Abstract
Three differing techniques were used to measure ambient black carbon (BC) aerosols in downtown Toronto through 20 December 2006 to 23 January 2007. These techniques were thermal analysis, as performed by a Sunset Labs OCEC Analyzer (OCEC); light attenuation, as performed by an Aethalometer (AE); and photoacoustic analysis, as performed by a Photoacoustic Instrument (PA). These measurements of ambient PM 2.5 were used to investigate the effects of coating thickness on BC Mass Absorption Cross-section (MAC). MAC values were determined by comparing 880 nm and 370 nm AE measurements and PA measurements of b abs (absorption coefficient, Mm–1) to the OCEC measurements. Based on mass size distributions and supporting criteria, the PM 2.5 was classified as fresh, semi-aged, or aged. The average MAC values in these categories, based on the PA measurements, were 9.3 ± 1.8, 9.9 ± 2.0, and 9.3 ± 2.2 m 2 /g (mean ± standard deviation), respectively, suggesting that any difference in coating thickness as a result of aging, on the time scale observed, did not produce a difference in MAC. In a second type of experiment, a thermodenuder was installed upstream of the AE, PA, and OCEC and samples were heated to 340°C in order to evaporate volatile and semi-volatile components within the coating. Based on the PA measurements, the average MAC values of these heated samples, for the fresh, semi-aged, and aged categories were 7.7 ± 2.2, 6.9 ± 2.2, and 9.1 ± 2.0 m 2 /g, respectively. Similar differences in MAC were also observed by the AE. The decrease in MAC in the fresh and semi-aged samples was interpreted in terms of the degree of coating of the PM 2.5 . Results agreed well with predictions made by absorption amplification theory and had ramifications for calibration of filter-base attenuation and photoacoustic instruments.
INTRODUCTION
Black Carbon (BC) is an aerosol species generated during incomplete combustion of carbonaceous fuels. Its presence in the atmosphere has effects on the health of living organisms (CitationJerret et al. 2005; CitationVogel et al. 2005) and on the radiation budget of the Earth (CitationJacobson 2001; CitationRamanathan et al. 2001). BC is an extremely efficient light absorber, so much so that per mass, it has a greater effect on Earth's radiative forcing than CO2 (CitationBond et al. 2006a).
Primary BC particles from combustion emissions such as on-road vehicles are 20–50 nm in size and usually approximately spherical in shape (CitationSmallwood et al. 2003). These primary particles agglomerate very shortly after emission to form the aggregate BC particles seen in the environment (CitationLiousse et al. 1993, CitationMartins et al. 1998; CitationBond 2006a).
BC aggregates are rarely emitted as pure BC, but are encapsulated, or “mixed,” with other aerosol species. Even upon emission, BC aggregates are thought to be covered in layers of carbonaceous organic matter and other species within the combustion plume (CitationJacobson 2000). Over the course of their atmospheric lifetimes, BC containing particles will gather more layers of coating, mostly through condensation of secondary species (CitationCachier 1998; CitationMartins et al. 1998; CitationSaathoff et al. 2003; CitationSchnaiter et al. 2003, Citation2005; CitationBond 2006a; CitationShiraiwa et al. 2007). The degree of coating of BC particles is referred to as “mixing state” (CitationBond et al. 2006a). All BC particles relevant to this study were assumed to be “internally mixed” (with coating), as opposed to “externally mixed” (without coating), with mixing state referring to the extent of coating.
Light absorption by BC is often described using the term “absorption coefficient” (babs, Mm–1). babs is the cross-section of BC available to absorb light per volume in which that BC exists (CitationLiousse et al. 1993; CitationMartins et al. 1998; CitationBond 2006a). BC absorption per mass is described using the term “mass absorption cross-section” (MAC).
BC MAC is reported to be dependent on BC mixing state in many studies (CitationGunter et al. 1993; CitationMartins et al. 1998; CitationSaathoff et al. 2003; CitationSchnaiter et al. 2003, Citation2005; CitationDoran et al. 2007). The coatings that build up on BC can act as a lens, focusing light into the BC coreFootnote 1 of the particle, increasing absorption per mass of BC (CitationBergstrom et al. 1982). Mixing state of BC particles is a function of particle age and other factors. BC particles that have travelled for long periods before reaching a sampling site will have had ample opportunity to gather layers of coating (CitationShiraiwa et al. 2007). Freshly emitted BC containing particles will have had less opportunity to gather a coating beyond that imparted upon emission. Differences in mixing state are related to particle history. These differences may change MAC substantially.
Increases in absorption by coating can be described in terms of “absorption amplification.” Absorption amplification multiplies the MAC of a BC particle. For example, if a completely externally mixed BC particle with a MAC of 10 m2/g were to undergo mixing resulting in absorption amplification of 1.5, it would have its MAC increased to 15 m2/g.
Presented here are the results of a study that used optically based instruments to quantify BC light absorption, thermal carbon analysis to quantify mass concentrations of BC, and a combination of the two methods to determine BC MAC. This study employed a Photoacoustic Instrument (PA) in addition to traditional filter-optical type instruments. As well, non-BC data was used to support inferences about air mass history, and subsequently, the effects of that history on MAC were examined. Non-BC data included concentrations of inorganic aerosols, size distribution data, and modeled air mass back trajectories. The experiments were performed in January and February 2007 as part of Seasonal Particle Observations in the Region of Toronto (SPORT).
Previous studies have used thermodenuders to volatilize BC coating and thereby alter MAC. While heated sample inlets have been used in BC studies (CitationArnott et al. 2005), no study of BC MAC has employed both a heated inlet and a PA (which would be less susceptible to the scattering artefacts that affect filter-optical instruments), or explored the relationship of MAC with air mass history.
EXPERIMENTAL
The pertinent aspects of the experimentation included the nature of the sampling site, the BC measurement instrumentation used, the methods used to determine the atmospheric age of samples, and the thermal treatment applied to selected samples.
Sampling Site
The sampling site was the field-sampling facility of the Southern Ontario Centre for Atmospheric Aerosols Research (SOCAAR) at the University of Toronto. The facility is located at 200 College St., in the downtown core of Toronto, Ontario, an area of high urban density. The multiple local and regional emission sources that affect the facility create an ideal situation for comparing locally generated particles to long-range-transport particles. The concentrations of BC seen in Toronto are lower, on average, than those observed in other major metropolitan areas.
Sources of Particulate Matter
College St. is a high traffic route through Toronto's downtown, meaning that vehicle emissions can be responsible for a significant portion of the PM2.5 seen at SOCAAR when the synoptic meteorology brings in clean air masses from the north (CitationBuset et al. 2006; CitationOwega et al. 2006). Receptor modeling showed that restaurants, and street food vendors also contributed to the organic mass observed during SPORT (CitationSlowik et al. 2009). Long-range transport of air pollutants from the south can increase concentrations of gaseous and particulate pollutants in Toronto. The SOCAAR facility's situation, with many sources of polluted air to the south, and very few to the north, allows for comparison between air samples that are heavily influenced by long range, aged emissions, and those that are mainly locally influenced. All instruments used in this study drew samples from the same inlet, located 5 m above ground level and 15 m from College St. The measurement of aerosol composition was for PM2.5 except for the Aerosol Mass Spectrometer, which measured PM1.0.
Typical Black Carbon Concentrations
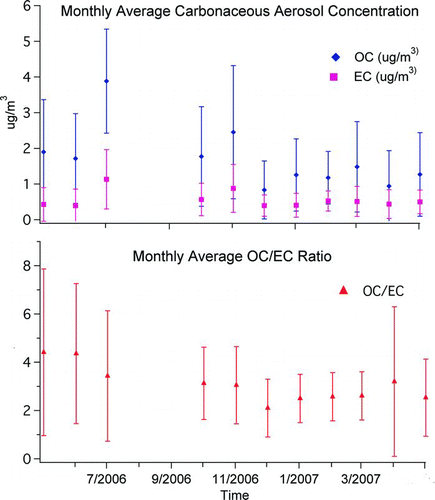
Monthly averagedFootnote 2 BC concentrations (analogous to Thermal EC, or thermally analyzed Elemental Carbon, concentrations) for downtown Toronto ranged from 0.44 ± 0.40 μ gC/m3 to 1.19 ± 0.82 μ gC/m3 for the period May 2006 to May 2007 (excluding July and August 2006) as shown in . As compared to other major cities in the world, Toronto's BC concentrations are low: Winter averages have been observed to be 6.3 μ g/m3, 12.2 μ g/m3, and 2.4 μ g/m3 in Beijing (CitationHe et al. 2001), Athens (CitationValaoraas et al. 1988), and Chattanooga, Tennessee (CitationTanner et al. 2004), respectively. The ratio of OC/EC varied seasonally, suggesting less material was available for coating of BC during winter months, consistent with less photochemical production of secondary organic compounds during the winter. The low OC/EC ratio during the winter study period, as seen in , may have had an impact on the results. The limited material (OC) available for coating, may have led to minimal coating, which, in turn may have been responsible for changes being more easily observable in MAC values.
FIG. 1 Monthly averaged carbonaceous aerosol concentration and OC/EC ratio for downtown Toronto, based on OCEC analyzer data. Data below detection limit replaced by one half of the detection limit. Data missing in August and September due to a field campaign. The figure shows that EC and OC concentrations are generally lower in Toronto than in other major cities. Low levels resulted in many data below detection limit.
Black Carbon Instrumentation
The BC measurement instruments used were an AE-21 Aethalometer (AE), a DMT Photoacoustic Instrument (PA), and a Sunset Laboratory Inc., Semi-Continuous OCEC Analyzer (OCEC). The AE operated at two wavelengths, 880 nm in the infra-red and 370 nm in the UV. The PA's laser emitted 760 nm light. Absorption coefficient (babs [Mm–1]) was obtained using the optical instrumentation and the Thermal EC (thermally analyzed Elemental CarbonFootnote 3 ) concentration was taken to be the concentration of BC. MAC was determined using the following:
While the AE was operated with a time resolution of 5 min and the PA with time resolution of 1 second, data was averaged to 105 minutes in the case of the AE and 108 minutes in the case of the PA in order to match the 108 minute time resolution of the OCEC. While it is possible to use finer time resolution with the OCEC, the generally low concentrations of BC at the SOCAAR facility (ranging from 0.44 ± 0.40 μ gC/m3 to 1.19 ± 0.82 μ gC/m3 for the period May 2006 to May 2007) made 108 minutes a prudent time resolution.
Quality Assurance/Quality Control (QA/QC) work on the OCEC determined that Thermal EC (μ g) data below the EC uncertainty (ECunc as provided by the OCEC for every measurement) were unreliable. The basis of this determination was that Thermal EC showed no relationship with readings from the AE or PA when its magnitude was less than ECunc (R2 = 0.016 and 0.034, respectively, versus 0.61 and 0.59 when Thermal EC was greater than ECunc). As well, measurement precision was poor; the average value and standard deviation of Thermal EC, when less than ECunc, was 0.002 ± 0.07 μ g/m3. Finally, comparisons with 24 h filter sample showed good agreement when Thermal EC (μ g) was greater than ECunc. These tests prompted the use of ECunc as an effective detection limit and the analysis presented here was limited to data with Thermal EC (μ g) greater than the ECunc. ECunc is calculated by the OCEC using the following equation:
Sample Age Determination
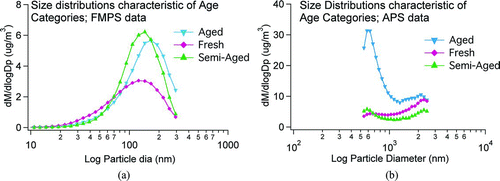
Mass size distributions, acquired by a Fast Mobility Particle Sizer (FMPS) and an Aerodynamic Particle Sizer (APS), were the primary source of information on air mass and sample age. Certain mass size modes, detailed below, were understood to be indicative of sample age. The term “age” referred to a time integrated measure of the duration and rate of change of the particle composition, predominantly due to coating. However, it was not the mere presence of marker modes that led to a classification of a sample as fresh, semi-aged, or aged, but the predominance of certain modes in relation to others. shows size distributions that are characteristic of the age categories used.
FIG. 2 Mass size distributions indicating various age influences on aerosol sample. (a) on the left shows mass size distributions in the smaller size range, as measured by the FMPS (which measures mobility diameter), and (b) on the right shows mass size distributions in the larger size range, as measured by the APS (which measures aerodynamic diameter). Densities of 1 g/cm3 were assumed.
Other indicators of age were also employed. One such indicator was air mass back trajectory as modeled by the NOAA HYSPLIT (National Oceanic and Atmospheric Administration's HYbrid Single-Particle Lagrangian Integrated Trajectory) model. Sulphate concentration, which increased as aged air masses influenced by the power generation stations to the south and southwest arrived in the vicinity of the SOCAAR facility, was also used. The non-size based information was helpful when sample age related clues from the FMPS and APS did not indicate any one age category conclusively.
Use of FMPS Data (10–500 nm Range)
The 10–500 nm range, almost without exception, contained modes in the 100–200 nm range. This range has been identified with urban particulate (CitationAlfarra et al. 2004; CitationZhang et al. 2005). Modes in the 100–200 nm range were taken to be indicative of local emissions, and of the two often observed modes, ∼ 120 nm and ∼ 160 nm, were used as indicators of fresh and semi-aged particles, respectively, based on the assumption of increased growth with aging. The distinction between the fresh and semi-aged particles was at times unclear. However, including the semi-aged category allowed better resolution between the fresh and aged categories.
Use of APS Data (Focusing on 523–2500 nm Range)
The aerosol accumulation mode, usually observed between 400 and 600 nm is, in many cases, dominated by aged particles (CitationAlfarra et al. 2004; CitationZhang et al. 2004, Citation2005). It was decided, based on the observations in the literature cited, that mass modes in the 400–600 nm range, as detected by the APS, were indicative of aged aerosol. Mass modes near 1.5–2.5 microns were indicative of a local influence on aerosol samples. When the 400–600 nm mode was dominant, samples were thought to be aged. When the micron range mode was dominant, samples were thought to be fresh. The fresh and semi-aged samples very rarely contained a 400–600 nm mode that was large relative to the modes in the 100–200 nm range.
Use of Non-Size Distribution Age Indicators
The NOAA HYSPLIT model was used to obtain air mass back trajectories for each of the samples. As shown by the work of CitationOwega et al. (2006), air masses arriving at the SOCAAR facility from the north tend to be cleaner than those that arrive from the south. When back trajectories showed air coming in from the north it was assumed that, with the incoming air being clean, the major influences on samples would have been more local and PM2.5 would therefore tend to be fresh. Air coming from the south was assumed to contain PM2.5 from coal fired power generation stations and other industrial emission sources from 100 to 500 km upwind. This PM2.5, having travelled appreciable distances, would be likely to be well aged. In this way, the HYSPLIT back trajectories aided in sample age classification.
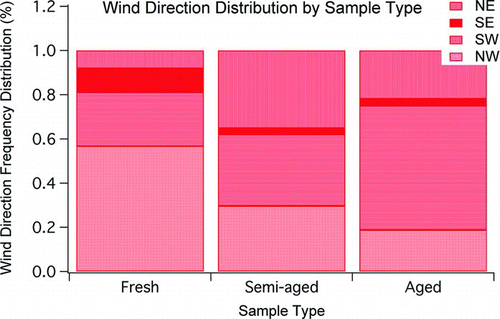
, based on wind directions, shows relative frequency of direction of origin of samples of the different age categories. While wind direction does not give as complete an air mass history as back trajectory would, the wind direction data do point to the same conclusion, that fresh samples were often found in air masses from the north and northwest, while aged samples were often found in with air masses from the south and southwest.
FIG. 3 Relative frequency of wind direction by sample age category. The figure shows that fresh samples were most often from the northwest, while aged samples were most often from the southwest.
Sulphate concentrations, as measured by a Gas-Particle Ion Chromatograph (GPIC), were used as another indicator of air mass age. Further details regarding GPIC operation details have been described elsewhere (CitationGodri et al. 2008). Associated with emissions from coal fired power generation facilities such as those to the southwest of the SOCAAR facility (CitationBuset et al. 2006), high sulphate concentrations were an indication that emissions from the southwest were influencing the samples collected. Particles from these southwestern emission sources would have aged on their way to the facility. Thus, sulphate concentrations that were high relative to those associated with fresh samples, were indicative of aged samples. Average SO4 2– concentrations were 0.8 ± 0.54 μ g/m3, 1.35 ± 0.81 μ g/m3, 2.16 ± 1.35 μ g/m3 for the fresh, semi-aged, and aged categories, respectively. Overall, the non-BC to BC ratios of the fresh and aged samples were 6.9 and 14.3, respectively, confirming the higher degree of coating on the aged samples.
Thermal Treatment of Samples
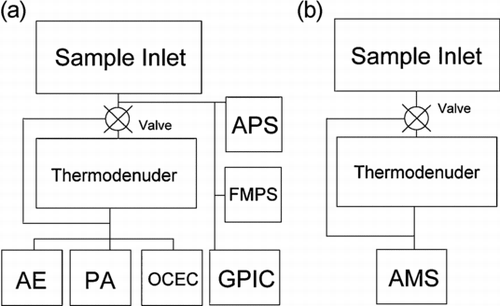
A Dekati TD3 Thermodenuder (TD) was used to volatilize the components coating the BC so that the effect of coating on MAC could be examined. All BC instruments and the AMS used were downstream of the TD, as shown in and b, whereas other instrumentation (e.g., the GPIC) was not affected by the TD. The flow rate through the TD was 13 lpm, giving samples a residence time within the heated section of 0.56 s. The TD was operated at 340°C. A valve was in place so that sample air could either bypass or flow through the TD. The valve was typically switched every 2 h to collect heated and unheated PM2.5 in each age category. Valve switching occurred during the 12 min analysis phase of the OCEC, when the instrument did not collect sample, and data taken by the AE and PA during periods when the valve was switched were not included in the data averaging process. BC, being highly refractory, would not be volatilized by 340°C temperatures. The MAC results were based on light absorption per mass, so any particle losses in the TD, which may have affected mass, but not absorption per mass, would not have altered the final results.
FIG. 4 Configuration of instruments, showing the Aethalometer (AE), Photoacoustic Instrument (PA), Sunset OCEC Analyzer (OCEC), Aerodynamic Particle Sizer (APS), Fast Mobility Particle Sizer (FMPS), Gas-Particle Ion Chromatograph (GPIC) and Aerosol Mass Spectrometer (AMS). (a) At left shows the configuration during the main experiment, while (b), at right, shows the configuration during the AMS volatilization experiment.
To ensure that the TD was volatilizing non-BC aerosol as expected, a test was performed using an Aerosol Mass Spectrometer (AMS). (Instrument configuration is shown in .) In this test, the AMS was placed downstream from the TD. Air bypassed or flowed through the TD alternately every ten minutes and the temperature was raised incrementally after each pair of samples were taken. A temperature range of 100°C to 340°C was covered in increments of 20°C. Concentrations were averaged over the ten-minute periods and the ratios of the average concentrations from consecutive unheated and heated periods were calculated to determine the percentage of non-BC species volatilized by heating at the given temperature. shows that at 340°C more than 80% of each non-BC species measured was volatilized.
FIG. 5 AMS results showing the removal from the particle phase of non-BC species. Heated and unheated samples not taken concurrently. Each point is a ratio of two consecutive 10 min averages.
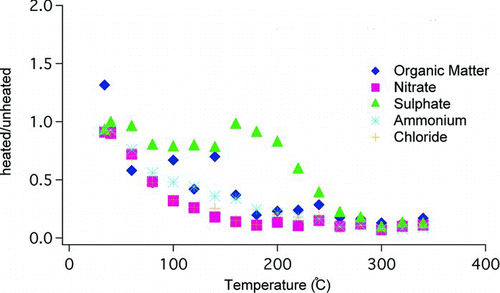
OCEC data also made apparent the effects of heating on the carbonaceous aerosol. The effect of heating on OC was a dramatic reduction in concentration. The average ratio of heated OC to unheated OC in consecutive pairs was 0.11 ± 0.22. The effect of heating on EC (analogous to BC) concentration was not significant; the ratio of heated EC to unheated EC in consecutive pairs was 0.89 ± 0.29. For consecutive pairs with EC concentrations within the coefficient of variance for EC (9.4%) this ratio was 1.0 ± 0.05. While heating volatilized much OC, it did not affect EC appreciably, showing that BC coatings were altered, but BC cores were left intact.
Comparison with Theory
CitationBond et al. (2006b) describe a method of calculating theoretical absorption amplifications for individual particles based on the sizes of their BC cores and non-BC coatings. This method provided a theoretical basis for interpretation of data.
Hypothetical particles were chosen to represent, alone or in combination, the age categories defined above. The CitationBond et al. (2006b) absorption amplification equations were applied (). Particle structures were assumed to be the same as used in the absorption amplification theoretical analysis of CitationBond et al. (2006b), i.e., comprised of a spherical BC core and a concentric spherical, non-BC, negligibly light absorbing shell. It is recognized that these assumptions do not hold true in actual particles, but they are useful in describing absorption behavior. The vital characteristics of the theoretical particles were the core diameter, and “shell diameter,” or total particle diameter. Core and shell diameters were calculated for unheated and heated particles. The core and shell diameters of the theoretical particles were then used as inputs in the absorption amplification equations so that the differences between the absorption amplification of unheated and heated particles could be calculated.
TABLE 1 Equations used to calculate theoretical absorption amplification, reproduced from Bond et al. 2006b. xcore is the core's size parameter (xcore = π d/λ, where d is the core diameter and λ is the wavelength of incident radiation). Xshell is the size parameter of the particle with shell (xshell = π d/λ where d is the shell diameter and λ is the wavelength of incident radiation). ts is relative shell thickness (ts = (xshell-xcore)/xcore)
Hypothetical “unheated” particles representative of the fresh age category were given 120 nm shell diameters based on the observed 120 nm size modes for that age category. Unheated semi-aged samples were represented by particles with 160 nm shells, based on the often observed 160 nm mode. A combination of 160–200 nm and 580 nm shell diameters were used to represent the unheated aged samples. The mode believed to most strongly affect absorption among the aged sample was the 580 nm mode, but there was rarely a case when no mode in the 160–200 nm range was present in the size distributions.
The ratio of non-BC to BC among the hypothetical particles was based on average mass concentrations of the major aerosol species for each age category. Thermal OC concentrations were measured by the OCEC and major inorganic species concentrations were measured using a GPIC. The averages were based on the unheated data only, so that the heated Thermal OC data would not artificially reduce the ratios of non-BC to BC mass. Assumed densities (1.8 g/cm3 for BC and 1.5 g/cm3 for non-BC species) were used to convert the average masses, including the average mass of BC, to volumes. The relative amount of BC volume to total particle volume (based on the particle diameter) gave the volume of the BC core. Core diameter was backsolved from the volume, assuming a spherical core.
In calculating the shell diameters of the “heated” hypothetical particles, a loss of mass was assumed. A loss of 80% of particle mass was applied to the 120 nm and 160 nm particles. This percent loss was conservative as compared with the slightly higher mass losses depicted in and the difference between the average values for heated and unheated Thermal OC. A loss of 70% of particle mass was applied to the 580 nm particles to account for some surface hardening associated with particle age. The assumption that fresh samples lost more coating than aged samples was born out in the data. The OCEC's Thermal OC data showed that OC concentrations among heated fresh samples, on average, was 14% of the concentration of OC among unheated fresh samples. In the aged category, average residual OC among heated samples was 20% of the average concentration for unheated samples. The mass losses applied were losses of total particle mass, but it was assumed that none of the mass lost was BC. The sizes of the BC cores of the hypothetical heated particles, therefore, were assumed to be no different from those of the unheated particles. The shell sizes were calculated based on the residual mass after the assumed mass loss. The resulting core and shell combinations are shown in .
TABLE 2 Non-BC to BC ratios per age category and the implications for the modeling work
RESULTS
Pre-heating samples to 340°C affected OC concentrations substantially, but had little effect on BC concentrations. Average MAC values were significantly lower among the heated fresh and semi-aged samples than in the unheated samples. The average MAC values for the unheated samples showed no statistically significant difference between age categories. The average MAC for heated aged samples was not statistically significantly different than the average MAC for unheated aged samples. Examining the data in the light of the CitationBond et al. (2006b) absorption amplification theory helped to explain why heating did not produce a change in MAC of aged samples, and confirmed that heating induced reductions in the average MAC values of fresh and semi-aged samples were in accordance with theoretical predictions.
MAC Values in the Fresh and Semi-Aged Categories
The average MAC values observed for each age category and heating condition are shown in . In the fresh and semi-aged categories the average MAC of heated samples were significantly lower than values of unheated samples. As has been shown, heating volatilized the species that would coat BC, thereby changing the mixing state of BC, reducing the amount of coating. That a reduction in coating resulted in a reduction in MAC proved the hypothesis that, at least in the fresh and semi-aged categories, BC coating increased MAC. The data also show that the optical instruments used were both affected by BC mixing condition. Based on the PA measurements, heating caused a 17% and 30% reduction in the MAC values for fresh and semi-aged samples respectively. The reduction in the AE MAC values for the fresh and semi-aged samples were, respectively, 25 and 24% at 880 nm and 29 and 26% at 370 nm, showing reasonable agreement with the PA values; the reason for the lower (17%) reduction in the PA-MAC value for the fresh samples is not known.
TABLE 3 Influence of heating on the mass absorption cross-section of aerosol by age category
It is worth noting that heating influenced the PA and AE MAC values to a similar extent. Interpolation between the 880 nm AE and the 370 nm AE MAC values allowed 760 nm AE MAC values to be estimated for comparison with the PA MAC values. A constant ratio of 2.4 ± 0.15 between the 760 nm AE and PA MAC values was found for both the heated and unheated samples which was likely predominantly due to filter based light scattering artefacts in the AE.
Theoretical predictions agree with the observations in the fresh and semi-aged category. The hypothetical fresh and semi-aged particles showed a predicted decrease in absorption amplification of over 30% as a result of “heating.” These smaller particles are in region 3 () prior to heating, where the absorption amplification is a strong function of coating thickness. Although the PA data of the fresh samples showed only a 17% decrease in absorption amplification, compared to the predicted 32%, the semi-aged samples showed a decrease of 30%, which was very close to the predicted 31%. The heating-introduced-reductions in absorption amplification seen in the AE data (24–25%) also were in reasonable agreement with the model.
Among unheated samples, there was not a statistically significant difference between the average MAC values of the three age categories. It is hypothesized that while the levels of coating of these samples of different ages was different, the coating in each category was sufficient to impart similar MACs. Theoretical predictions support this hypothesis. The theoretical absorption amplifications for the “unheated shells” () of the three age categories only varied from 1.6 to 1.9 for the three core and shell combinations examined. The difference in absorption amplification of 0.3 was less than the predicted difference between unheated and heated samples in the fresh and semi-aged categories (0.5 and 0.6, respectively). The smaller variance in the theoretical model is consistent with the lack of detectable variance in the observations.
TABLE 4 Calculated absorption amplification and difference in absorption amplification as a result of heating. Incident radiation is assumed to be 550 nm, to make use of the equations in Bond et al., 2006b
Average MAC Values in the Aged Category
There was no statistically significant difference between the unheated MACs of the different age categories. This observation is not an indication that MAC is independent of mixing state, but does support the theoretical predictions of CitationBond et al. (2006b), that beyond a certain coating thickness, MAC and mixing state are only weakly related. It is hypothesized here that in this experiment, the residual coating remaining after heating of aged samples was enough to impart an absorption amplification similar to that of the unheated samples.
The lack of observed difference between unheated and heated aged samples was not the result of a failure of the heating process. The aged samples were affected by heating as evidenced by measurements of OC and scattering cross-section. The OCEC confirmed removal of OC, a major constituent of BC coating (CitationCachier 1998), as a result of heating. The OC concentration was 0.2 ± 0.2 μ gC/m3 for heated aged samples, as compared to 1.0 ± 0.8 μ gC/m3 for unheated, indicating coating removal. It has been observed that coated BC particles scatter more than uncoated BC particles (CitationReid et al. 1998; CitationSaathoff et al 2003; CitationSchnaiter et al. 2003) and the average scattering cross-section (bsca) for the heated samples was 2.1 ± 0.9 Mm–1, as compared to 11.6 ± 5.7 Mm–1 for unheated. Hence, two independent measurements indicated that there was a removal of much, but not necessarily all, of the coating of aged samples.
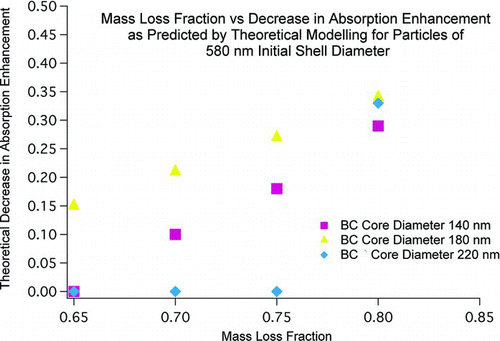
The theoretical modeling of absorption amplification suggests that this residual coating could have been sufficient to cause the observed lack of difference between the unheated and heated aged samples. The calculated changes in absorption amplification are shown in . Based on the equations in , these predicted changes show that the 580 nm particles should undergo little change in absorption amplification. The modeling suggests that the large size of these particles places them in a region where absorption amplification is nearly independent of coating thickness. The base assumptions for the modeling of aged samples (220 nm core diameter, 70% mass loss) result in no change in predicted absorption amplification.
Sensitivity analysis was performed for 580 nm particles to investigate the effect of different core sizes and mass losses. The results of this analysis showed that changes in absorption amplification were predicted in some cases (). Most of the predicted changes were small (< 0.35) as compared to those predicted for 120 and 160 nm particles (changes of 0.5 and 0.6, respectively). The largest changes shown, those resulting from an 80% mass loss, are 0.29, 0.34, and 0.33 for core diameters of 140 nm, 180 nm, and 220 nm, respectively. These changes were too small to be detected experimentally and comparable to the theoretical difference between unheated particles in the aged and fresh category (0.3).
FIG. 6 Changes in predicted absorption amplification for 580 nm particles with various core sizes and degrees of mass loss.
The absorption amplification theory of CitationBond et al. (2006b) agreed with the observed insignificant change in MAC of aged particles. The theory predicted that many of the particles in the larger size range would not undergo changes in absorption amplification on the scale that smaller particles would. The aged samples also contained some particles with a mode in the 160–200 nm range. The observed lack of change in average MAC is consistent with, but does not demonstrate that, the 580 nm mode was responsible for the majority of the absorption. This interpretation was considered more credible than two alternate hypotheses: that the 160–200 nm particles contained most of the BC and that (1) any coating on these particles was not influenced by heating or (2) that they were uncoated and contained BC with an “un-amplified” MAC that happened to match that of the fresh and semi-aged samples.
DISCUSSION
In the fresh and semi-aged categories, it was shown that heating, which altered the mixing state of BC, reduced the MAC of the samples. A similar reduction was not detected in the aged category, but such a result was in keeping with available theory. The evidence gathered in this study provides experimentally derived support for the absorption amplification theory of CitationBond et al. (2006b). The observations suggest that existing theory is correct in predicting a strong relationship between coating thickness and absorption amplification for low coating thickness, and a weaker relationship for high coating thickness. By showing that removal of BC coating reduced the MAC of BC, this work provides direct evidence that mixing with non-BC species can increase the effective MAC of BC for atmospheric aerosol.
The relatively low concentrations of BC in Toronto increased the uncertainty in the measured BC concentrations. Consequently, MAC data could only be presented as bulk averages. To create a body of data that could be developed further, a long sampling campaign, or an area with higher ambient BC concentrations, would be useful. Alternatively, BC measurement instrumentation that is not affected by coating and has a lower detection limit, such as LII (Laser Induced Incandescence), could replace thermal analysis. Another advantage of LII is that, coupled with a Photoacoustic Instrument, the time resolution could allow for monitoring changes during the coating process, as well as finer data on the temporal trends of absorption amplification.
Much work has been done in the past to determine the proper algorithm for correcting filter based light attenuation measurements of BC (CitationLiousse et al. 1993; CitationBond et al. 1999; CitationWeingartner et al. 2003). The findings of CitationWeingartner et al. (2003) showed that mixing state had an effect on readings from filter based BC measurement instruments such as the AE, and it was suggested that information on the light scattering properties of BC be known if filter based methods are to be calibrated properly. This work showed that both the AE and the PA were affected by mixing state, indicating that mixing state affects light attenuation and absorption methods in ways that are not solely attributable to filter artefacts. It is suggested here that both filter based light attenuation instruments and photoacoustic instruments require information on scattering and absorption amplification resulting from mixing state if they are to be calibrated properly. The research that would facilitate development of quantifiable calibration methods based on mixing state would be aided in large part by the LII-based work suggested above.
CONCLUSIONS
A combination of size distribution spectra measured by two particle sizers, air mass back trajectory as measured by the HYSPLIT model, and sulphate concentrations was used to classify air masses arriving in downtown Toronto into fresh, semi-aged, and aged categories. The average MAC values of the ambient samples observed were 9.3 ± 1.8, 9.9 ± 2.0, and 9.3 ± 2.2 m2/g, for the fresh, semi-aged, and aged categories, respectively. The differences between these MAC values was not statistically significant, likely due to coating of BC by organic and inorganic components within vehicle plumes creating a coating on fresh BC particles that imparted an absorption amplification similar to that seen in aged particles.
The removal of coating using a thermodenuder resulted in differences in average MAC, reducing the average MAC of the fresh particles from 9.3 ± 1.8 m2/g to 7.7 ± 2.2 m2/g and that for the semi-aged particles from 9.9 ± 2.0 m2/g to 6.9 ± 2.2 m2/g. There was no such statistically significant change among aged samples, an observation that can be attributed to the larger, heavily coated BC containing particles associated with this age category. Measurements confirmed that these aged particles did lose coating as a result of the heating process, but the coating loss was not sufficient to produce a detectable change in MAC. For these larger particles, the initial coating thickness was so great that it had surpassed the point where MAC was strongly dependent on coating thickness. These observations and explanations were shown to be largely consistent with the theoretical model put forward by CitationBond et al. (2006b).
The close agreement between the results and theoretical predictions demonstrated that the methods used are well suited for further experimental exploration of absorption amplification by particle coating.
The low OC/EC concentrations during the study period were likely major factors in the observation of changes in MAC. Were there more coating material available, all of the BC cores in the samples, not just those in the fresh and semi-aged categories, may have been coated so heavily that they would have been in the regions where absorption amplification becomes largely independent of coating thickness. If this is the case, then in most urban situations, absorption amplification can be assumed nearly always be the maximum for the calibration of instruments.
Acknowledgments
Operational funding for this study was provided by Environment Canada and the Canadian Foundation for Climate and Atmospheric Sciences. Canada Foundation for Innovation, Ontario Innovation Trust and the Ministry of Economic Development and Trade and Environment Canada provided the funding for the instrumentation, as part of the support to create the Southern Ontario Centre for Atmospheric Aerosol Research (SOCAAR).
Notes
1 Terms such as “core” and “coating” may carry connotations of concentricity, implying that BC is spherical and at the center of a larger spherical particle. However, it is not the case that BC cores are spherical, nor is it true that their coatings are always spherical. It is also rare for the BC core to be exactly at the centre of a particle. The terms are merely used for convenience and should not be taken to imply concentricity.
2. Throughout this work, “average” refers to the mean of a set of values. Error terms are standard deviations of those values.
3. Despite its slightly different connotations (explained by CitationBond 2006a), “Elemental Carbon” is often used synonymously with BC, and, for the purposes of this study, EC and BC are similar enough to be treated as the same substance.
Related Research Data
REFERENCES
- Alfarra , M. R. , Coe , H. , Allan , J. D. , Bower , K. N. , Boudries , H. , Canagaratna , M. R. , Jimenez , J. L. , Jayne , J. T. , Garforth , A. A. , Li , S.-M. and Worsnop , D. R. 2004 . Characterization of Urban and Rural Organic Particulate in the Lower Fraser Valley using Two Aerodyne Aerosol Mass Spectrometers . Atmos. Environ. , 38 : 5745 – 5758 .
- Arnott , W. P. , Moosmüller , H. and Rogers , C. F. 1999 . Photoacoustic Spectrometer For Measuring Light Absorption by Aerosol: Instrument Description . Atmos. Environ. , 33 : 2845 – 2852 .
- Arnott , W. P. , Zielinska , B. , Rogers , C. F. , Sagebiel , J. , Park , K. , Chow , J. , Moosmüller , H. and Watson , J. G. 2005 . Evaluation of 1047-nm Photoacoustic Instruments and Photoelectric Aerosol Sensors in Source-Sampling of Black Carbon Aerosol and Particle-Bound PAHs from Gasoline and Diesel Powered Vehicles . Environ. Sci. Technol. , 39 : 5398 – 5406 .
- Bergstrom , R. W. , Ackerman , T. P. and Richards , L. W. 1982 . “ Optical Properties of Particulate Elemental Carbon ” . In Particulate Carbon: Atmospheric Life Cycle , Edited by: Wolff , G. T. and Klimsch , R. Lc. New York : Plenum Press .
- Bond , T. C. , Anderson , T. L. and Campbell , D. 1999 . Calibration and Intercomparison of Filter-Based Measurements of Visible Light Absorption by Aerosols . Aerosol Sci. Technol. , 30 : 582 – 600 .
- Bond , T. C. and Bergstrom , R. W. 2006a . Light Absorption by Carbonaceous Particles: An Investigative Review . Aerosol Sci. Technol. , 40 : 27 – 67 .
- Bond , T. C. , Habib , G. and Bergstrom , R. W. 2006b . Limitations in the Enhancement of Visible Light Absorption Due to Mixing State . J. Geophys. Res. , 111 : D20211
- Buset , K. C. , Evans , G. J. , Leaitch , W. R. , Brook , J. R. and Toom-Sauntry , D. 2006 . Use of Advanced Receptor Modeling for Analysis of an Intensive 5-Week Aerosol Sampling Campaign . Atmos. Environ. , 40 : S482 – S499 .
- Cachier , H. 1998 . “ Carbonaceous Combustion Aerosols ” . In Atmospheric Particles , Edited by: Harrison , R. M. and vanGrieken , R. E. Toronto : John Wiley & Sons .
- Chow , J. C. , Watson , J. G. , Chen , A. L.-W. , Arnott , W. P. , Moosmüller , H. and Fung , K. 2004 . Equivalence of Elemental Carbon by Thermal/Optical Reflectance and Transmittance with Different Temperature Protocols . Environ. Sci. Technol. , 38 : 4414 – 4422 .
- Doran , J. C. , Barnard , J. C. , Arnott , W. P. , Cary , R. , Coulter , R. , Fast , J. D. , Kassianov , E. I. , Kleinman , L. , Laulainen , N. S. , Martin , T. , Paredes-Miranda , G. , Pekour , M. S. , Shaw , W. J. , Smith , D. F. , Springston , S. R. and Yu , X.-Y. 2007 . The T1-T2 Study: Evolution of Aerosol Properties Downwind of Mexico City . Atmos. Chem. Phys. , 7 : 1585 – 1598 .
- Godri , K. , Evans , G. , Slowik , J. , Knox , A. , Abbatt , J. , Brook , J. R. , Dann , T. and Dabek-Zlotorzynska , E. 2008 . Evaluation of a Semi-Continuous Chemical Characterization System for Water Soluble Inorganic PM2.5 and Associated Precursor Gases . Atmospheric Measurement Techniques Discussions , 1 : 1 – 44 .
- Gunter , R. L. , Hansen , A. D. A. , Boatman , J. F. , Bodhaine , B. A. , Schnell , R. C. and Garvey , D. M. 1993 . Airborne Measurements of Aerosol Optical Properties Over South-Central New Mexico . Atmos. Environ. , 27A : 1363 – 1368 .
- He , K. , Yang , F. , Ma , Y. , Zhang , Q. , Yao , X. , Chan , C. K. , Cadle , S. , Chan , T. and Mulawa , P. 2001 . The Characteristics of PM2.5 in Beijing, China . Atmos. Environ. , 35 : 4959 – 4970 .
- Jacobson , M. Z. 2000 . A Physically Based Treatment of Elemental Carbon Optics: Implications for Global Direct Forcing of Aerosols . Geophys. Res. Lett. , 27 : 217 – 220 .
- Jacobson , M. Z. 2001 . Strong Radiative Heating Due to the Mixing State of Black Carbon Inatmospheric Aerosols . Nature , 409 : 695 – 697 .
- Jerret , M. , Burnett , R. T. , Ma , R. , Pope , C. A. , Krewski , D. , Newbold , K. B. , Thurston , G. , Shi , Y. , Finkelstein , N. , Calle , E. E. and Thun , M. J. 2005 . Spatial Analysis of Air Pollution and Mortality in Los Angeles . Epidemiol. , 16 : 727 – 736 .
- Liousse , C. , Cachier , H. and Jennings , S. G. 1993 . Optical and Thermal Measurement of Black Carbon Aerosol Content in Different Environments: Variation of the Specificattenuation Cross-Section, Sigma (σ) . Atmos. Environ. , 27A : 1203 – 1211 .
- Martins , J. V. , Artaxo , P. , Liousse , C. , Reid , J. S. , Hobbs , P. V. and Kaufman , Y. J. 1998 . Effects of Black Carbon Content, Particle Size and Mixing on Light Absorption Byaerosols from Biomass Burning in Brazil . J. Geophys. Res.—Atmospheres , 103 : 32041 – 32050 .
- Owega , S. , Khan , B. U.-Z. , Evans , G. J. , Jervis , R. E. and Fila , M. 2006 . Identification of Long-Range Aerosol Transport Patterns to Toronto via Classification of Back Trajectories by Cluster Analysis and Neural Network Techniques . Chemometrics and Intelligent Laboratory Systems , 83 : 26 – 33 .
- Petzold , A. and Schönlinner , M. 2004 . Multi-Angle Absorption Photometry—A New Method for the Measurement of Aerosol Light Absorption and Atmospheric Black Carbon . J. Aerosol Sci. , 35 : 421 – 441 .
- Petzold , A. , Schloesser , H. , Sheridan , P. J. , Arnott , W. P. , Ogren , J. A. and Virkkula , A. 2005 . Evaluation of Multiangle Absorption Photometry for Measuring Aerosol Light Absorption . Aerosol Sci. Technol. , 39 : 40 – 51 .
- Ramanathan , V. , Crutzen , P. J. , Kiehl , J. T. and Rosenfeld , D. 2001 . Aerosols, Climate, and the Hydrological Cycle . Science , 294 : 2119 – 2124 .
- Reid , J. S. , Hobbs , P. V. , Liousse , C. , Martins , J. V. , Weiss , R. E. and Eck , T. F. 1998 . Comparisons of Techniques for Measuring Shortwave Absorption and Black Carbon Content of Aerosols from Biomass Burning in Brazil . J. Geophys. Res. , 103 ( D24 ) : 32031 – 32040 .
- Saathoff , H. , Naumann , K.-H. , Schnaiter , M. , Schöck , W. , Möhler , O. , Schurath , U. , Weingartner , E. , Gysel , M. and Baltensperger , U. 2003 . Coating of Soot and [NH4)2SO4 Particles by Ozonolysis Products of α -Pinene . Aerosol Sci. , 34 : 1297 – 1321 .
- Schnaiter , M. , Horvath , H. , Möhler , O. , Naumann , K.-H. , Saathoff , H. and Schöck , O. W. 2003 . UV-VIS-NIR Spectral Optical Properties of Soot and Soot-Containing Aerosols . Aerosol Sci. , 34 : 1421 – 1444 .
- Schnaiter , M. , Linke , C. , Möhler , O. , Naumann , K.-H. , Saathoff , H. , Wagner , R. , Schurath , U. and Wehner , B. 2005 . Absorption Amplification of Black Carbon Internally Mixed with Secondary Organic Aerosol . J. Geophys. Res.—Atmospheres , 110 : D19204
- Sheridan , P. J. , Arnott , W. P. , Ogren , J. A. , Andrews , E. , Atkinson , D. B. , Covert , D. S. , Moosmüller , H. , Petzold , A. , Schmid , B. , Strawa , A. W. , Varma , R. and Virkkula , A. 2005 . The Reno Aerosol Optics Study: An Evaluation of Aerosol Absorption Measurement Methods . Aerosol Sci. Technol. , 39 : 1 – 16 .
- Shiraiwa , M. , Kondo , Y. , Moteki , N. , Takegawa , N. , Miyazaki , Y. and Blake , D. R. 2007 . Evolution of Mixing State of Black Carbon in Polluted Air from Tokyo . Geophys. Res. Letters , 34 : L16803 doi:10.1029/2007GL029819
- Slowik , J. G. , Vlasenko , A. , McGuire , M. , Evans , G. J. and Abbatt , J. P. D. 2009 . Simultaneous Factor Analysis of Organic Particle and Gas Measurements in Downtown Toronto, Submitted to Environ. Sci. Technol.
- Smallwood , G. J. , Bachalo , W. D. and Sankar , S. V. 2003 . “ Particulate Measurement Methods ” . In Optical Metrology for Fluids, Combustion, and Solids , Edited by: Mercer , C. Dordrecht : Kluwer Academic .
- Subramanian , R. , Khlystov , A. Y. and Robinson , A. L. 2006 . Effect of Peak Inert-Mode Temperature on Elemental Carbon Measured Using Thermal-Optical Analysis . Aerosol Sci. Technol. , 40 : 763 – 780 .
- Tanner , L. , Parkhurst , W. J. , Valente , M. L. and Phillips , W. D. 2004 . Regional Composition of OM2.5 Aerosols Measured at Urban, Rural and “Background” Sites in the Tennessee Valley . Atmos. Environ. , 38 : 3143 – 3153 .
- Valaoraas , G. , Huntzicker , J. J. and White , W. H. 1988 . On the Contribution of Motor Vehicles to the Athenian “Nephos”: An Application of Factor Signatures . Atmos. Environ. , 22 : 965 – 971 .
- Vogel , C. F. A. , Sciullo , E. , Wong , P. , Kuzmicky , P. , Kado , N. and Matsumura , F. 2005 . Introduction of Proinflamatory Cytokines and C-Reactive Protein in Humanmacrophage Cell Line U937 Exposed to Air Pollution Particulates . Environ. Health Perspect. , 113 : 1536 – 1541 .
- Weingartner , E. , Saathoff , H. , Schnaiter , M. , Streit , N. , Bitnar , B. and Baltensperger , U. 2003 . Absorption of Light by Soot Particles: Determination of the Absorption Coefficient by Means of Aethalometers . J. Aerosol Sci. , 34 : 1445 – 1463 .
- Zhang , Q. , Stanier , C. O. , Canagaratna , M. R. , Jayne , J. T. , Worsnop , D. R. , Pandis , S. N. and Jimenez , J. L. 2004 . Insights into the Chemistry of New Particle Formation and Growth Events in Pittsburgh Based on Mass Spectrometry . Environ. Sci. Technol. , 38 : 4797 – 4809 .
- Zhang , Q. , Canagaratna , M. R. , Jayne , J. T. , Worsnop , D. and Jimenez , J.-L. 2005 . Time and Size Resolved Chemical Composition of Submicron Particles in Pittsburgh: Implications for Aerosol Sources and Processes . J. Geophys. Res. , 110 : DO750 doi: 10.1029/2004JD004649