipolar charger. Using this setup, updraft and downdraft particles were collected in separate samples and analyzed separately. In principle, the turbulent flux FREA of individual chemical compounds can be derived from this information by comparing the updraft and downdraft particulate concentrations using
In our preliminary field tests, two shortcomings precluded the calculation of turbulent particle fluxes: Firstly, updraft and downdraft samples should be collected at the same time under stationary conditions. Due to the fact that only one collection filament was present in the instrument, updraft and downdraft samples had to be collected in sequence and the stationarity requirement was violated in most cases. This technical problem can easily be solved by addition of another electrostatic precipitator to the instrument. Secondly, the concentration differences between updraft and downdraft samples are typically small and require highly precise measurements and a sufficient amount of collected particle mass. Due to the conditional sampling approach, aerosol particles are collected only during a fraction of the collection period. Therefore, it is highly desirable to increase the collection efficiency of particles in the electrostatic precipitator, optimize the ion transfer to the ion trap, and improve the ion trapping efficiency. In order to achieve these improvements, the optimum collection position of the filament in the electrostatic precipitator will be evaluated depending on particle size and sheath flow. Furthermore, the ion trajectories from the source chamber to the ion trap will be optimized using modified guiding lenses and electrical fields.
5. CONCLUSIONS
A new thermal desorption chemical ionization ion trap mass spectrometer for the chemical characterization of ultrafine aerosol particles has been developed and applied successfully in preliminary laboratory and field measurements. Size-classified particles are collected by electrostatic precipitation with an efficiency of up to 90–100%. Due to the operation principle and the physical size of the quadrupole ion trap, the sensitivity of the new instrument is about one to two orders of magnitude below the sensitivity of the original TDCIMS incorporating a triple quadrupole mass spectrometer. Using a larger quadrupole ion trap or a linear ion trap could improve the instrument's sensitivity which is critical with regard to the very small amounts of particle mass collected in the precipitator. There are several major advantages over the original instrument: The more compact design of the ion trap instrument facilitates transport and allows field measurements in previously inaccessible locations. In addition, fast scans of full mass spectra allow monitoring of many m/z values simultaneously. The potential to extend the mass range by axial modulation (e.g., CitationKaiser et al. 1991) and the analytical versatility of the ion trap mass spectrometer (e.g., MS n experiments) complement and expand the existing TDCIMS capabilities considerably. For example, application of the instrument in aerosol chamber studies can take full advantage of the ion trap tandem mass spectrometry capabilities to identify key organic compounds participating in secondary particle formation and elucidate relevant gas-to-particle conversion pathways under controlled laboratory conditions.
Acknowledgments
AH is indebted to the German Research Foundation for a DFG research fellowship grant (HE-5214/1-1). This research was supported by the Office of Science (BER), U.S. Department of Energy, Grant No. DE-FG-02-05ER63997, and by The Institute for Integrative and Multidisciplinary Earth Studies (TIIMES) at the National Center for Atmospheric Research (NCAR); NCAR is operated by the University Corporation for Atmospheric Research under the sponsorship of the National Science Foundation.
Related Research Data
REFERENCES
- Businger , J. A. and Oncley , S. P. 1990 . Flux Measurement with Conditional Sampling . J. Atmos. Ocean. Tech. , 7 : 349 – 352 .
- Canagaratna , M. R. , Jayne , J. T. , Jimenez , J. L. , Allan , J. D. , Alfarra , M. R. , Zhang , Q. , Onasch , T. B. , Drewnick , F. , Coe , H. , Middlebrook , A. , Delia , A. , Williams , L. R. , Trimborn , A. M. , Northway , M. J. , DeCarlo , P. F. , Kolb , C. E. , Davidovits , P. and Worsnop , D. R. 2007 . Chemical and Microphysical Characterization of Ambient Aerosols with the Aerodyne Aerosol Mass Spectrometer . Mass Spec. Rev. , 26 : 185 – 222 .
- Chen , D. R. and Pui , D. Y. H. 1999 . A High Efficiency, High Throughput Unipolar Aerosol Charger for Nanoparticles . J. Nanoparticle Res. , 1 : 115 – 126 .
- Eisele , F. L. and Tanner , D. J. 1993 . Measurement of the Gas-Phase Concentration of H2SO4 and Methane Sulfonic-Acid and Estimates of H2SO4 Production and Loss in the Atmosphere . J. Geophys. Res. , 98 : 9001 – 9010 .
- Harris , W. A. , Reilly , P. T. A. , Whitten , W. B. and Ramsey , J. M. 2005 . Transportable Real-Time Single-Particle ion Trap Mass Spectrometer . Rev. Sci. Instrum. , 76 doi:10.1063/1.1938607
- Harris , W. A. , Reilly , P. T. A. and Whitten , W. B. 2007 . Detection of Chemical Warfare-Related Species on Complex Aerosol Particles Deposited on Surfaces Using an Ion Trap-Based Aerosol Mass Spectrometer . Anal. Chem. , 79 : 2354 – 2358 .
- Hearn , J. D. and Smith , G. D. 2006 . Reactions and Mass Spectra of Complex Particles Using Aerosol CIMS . Int. J. Mass Spectrom. , 258 : 95 – 103 .
- Kaiser , R. E. , Cooks , R. G. , Stafford , G. C. , Syka , J. E. P. and Hemberger , P. H. 1991 . Operation of a Quadrupole Ion Trap Mass-Spectrometer to Achieve High Mass/Charge Ratios . Int. J. Mass Spectrom. , 106 : 79 – 115 .
- Kane , D. B. , Oktem , B. and Johnston , M. V. 2001 . An Electrostatic Lens for Focusing Charged Particles in a Mass Spectrometer . Aerosol Sci. Technol. , 35 : 990 – 997 .
- Kürten , A. , Curtius , J. , Helleis , F. , Lovejoy , E. R. and Borrmann , S. 2007 . Development and Characterization of an Ion Trap Mass Spectrometer for the On-Line Chemical Analysis of Atmospheric Aerosol Particles . Int. J. Mass Spectrom. , 265 : 30 – 39 .
- Murphy , D. M. 2007 . The Design of Single Particle Laser Mass Spectrometers . Mass Spec. Rev. , 26 : 150 – 165 .
- Phares , D. J. , Rhoads , K. P. and Wexler , A. S. 2002 . Performance of a Single Ultrafine Particle Mass Spectrometer . Aerosol Sci. Technol. , 36 : 583 – 592 .
- Reents , W. D. and Ge , Z. Z. 2000 . Simultaneous Elemental Composition and Size Distributions of Submicron Particles in Real time Using Laser Atomization/Ionization Mass Spectrometry . Aerosol Sci. Technol. , 33 : 122 – 134 .
- Smith , J. N. , Moore , K. F. , McMurry , P. H. and Eisele , F. L. 2004 . Atmospheric Measurements of sub-20 nm Diameter Particle Chemical Composition by Thermal Desorption Chemical Ionization Mass Spectrometry . Aerosol Sci. Technol. , 38 : 100 – 110 .
- Smith , J. N. , Moore , K. F. , Eisele , F. L. , Voisin , D. , Ghimire , A. K. , Sakurai , H. and McMurry , P. H. 2005 . The Chemical Composition of Atmospheric Nanoparticles During Nucleation Events in Atlanta . J. Geophys. Res. , 110 doi:10.1029/2005JD005912
- Smith , J. N. and Rathbone , G. J. 2008 . Carboxylic Acid Characterization in Nanoparticles by Thermal Desorption Chemical Ionization Mass Spectrometry . Int. J. Mass Spectrom. , In Press
- Sullivan , R. C. and Prather , K. A. 2005 . Recent Advances in Our Understanding of Atmospheric Chemistry and Climate Made Possible by On-Line Aerosol Analysis Instrumentation . Anal. Chem. , 77 : 3861 – 3886 .
- Voisin , D. , Smith , J. N. , Sakurai , H. , McMurry , P. H. and Eisele , F. L. 2003 . Thermal Desorption Chemical Ionization Mass Spectrometer for Ultrafine Particle Chemical Composition . Aerosol Sci. Technol. , 37 : 471 – 475 .
- Wang , S. , Zordan , C. A. and Johnston , M. V. 2006 . Chemical Characterization of Individual, Airborne Sub-10-nm Particles and Molecules . Anal. Chem. , 78 : 1750 – 1754 .
- Zhang , S.-H. , Akutsu , Y. , Russell , L. M. , Flagan , R. C. and Seinfeld , J. H. 1995 . Radial Differential Mobility Analyzer . Aerosol Sci. Technol , 23 : 357 – 372 .
- Zhang , S.-H. and Flagan , R. C. 1996 . Resolution of the Radial Differential Mobility Analyzer for Ultrafine Particles . J. Aerosol Sci. , 27 : 1179 – 1200 .