Abstract
The effects of molecular structure on the products and mechanisms of SOA formation from OH radical-initiated reactions of linear, branched, and cyclic alkanes in the presence of NO x were investigated in a series of environmental chamber experiments. SOA mass spectra were obtained in real time and off line using a thermal desorption particle beam mass spectrometer and used to identify reaction products. Real-time mass spectra were used to classify products according to their temporal behavior, and off-line temperature-programmed thermal desorption analysis of collected SOA was used to separate products by volatility prior to mass spectral analysis and to gain information on compound vapor pressures. A reaction mechanism that includes gas- and particle-phase reactions was developed that explains the formation of SOA products and is consistent with the various lines of mass spectral information. Results indicate that the SOA products formed from the reactions of linear, branched, and cyclic alkanes are similar, but differ in a few important ways. Proposed first-generation SOA products include alkyl nitrates, 1,4-hydroxynitrates, 1,4-hydroxycarbonyls, and dihydroxycarbonyls. The 1,4-hydroxycarbonyls and dihydroxycarbonyls rapidly isomerize in the particle phase to cyclic hemiacetals that then dehydrate to volatile dihydrofurans. This conversion process is catalyzed by HNO 3 formed in the chamber and is slowed by the presence of NH 3 . Volatile products can react further with OH radicals, forming multi-generation products containing various combinations of the same functional groups present in first-generation products. For linear and branched alkanes, the products are acyclic or monocyclic, whereas for cyclic alkanes they are acyclic, monocyclic, or bicyclic. Some of the products, especially those formed from ring-opening reactions of cyclic alkanes appear to be low volatility oligomers. The implications of the results for the formation of atmospheric SOA are discussed.
INTRODUCTION
Emissions of non-methane volatile organic compounds (NMVOCs) to the global atmosphere are dominated by biogenic sources, which contribute ∼ 90% of the estimated 1150 Tg emitted annually (CitationGuenther et al. 1995). In urban areas, however, anthropogenic sources dominate, with approximately one third of emissions being alkanes (CitationCalvert et al. 2000). In the atmosphere, alkanes only react at significant rates with OH radicals, with typical atmospheric lifetimes of a day (CitationAtkinson and Arey 2003). The products of these reactions consist of a variety of oxygenated compounds (CitationAtkinson 1997; CitationArey et al. 2001; CitationAtkinson and Arey 2003; CitationLim and Ziemann 2005); some of these, especially those with multiple functional groups, can condense to form secondary organic aerosol (SOA) (CitationLim and Ziemann 2005; CitationKroll and Seinfeld 2008). SOA comprises a significant fraction of the mass of atmospheric fine (diameter ≤ 2.5 μ m) particles (CitationZhang et al. 2007), which have important effects on visibility, global climate, and human health (CitationAndrea and Crutzen 1997; CitationSeinfeld and Pandis 1998; CitationEnglert 2004). The contribution of alkane reactions to atmospheric SOA is not yet known (CitationKanakidou et al. 2005), but recent studies indicate that emissions of high molecular weight alkanes (∼ C12–C30) with sufficient volatility to serve as precursors to SOA formation have been seriously underestimated (CitationRobinson et al. 2007), leading to increased interest in understanding the atmospheric chemistry of these compounds.
Previously, we investigated the chemistry of SOA formation from reactions of a few linear alkanes with OH radicals in the presence of NOx (CitationLim and Ziemann 2005), conditions representative of a polluted atmosphere. That was the first molecular level study of SOA formation from this class of compounds and identified a number of interesting and important new products and pathways to SOA formation from OH radical-initiated reactions of saturated hydrocarbons. Here, we present results of more detailed studies that included reactions of linear, branched, and cyclic alkanes. By using time profiles of mass spectra obtained during reactions, which was not done in our previous study, in addition to thermal desorption profiles of collected SOA, we were able to plausibly identify a much larger suite of compounds than before, including first- and multi-generation products formed by gas- and particle-phase reactions. The results were used to develop a chemical mechanism for the formation of SOA products and to investigate the effects of alkane molecular structure on this chemistry.
EXPERIMENTAL SECTION
Chemicals
The following chemicals, with purities (when available) and suppliers, were used: n-pentadecane (99+%), n-hexadecane (99%), and dioctyl sebacate (90%) [Sigma-Aldrich]; cyclododecane (98%), cyclopentadecane (98%), and 2,6,10-trimethyldodecane [ChemSampCo]; and NO and NH3 [Matheson Gas]. Methyl nitrite was synthesized (CitationTaylor et al. 1980) and stored in liquid nitrogen.
Environmental Chamber Method
Reactions of alkanes with OH radicals in the presence of NOx were performed in a 5900 L PTFE environmental chamber at ∼ 25°C and atmospheric pressure. The chamber was filled with clean, dry air (< 5 ppbv hydrocarbons, < 1% RH) and has blacklights covering two walls. Dioctyl sebacate (DOS) particles from an evaporation-condensation apparatus were flushed into the chamber to achieve a seed particle concentration of ∼ 200–400 μ g m−3. An alkane, methyl nitrite, and NO were then added to achieve concentrations of 1, 10, and 10 ppmv. NO suppresses O3 and NO3 radical formation. An experiment was also performed with n-hexadecane with 20 ppmv NH3 in dry air. Reactions were initiated by turning on the blacklights to form OH radicals by methyl nitrite photolysis (CitationAtkinson et al. 1981). The blacklights were turned off after 60 min. Typically, ∼ 70–85% of the alkane reacted, NO decreased by 1–2 ppmv, NO x stayed approximately constant, and SOA formed within a minute. The average OH radical concentration estimated from the amount of alkane that reacted and its OH radical rate constant (CitationAtkinson and Arey 2003) was ∼ 3 × 107 cm−3.
Particle Composition Analysis
A thermal desorption particle beam mass spectrometer (TDPBMS) was used to analyze particle composition in real-time (CitationTobias et al. 2000) and by temperature-programmed thermal desorption (TPTD) (CitationTobias and Ziemann 1999). Aerosol was sampled into an aerodynamic lens to form a particle beam that impacted on a polymer-coated metal vaporizer rod (CitationChattopadhyay and Ziemann 2005). For real-time analysis, the rod was resistively heated to 180°C. Particles vaporized upon impact and the vapor was ionized by 70 eV electrons and analyzed in a quadrupole mass spectrometer. For TPTD analysis, the rod was cooled to −40°C and particles were collected for 30 min. The rod was allowed to warm to −5°C and then heated to 200°C using a 2°C min−1 ramp to desorb and separate compounds by volatility prior to mass analysis. The vaporizer was only heated to 200°C (except for periodic cleaning at higher temperatures) in order to avoid evaporation of the polymer coating, which complicates the mass spectral analysis. Therefore, products may be present that are less volatile than those reported here.
REACTION MECHANISM
In this discussion, abbreviations are introduced that are used throughout the text for the parent alkanes, and structures and combinations of functional groups frequently encountered in the reaction products. The structures of the linear, branched, and cyclic alkanes studied here, n-pentadecane (PD), 2,6,10-trimethyldodecane (2,6,10-TMDD), cyclopentadecane (CPD), and cyclododecane (CDD), are shown in . The mechanism of OH radical-initiated reactions of alkanes in the presence of NO x is shown in (CitationAtkinson and Arey 2003; CitationLim and Ziemann 2005). The mechanism is written explicitly for the reaction of an acyclic alkane, where R1 and R2 are alkyl groups, but the reaction of a cyclic alkane can be represented by simply connecting R1 and R2 with a C-C bond to form a single alkyl group, R. For the alkanes studied here, the identities of R1, R2, and R are constrained in different ways. For PD, R1 and R2 can be any combination of linear alkyl groups whose total carbon number is 10, whereas for CPD and CDD R is (CH2)10 and (CH2)7. As can be seen from and , the representation of the reaction of 2,6,10-TMDD depends on where on the molecule the OH radical attacks. One or two H atoms in the linear (CH2)5 chain that connects R1 and R2 in are replaced by CH3 groups (e.g., CH(CH3)(CH2)3CH(CH3)) and R1 and R2 are linear or branched alkyl groups that complete the 2,6,10-TMDD structure. The reaction products will be similar to those in , but with one or two additional CH3 groups.
FIG. 1 Structures of linear, branched, and cyclic alkane precursors employed in SOA studies.
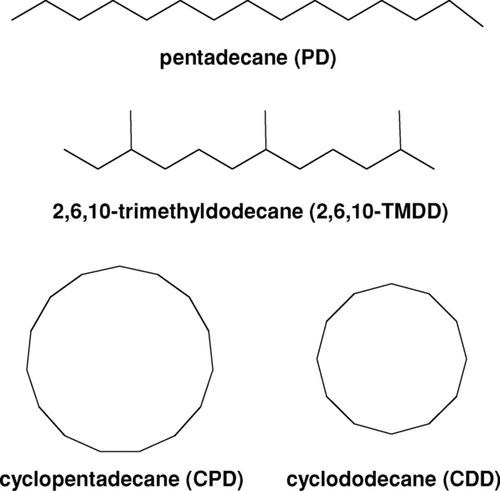
FIG. 2 Mechanism of OH radical-initiated reaction of alkanes in the presence of NOx.
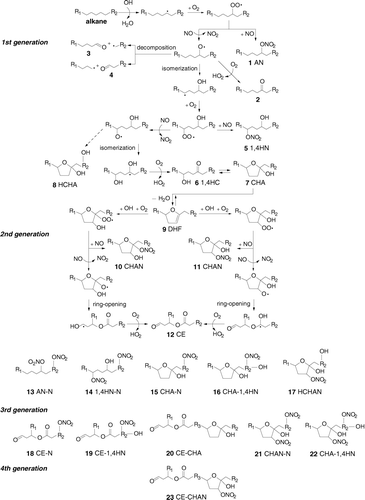
The OH radical reaction is initiated by abstraction of an H atom, forming an alkyl radical that adds O2 to form an alkylperoxy radical. Alkylperoxy radicals can react with NO or NO2. In the latter case, they form peroxynitrates, but these rapidly decompose back to the alkylperoxy radical and NO2 and so are not important here and not shown in . Reaction with NO forms an alkyl nitrate (AN) [1] or alkoxy radical. Alkoxy radicals can react with O2, decompose, or isomerize. Reaction with O2 forms a carbonyl [2] and decomposition creates two carbonyl [3, 4] plus alkyl radical pairs, with the alkyl radicals then reacting as described above. For linear alkanes with Cn > 6, reactions with O2 and decomposition are too slow to compete with isomerization (CitationArey et al. 2001; CitationAtkinson 2007), so carbonyl formation is negligible. Isomerization through a six-membered ring followed by reaction with O2 forms 1,4-hydroxyperoxy radicals that react with NO similarly to alkylperoxy radicals. The products are 1,4-hydroxynitrates (1, 4HN) [5] and 1, 4-hydroxyalkoxy radicals that primarily undergo a reverse isomerization and react with O2 to form 1,4-hydroxycarbonyls (1,4HC) [6]. AN, 1,4HN, and 1,4HC have been measured in the gas-phase for reactions of C7–C10 alkanes, with yields of approximately 0.2, 0.05, and 0.5 (CitationReisen et al. 2005) and with no other observed products. AN and 1,4HN yields are probably closer to ∼ 0.3 (CitationArey et al. 2001) and ∼ 0.15 (CitationLim and Ziemann 2005) for reactions of larger alkanes. 1,4HC partition to particles and chamber walls where they isomerize to cyclic hemiacetals (CHA) [7] (note that the structure of the cyclic hemiacetal is that of an alkyl substituted 2-hydroxytetrahydrofuran). Though the reaction mechanism is not shown in , calculations made using the method of CitationAtkinson (2007) indicate that a small fraction (∼ 5–10%) of the 1,4-hydroxyalkoxy radicals can isomerize by abstracting an H atom from the R1 group, instead of from the CHOH group, thereby forming dihydroxyalkyl radicals instead of 1,4-HC [6]. These radicals then react similarly to 1,4-hydroxyalkyl radicals, eventually forming mostly dihydroxycarbonyls (1,4HC with an additional OH group) that can then isomerize to hydroxy CHA (HCHA) [8].
The CHA (and HCHA) can dehydrate to dihydrofurans (DHF) [9] that then evaporate (CitationMartin et al. 2002; CitationHolt et al. 2005). The DHF react rapidly with OH radicals (in the atmosphere they also react with O3 and NO3 radicals), O2, and NO to form second-generation products, low volatility isomers referred to here as CHA nitrates (CHAN) [10, 11] and, via alkoxy radical decomposition, carbonylesters (CE) [12] (CitationMartin et al. 2002). Calculations performed using the structure-reactivity method of CitationAtkinson (2007) indicate that the furan ring opens solely as shown, with negligible scission of the C-O bond in the ring. The CE yield from the reaction of the DHF formed from 2,5-hydroxypentanone is 0.72 (CitationMartin et al. 2002); those formed in this study were apparently too volatile to form SOA.
AN, CE, sufficiently small 1,4HN, and other products that do not partition entirely to the particle phase, can react further with OH radicals in the gas phase to form less volatile multi-generation products. It is therefore expected that, in addition to products 1, 5, 7, 8, 10, and 11, SOA can also contain these compounds with an added N, CE, or 1,4HN group. Those proposed to be present in SOA formed here are shown in . In these experiments, reactions of OH radicals with 1,4HC appear not to compete significantly with particle-phase isomerization, and heterogeneous reactions with OH radicals are also not important.
As noted above, the mechanisms of the reactions of branched and cyclic alkanes are similar to those shown in for linear alkanes; however, the different structures of these compounds can lead to different branching ratios for some reaction pathways and, therefore, different product yields. Most important are the effects of branching and ring strain on rates of decomposition of alkoxy radicals. For branched alkanes, alkoxy radicals with the radical site located adjacent to a branch point decompose much faster than those formed from n-alkanes (CitationAtkinson 2007), leading to fragmentation and therefore higher yields of smaller, more volatile carbonyls and alkyl radicals at the expense of lower volatility multifunctional isomerization products. For cyclic alkanes, ring strain enhances the rates of decomposition of cyclic alkoxy radicals (CitationAtkinson 2007), but rather than fragmenting to a pair of smaller carbonyls and alkyl radicals, ring-opened carbonylalkyl radicals are formed. Their subsequent reactions should be similar to those of alkyl radicals formed from the corresponding linear alkane, but the products should be less volatile because of the presence of an added carbonyl group. The products shown in should therefore either have an additional ring or carbonyl (for the cyclic alkanes studied here it will be an aldehyde) group when formed from cyclic alkanes.
TDPBMS DATA INTERPRETATION
SOA products are assigned using mass spectral information. The assignments are consistent with potential products formed via the reaction mechanism shown in , established electron ionization fragmentation pathways that lead to characteristics ions, and thermal desorption and time profiles of these ions.
Thermal Desorption Profiles
Two general principles are used to interpret thermal desorption profiles. The first is that ion fragments associated with the same compound must exhibit maxima at the same desorption temperature (designated here as Tdes). However, although ion fragments with the same Tdes are often associated with the same compound, they may also come from different isomers or compounds with similar vapor pressures. The profiles are therefore similar to single ion chromatograms obtained with gas chromatography (although retention times also depend on compound-column interactions). The second general principle is that compound vapor pressures decrease as the number of functional groups increase, leading to the expectation that more functionalized products will desorb at higher temperatures. For the functional groups expected to be present in the products of alkane reactions, hydroxy and nitrate (also called nitrooxy) groups should reduce vapor pressures and therefore increase Tdes more than carbonyls and esters (CitationPankow and Asher 2008). Also, for reasons that are not understood, but may involve particle-phase oligomer-forming reactions, products containing a CHA structure desorb at much higher temperatures than expected on the basis of vapor pressures calculated from structure–activity relationships (CitationJordan et al. 2008).
Time Profiles
For the purposes of interpreting the time profiles of characteristic ions observed here, it is useful to classify the products into five groups according to the chemical mechanisms by which they are formed. Approximate model time profiles expected for these five groups are shown in . These were obtained using a model described elsewhere (CitationLim and Ziemann 2009a), which incorporates gas-phase reactions of OH radicals with the alkane and reaction products, gas-particle partitioning of reaction products, and particle-phase isomerization of 1,4HC to CHA and dehydration of CHA and CHAN to DHF. The profiles make intuitive sense, given the underlying reaction mechanisms. In this discussion, first- second-, and third-generation products refer to those whose formation mechanisms involve one, two, and three OH radical reactions. Profile 1 is characteristic of first-generation products, such as AN [1] and 1,4HN [5], which are formed in the gas phase by the OH radical-initiated reaction of the alkane and then partition to the particle phase, with reactive loss rates being significantly slower than formation rates. Product concentrations increase most rapidly at the beginning of the reaction when alkane and OH radical concentrations are highest, then monotonically approach a plateau as reactant concentrations drop. The steepness of the profile depends on formation and loss rates as well as on the extent of gas-to-particle partitioning, which increases with time due to increasing aerosol mass concentrations. Profile 2 is characteristic of first-generation products that subsequently undergo substantial and rapid reactive losses. Product concentrations increase to a maximum, at which point rates of formation and loss are equal, and thereafter decrease as loss processes dominate. Here, first-generation products that fit into this class include CHA [7] and HCHA [8] formed from rapid particle-phase isomerization of 1,4HC and dihydroxycarbonyls, which are then lost by dehydration to DHF [9]. Profile 3 is characteristic of second-generation products such as CHAN [10, 11], whose formation involves fast gas-phase reactions of first-generation DHF products with OH radicals, followed by partitioning to the particle phase. The rate of OH radical reactions with DHF is ∼ 10 times faster than with the other reaction products (CitationMartin et al. 2002). The time lag in Profile 3 relative to Profile 1 for first-generation products depends on the rate of particle-phase conversion of 1,4HC to DHF. The faster the conversion the smaller the lag. There are two profiles labeled as 4 because of their similarity. The upper one is characteristic of second-generation products formed by gas-phase reactions of first-generation products such as AN or 1,4HN with OH radicals, which then partition to the particle phase. Product concentrations increase monotonically, but more slowly than first-generation products and with a time lag during which first-generation product concentrations build up. An example is 1,4HN-N [14]. The lower Profile 4 is characteristic of third-generation products formed through a mechanism that includes rapid conversion of 1,4HC to gas-phase DHF via CHA, which then quickly react with OH radicals. This set of reactions is sufficiently fast that the time-dependence for the formation of third-generation products is similar to that for second-generation products formed by gas-phase reactions of first-generation products other than DHF. For example, these third-generation products can be formed when first-generation products such as AN or 1,4HN react with OH radicals and then follow the same reaction pathways that lead to CHAN from reactions of alkanes. The products are therefore CHAN that also contain the functional groups present in the first-generation products [21, 22]. The time lag in this profile relative to the upper Profile 4 depends on the rate of conversion of 1,4HC to DHF, but in this case is smaller than the lag between Profiles 1 and 3. These two profiles are essentially indistinguishable (hence the same Profile 4 label), and so are expected to be observed for all multi-generation products, with the exception of CHAN and HCHAN, which should follow Profile 3.
FIG. 3 Time profiles of the major types of SOA products obtained by modeling SOA formation from the OH radical-initiated reaction of n-pentadecane [PD] in the presence of NOx. Profile 1: first-generation products that have slow reactive losses. Profile 2: first-generation products that are lost rapidly through particle-phase reactions. Profile 3: second-generation products whose formation involves gas- and particle-phase reactions. Profile 4: (upper profile) second-generation products whose formation involves only gas-phase reactions, and (lower profile) third-generation products whose formation involves gas- and particle-phase reactions.
![FIG. 3 Time profiles of the major types of SOA products obtained by modeling SOA formation from the OH radical-initiated reaction of n-pentadecane [PD] in the presence of NOx. Profile 1: first-generation products that have slow reactive losses. Profile 2: first-generation products that are lost rapidly through particle-phase reactions. Profile 3: second-generation products whose formation involves gas- and particle-phase reactions. Profile 4: (upper profile) second-generation products whose formation involves only gas-phase reactions, and (lower profile) third-generation products whose formation involves gas- and particle-phase reactions.](/cms/asset/d71b13f4-0682-44db-a328-f258139a70f0/uast_a_380426_o_f0003g.gif)
RESULTS AND DISCUSSION
SOA Composition
The SOA products proposed to be formed from the reactions of the C15 alkanes, electron ionization fragmentation pathways (specified by neutral losses) that lead to characteristic mass spectral ion peaks, and Tdes are given in . Also shown are predicted and observed time profiles of characteristic ions measured during the PD reaction, classified as one of four types on the basis of the reaction mechanism as discussed above and shown in . The characteristic ions can be formed from the molecular ions of products shown in by the loss of simple, stable neutral species including H2, OH, H2O, (CH2)2, HCHO, NO2, NO3, and HNO3 as is commonly observed for compounds having these structures and functional groups (CitationMcLafferty and Turecek 1993; CitationGong et al. 2005; CitationLim and Ziemann 2005; CitationDocherty and Ziemann 2006). The assigned products are consistent with those expected from the reaction mechanism and with thermal desorption and time profiles.
TABLE 1 Proposed SOA products formed from OH radical-initiated reactions of linear, branched, and cyclic alkanes in the presence of NO x , and corresponding experimental evidence
In the following sections, the general features of mass spectra and profiles obtained for all alkanes are discussed, followed by profiles for individual ions that are used to identify specific SOA products. Results for the PD reaction are discussed in detail, using the information in to justify product assignments, followed by a brief discussion of the other alkanes, highlighting differences with the PD results. Note that to avoid crowding not all the characteristic ions that are discussed below are plotted in the figures; however, their Tdes values and time profiles are listed in .
Linear, Branched, and Cyclic Alkanes: General Trends
In this section, we discuss and compare the general features of the profiles observed for all the alkanes studied here, and later present profiles for ions that are used to identify individual reaction products. Real-time mass spectra obtained at the end of reactions of these compounds are shown in and . They cover the high-mass range above m/z 200 for the C15 alkanes and m/z 170 for CDD, where the mass spectral information is most useful for identifying products. One obvious and important observation is that the mass spectra of SOA formed from reactions of the three C15 compounds exhibit similar patterns (note the molecular weight of CPD is 210 compared to 212 for PD and 2,6,10-TMDD, so similar product peaks are shifted downwards by 2 mass units). For PD and 2,6,10-TMDD, distinctive peaks are present (in a few cases only in one spectrum) at m/z 211, 225, 239, 241, 253, 255, 258, 269, 272, 286, 300, 302, 316, 330, and 347 whereas for CPD peaks are present at m/z 209, 213, 223, 237, 239, 251, 253, 255, 267, 284, 300, and 345 (not shown). The close correspondence between many of these peaks indicates that the major SOA products of these reactions are similar. The mass spectrum of SOA formed from the reaction of CDD follows a pattern similar to that of CPD (shifted by 42 mass units) up to about m/z 225, and including m/z 242 and 258, but also has peaks at m/z 227, 230, 244, 260, and 272 for which corresponding peaks are not present in the CPD mass spectrum.
FIG. 4 Real-time mass spectra of SOA products formed from OH radical-initiated reactions of (a, b) n-pentadecane [PD] and (c) 2,6,10-trimethyldodecane [2,6,10-TMDD] in the presence of NOx. Contributions from DOS seed particles were subtracted.
![FIG. 4 Real-time mass spectra of SOA products formed from OH radical-initiated reactions of (a, b) n-pentadecane [PD] and (c) 2,6,10-trimethyldodecane [2,6,10-TMDD] in the presence of NOx. Contributions from DOS seed particles were subtracted.](/cms/asset/10cb9097-e8f2-4b4e-9503-0fd9cddbabf2/uast_a_380426_o_f0004g.gif)
FIG. 5 Real-time mass spectra of SOA products formed from OH radical-initiated reactions of (a) cyclopentadecane [CPD] and (b) cyclododecane [CDD] in the presence of NOx. Contributions from DOS seed particles were subtracted.
![FIG. 5 Real-time mass spectra of SOA products formed from OH radical-initiated reactions of (a) cyclopentadecane [CPD] and (b) cyclododecane [CDD] in the presence of NOx. Contributions from DOS seed particles were subtracted.](/cms/asset/6d7dca73-c6d3-4dc2-8000-5a9c7d2c69f7/uast_a_380426_o_f0005g.gif)
Thermal desorption profiles for m/z 46, 185, and the total ion (TI) signal from m/z 50–500 for SOA formed from the reactions of the C15 alkanes and CDD are shown in . The TI signal is proportional to organic mass (CitationCrable and Coggeshall 1958), m/z 46 (from NO2 +) identifies regions where organic nitrates are present, and m/z 185 is dominated by signal from DOS seed particles. The contribution of DOS to the TI signal was removed by multiplying the m/z 185 desorption profile by the ratio of the TI/(m/z 185) signals obtained from real-time mass spectra and then subtracting the resulting profile from the TI profile. The consistent desorption behavior of DOS (range of Tdes∼ 65–73°C, with differences probably reflecting effects of particle deposit size and the nature of the particle matrix) throughout these experiments indicates that desorption temperatures can be used to compare the volatility of products formed from different alkanes.
FIG. 6 Thermal desorption profiles of SOA products formed from OH radical-initiated reactions of (a) n-pentadecane [PD], (b) 2,6,10-trimethyldodecane [2,6,10-TMDD], (c) cyclopentadecane [CPD], and (d) cyclododecane [CDD] in the presence of NOx. The m/z 46 signal is from organic nitrates, m/z 185 is from DOS seed particles, and TI is the total signal from m/z 50–500, which is proportional to aerosol mass. The contribution of DOS seed particles to TI signal was subtracted and profiles were normalized to maximum intensities.
![FIG. 6 Thermal desorption profiles of SOA products formed from OH radical-initiated reactions of (a) n-pentadecane [PD], (b) 2,6,10-trimethyldodecane [2,6,10-TMDD], (c) cyclopentadecane [CPD], and (d) cyclododecane [CDD] in the presence of NOx. The m/z 46 signal is from organic nitrates, m/z 185 is from DOS seed particles, and TI is the total signal from m/z 50–500, which is proportional to aerosol mass. The contribution of DOS seed particles to TI signal was subtracted and profiles were normalized to maximum intensities.](/cms/asset/8c738b8d-a088-47ae-90de-f0df6821a8cc/uast_a_380426_o_f0006g.gif)
Peaks (maxima or shoulders) in TI profiles are present at about 33, 53, 95, and 131°C for PD, 42 and 86°C for 2,6,10-TMDD, at 37, 59, 128, and 179°C for CPD, and at 39, 123, and 188°C for CDD. Since the fraction of the total area under each peak in a TI profile is proportional to the contribution of the associated products to the total SOA mass, these curves indicate that SOA formed from the reactions of PD, 2,6,10-TMDD, and CPD contains significant amounts of products covering a large range in volatility from semi-volatile to low volatility (discussed further below). Conversely, SOA formed from the reaction of CDD is composed almost entirely of low volatility products, with most being less volatile than those formed from reactions of the three C15 alkanes. This difference and those noted above in the CPD and CDD mass spectra are probably due to differences in the tendencies of these two cyclic alkanes to form ring-opened products that can form oligomers.
Linear Alkanes
First-Generation Products: AN, 1,4HC, and 1,4HN
As shown in , at low temperatures the m/z 46 and TI profiles overlap completely, indicating that the most volatile SOA products contained nitrate. Since the more volatile carbonyls and CE should desorb before either of the potential nitrate-containing products, AN or 1,4HN, this observation supports statements made above that carbonyls and CE were too volatile to form SOA. The most volatile nitrate-containing products are AN [1], and are assigned to the first desorption peak in , which occurs at 12°C in the m/z 210 and 211 profiles. These ions are formed by loss of HNO3 and NO3 from the parent ion. No 1,4HC [6] were observed (except when NH3 was added before reacting n-hexadecane, as described below, in which case they desorbed at the same temperature as AN), because upon partitioning to the particle phase they rapidly isomerize to CHA [7]. 1,4HN [5] are assigned to peaks in the m/z 208, 215, and 225 profiles at 32°C. These ions are formed by loss of HNO3 + H2O, NO2 + (CH2)2, and NO2 + H2O from the parent ion. In , the m/z 210 and 215 time profiles are both similar to Profile 1, consistent with their assignments as first-generation products. The thermal desorption profiles in show that the m/z 215 signal comes only from 1,4HN, whereas the m/z 210 signal has contributions from other products that may have impacted the time profile. The time profile for m/z 46 is also similar to Profile 1, which is reasonable since the thermal desorption profile indicates that most of the signal comes from AN and 1,4HN. The TI time profile has a similar shape, but initially increases more rapidly than the others, apparently because of the many other products contributing to the TI ion signal.
FIG. 7 Thermal desorption profiles of SOA products formed from OH radical-initiated reaction of n-pentadecane [PD] in the presence of NOx. Profiles were normalized to maximum intensities.
![FIG. 7 Thermal desorption profiles of SOA products formed from OH radical-initiated reaction of n-pentadecane [PD] in the presence of NOx. Profiles were normalized to maximum intensities.](/cms/asset/cd974333-6a3a-4882-90ea-e125741ad230/uast_a_380426_o_f0007g.gif)
FIG. 8 Time profiles of SOA products formed from OH radical-initiated reaction of n-pentadecane [PD] in the presence of NOx. Profiles were corrected for wall losses by dividing by the DOS seed particle m/z 185 signal. Profiles were normalized to an average signal of unity in the neighborhood of 60 min. Smooth curves were drawn through m/z time profiles representative of first-generation 1,4-HN products.
![FIG. 8 Time profiles of SOA products formed from OH radical-initiated reaction of n-pentadecane [PD] in the presence of NOx. Profiles were corrected for wall losses by dividing by the DOS seed particle m/z 185 signal. Profiles were normalized to an average signal of unity in the neighborhood of 60 min. Smooth curves were drawn through m/z time profiles representative of first-generation 1,4-HN products.](/cms/asset/9fd0ff77-251a-4c4d-9c69-e308bde10572/uast_a_380426_o_f0008g.gif)
First-Generation Products: CHA and HCHA
The CHA [7] and HCHA [8] are assigned to peaks at 90 and 92°C in the m/z 225 and 241 profiles in . These characteristic ions are both formed by loss of OH from the parent ion, which is a major fragmentation pathway for hydroxytetrahydrofurans and other cyclic hydroxyethers that have an OH group in the 2 position (CitationMcLafferty and Turecek 1993; CitationGong et al. 2005). The m/z 225 and 241 time profiles in are both similar to Profile 2, consistent with products formed by particle-phase isomerization of 1,4HC that then disappear when they dehydrate to DHF. The thermal desorption profiles in indicate that most of the signals from these ions come from these compounds, with smaller contributions from other products. The time profile of m/z 241 increases and decreases more slowly than that for m/z 225, possibly because the additional hydroxy group in HCHA affects rates of cyclization and dehydration.
Multi-Generation Products
A large number of multi-generation reaction products are also proposed to be present in SOA. It is important to note that their Tdes are consistent with expectations based on structure, the time profiles can be explained by the reaction mechanism, and the electron ionization fragmentation patterns are consistent with the neutral losses observed for AN, 1,4HN, and CHA.
The multi-generation products are assigned in order of increasing Tdes as follows: 26°C (m/z 241) = CE-N [18]; 82°C (m/z 269) = CE-1,4HN [19], 1,4HN-N [14], and CE-CHA [20]; 92°C (m/z 255) = CHA-1,4HN [16]; 95 (m/z 239) = CHAN [10] and CHA-N [15] (although they are isomers, these are formed by a different mechanism than CHAN); 100°C (m/z 286) = CHAN [11] and CHA-N [15]; 98°C (m/z 330) = CE-CHAN [23], 103°C (m/z 300 and 302) = HCHAN [17], 116°C (m/z 347) = CHAN-N [21], and 143°C (m/z 363) = CHAN-1,4HN [22]. The order of Tdes of these compounds is consistent with the expected reductions in vapor pressures due to the addition of functional groups (CitationPankow and Asher 2008): hydroxy + nitrate > hydroxy ≅ nitrate > carbonyl + ester. AN-N, which would be expected to desorb close to 1,4HN, were not assigned a Tdes because the potential characteristic ions could also be explained as coming from other, more abundant products. Also, no product was assigned to the 53°C peak in the m/z 258 profile, but the time profile (shown and discussed below) and the behavior in the presence of NH3 (shown below for other ions) suggest it is formed through a mechanism similar to the one leading to CHAN.
The m/z 239, 286, 300, and 302 time profiles in are similar to Profile 1, rather than to Profiles 3 and 4, as expected for the assigned products: CHAN [10, 11] and HCHAN [17] (Profile 3), and CHA-N [15] (Profile 4). It is highly unlikely, however, that these ions are associated with first-generation products, since those have already been assigned and cannot logically explain the observed ions or Tdes. Furthermore, as will be shown below, when NH3 was added to the reaction mixture there was a dramatic reduction in the formation of these ions, but not those associated with first-generation products such as AN and 1,4HN. The reduction is consistent with products whose formation is slowed because NH3 neutralizes the HNO3 catalyst of the 1,4HC isomerization reaction. If these ions were primarily associated with CHAN and HCHAN, they would all be expected to have time profiles similar to Profile 3. In the present case, the agreement between the measured and modeled profiles is good at longer times, but there is a discrepancy at early times. Such a discrepancy is not unreasonable, since the parameters used in the model were obtained by fitting the model to the time profile of CHA, and so may not be entirely appropriate for CHAN and HCHAN. For example, similar to CHA, it appears that CHAN can dehydrate. This is most apparent in time profiles of these ions in the 2,6,10-TMDD reaction (m/z 286 is shown in ; the others behave similarly), which look like Profile 2. If dehydration rates differ significantly for CHA and CHAN, the time profiles will also differ.
The m/z 255, 269, 330, 347, and 363 time profiles in are all similar to Profile 4, with a significant lag relative to time profiles of first-generation products, consistent with their assignments to various multi-generation products [14, 16, 19–23]. The narrowness of the thermal desorption profiles of some of these ions in indicates that the majority of the signals come from single compounds or isomers, but some clearly have significant contributions from more than one product. Even though the products were formed by different reaction mechanisms, the modeled and measured time profiles are similar, indicating that it is difficult to distinguish different types of multi-generation products solely on the basis of time profiles.
Effect of Ammonia
An experiment in which NH3 was added to the environmental chamber prior to initiating the reaction provides additional support for the assignment of products in , and also allows the identification of 1,4HC in the SOA. Profiles of a few characteristic ions of products of the reactions of the C16 alkane n-hexadecane (HD) in the absence and presence of NH3 are shown in . This alkane was used because the 1,4HC formed in the PD reaction were too volatile to form SOA. Note that the m/z values are all shifted upwards by 14 mass units relative to those given in for PD reaction products. The thermal desorption and time profiles of m/z 224 and 229, which are associated with first-generation AN and 1,4HN products whose formation and loss involves only gas-phase reactions, were essentially unchanged by the addition of NH3. For example, Tdes = 19 and 17°C for m/z 224 and 37 and 35°C for m/z 229. Conversely, m/z 238, 239, and 253, which are associated with 1,4HC, CHA, and CHAN whose formation and/or loss processes involve particle-phase isomerization and/or dehydration reactions, were affected by the addition of NH3. When NH3 was added a new peak appeared in the thermal desorption profile of m/z 238 at 17°C (same Tdes as AN), which is assigned to the 1,4HC of mass 256. This compound should lose H2O when ionized since this is a major fragmentation pathway for carbonyls and alcohols (CitationMcLafferty and Turecek 1993). It is also reasonable that a compound having a carbonyl group and hydroxy group desorbed at the same temperature as AN but before 1,4HN (35°C), since hydroxy and nitrate groups reduce vapor pressure more than carbonyl groups (CitationPankow and Asher 2008). The addition of NH3 also led to the disappearance of peaks at 86, 94, and 98°C in the thermal desorption profiles of m/z 238, 239, and 253, which are associated with CE-1,4HN, CHA, and CHAN. The rates of formation of these compounds were also reduced, as shown by the time profiles. The time profile of m/z 238 did not change significantly, because, as shown in the thermal desorption profiles, although signal from the 1,4HN-CE decreased in the presence of NH3, signal from the first-generation 1,4HC increased.
FIG. 9 Thermal desorption and time profiles of SOA products formed from OH radical-initiated reactions of n-hexdecane [HD] in the presence of NOx. (a) and (c) are for reactions performed in the absence of NH3, and (b) and (d) are for reactions performed in the presence of NH3. Profiles were treated as described in and .
![FIG. 9 Thermal desorption and time profiles of SOA products formed from OH radical-initiated reactions of n-hexdecane [HD] in the presence of NOx. (a) and (c) are for reactions performed in the absence of NH3, and (b) and (d) are for reactions performed in the presence of NH3. Profiles were treated as described in Figure 7 and Figure 8.](/cms/asset/cf443137-9f02-4752-acf2-58456ef68aff/uast_a_380426_o_f0009g.gif)
The most likely explanation for these results is that the presence of NH3 slowed the rate of isomerization of 1,4HC to CHA in particles by neutralizing HNO3 (formed in the chamber from the reaction of OH radicals with NO2), which is known from solution studies to catalyze this isomerization (CitationBruckner 2002). A similar effect was observed for the conversion of smaller 1,4HC to DHF on chamber walls when pyridine was added (CitationAtkinson et al. 2008). It has been shown that HNO3 adsorbs and dissociates on hydrophobic organic surfaces (CitationHandley et al. 2007), so it should be a source of catalytic protons on organic particles and chamber walls. The only other known potential effect of NH3 on the reaction mechanism shown in is that it can bind to hydroxyperoxy radicals and thereby increase the formation of hydroxyalkoxy radicals from the reaction with NO (CitationMatsunaga and Ziemann 2009), in this case at the expense of 1,4HN, the reaction co-product. This would increase the yield of 1,4HC (by possibly 10–20% from ∼ 0.5 if no 1,4-HN were formed (CitationReisen et al. 2005; CitationLim and Ziemann 2005)) and also enhance CHA formation. Although we cannot rule out the possibility that NH3 altered the reaction of 1,4-hydroxyperoxy radicals with NO, any impact appears to have been minor. In the presence of NH3, there was no obvious decrease in the formation of 1,4HN, CHA formation was dramatically reduced (possibly eliminated, with the remaining m/z 239 signal being due to another compound), and 1,4-HC appeared in SOA for the first time. All these observations are consistent with neutralization of HNO3 catalyst by NH3.
Branched and Cyclic Alkanes
The thermal desorption and time profiles of some of the same characteristic ions shown and discussed above for the PD reaction are shown in and for the 2,6,10-TMDD and CPD reactions. The products assigned to these ions are the same as those assigned to the corresponding ions for the PD reaction, as shown in . In general, the profiles are consistent with the assignments. There are also a few noteworthy differences in the results obtained for the reactions of these three alkanes. The time profiles of CHAN and HCHAN associated with m/z 239, 286, 300, and 302 (only m/z 286 is shown) for the 2,6,10-TMDD () reaction are similar to Profile 2, whereas the corresponding ions for the PD () and CPD () reactions are not. This suggests that these compounds have a stronger tendency to dehydrate when they are formed from reactions of branched alkanes. One reason could stem from the substitution of a CH3 group for an H atom in a CH2 group, which weakens the remaining C-H bond since the H atom is then tertiary instead of secondary. If this group were located adjacent to the OH group in the furan ring, as in CHAN 10, then the H atom would combine more easily with the adjacent OH group to form H2O in the dehydration reaction. It is also worth noting that there were no AN in the SOA formed from the CPD reaction, as indicated by the absence of a low temperature peak in the m/z 209 profile (the peak at 35°C is from 1,4HN). This is different from the PD and 2,6,10-TMDD results, and is probably due to insufficient gas-to-particle partitioning. The vapor pressures of cyclic AN may be higher than those formed from linear and branched alkanes because the nitrate group is never near the end of a carbon chain where it has the greatest effect on vapor pressure. This result is interesting, since Tdes of all products formed from reactions of CPD, other than 1,4HN, are ∼ 30–40°C higher than those formed from reactions of PD and 2,6,10-TMDD. The presence of a CHA ring structure also appears to dramatically lower vapor pressures, as indicated by the much higher Tdes of these products in all reactions. For example, as shown in , Tdes = 17°C for 1,4HC formed from the reaction of HD, but increased to 94°C when they isomerized to CHA. For products of the CPD reactions, some of which are bicyclic, the effect is greater. The reason for this is not understood, but could involve the formation of oligomers that dissociate at sufficiently high temperatures to monomers that subsequently evaporate and are analyzed.
FIG. 10 (a, b) Thermal desorption profiles and (c, d) time profiles of SOA products formed from OH radical-initiated reaction of 2,6,10-trimethyldodecane [2,6,10-TMDD] in the presence of NOx. Profiles were treated as described in and .
![FIG. 10 (a, b) Thermal desorption profiles and (c, d) time profiles of SOA products formed from OH radical-initiated reaction of 2,6,10-trimethyldodecane [2,6,10-TMDD] in the presence of NOx. Profiles were treated as described in Figure 7 and Figure 8.](/cms/asset/ca38e203-4d38-40fb-94de-9c4273f9ed9b/uast_a_380426_o_f0010g.gif)
FIG. 11 (a, b) Thermal desorption profiles and (c, d) time profiles of SOA products formed from OH radical-initiated reaction of cyclopentadecane [CPD] in the presence of NOx. Profiles were treated as described in and .
![FIG. 11 (a, b) Thermal desorption profiles and (c, d) time profiles of SOA products formed from OH radical-initiated reaction of cyclopentadecane [CPD] in the presence of NOx. Profiles were treated as described in Figure 7 and Figure 8.](/cms/asset/6af42808-e4dc-4ae8-a9f7-e937343c7f04/uast_a_380426_o_f0011g.gif)
Thermal desorption and time profiles for the major ions observed in the mass spectrum of SOA formed from the reaction of CDD () are shown in . Unlike the profiles measured for the reactions of C15 alkanes, which have many distinctive features that allow for the identification of a variety of different products, these profiles exhibit a fairly narrow range of behaviors. Except for m/z 171, whose thermal desorption profile has a small peak at 31°C that is probably due to 1,4HN, all other ions have one of the three profiles shown in for m/z 195 (same for m/z 197 and 209), m/z 211 (same for m/z 213, 225, and 227), and m/z 230 (same for m/z 242, 244, 258, 260, and 272). As mentioned previously, the major ions up to m/z 225 are similar to those observed for the CPD reaction (shifted by 42 mass units because of the difference in molecular weights of the parent compounds), indicating some similarities in products. From m/z 227 to 272, however, much of the similarity disappears. The time profiles of the major ions all fall within the range of profiles shown in for m/z 197 and 209, which are most similar to Profiles 1 and 4.
FIG. 12 (a) Thermal desorption profiles and (b) time profiles of SOA products formed from OH radical-initiated reaction of cyclododecane [CDD] in the presence of NOx. Profiles were treated as described in and .
![FIG. 12 (a) Thermal desorption profiles and (b) time profiles of SOA products formed from OH radical-initiated reaction of cyclododecane [CDD] in the presence of NOx. Profiles were treated as described in Figure 7 and Figure 8.](/cms/asset/23b50543-c5f5-4d98-8382-febe8635ee04/uast_a_380426_o_f0012g.gif)
One important difference between the reactions of CDD and CPD is that alkoxy radicals formed in the CDD reaction are expected to decompose more rapidly, due to greater ring strain, leading to ring-opened products having an additional aldehyde group. According to calculations made using the method of CitationAtkinson (2007), approximately 16% of CDD alkoxy radicals decompose compared to less than 1% of those formed from CPD (CitationLim and Ziemann 2009b). One explanation for the similarity in the thermal desorption and time profiles from the CDD reaction, as well as the low volatility of most of the products, is that the ring-opened products form oligomers (possibly hemiacetals) through reactions of aldehyde groups with hydroxy groups present in all products having CHA structures. As noted above, aldehyde formation from reactions of linear alkanes is negligible.
ATMOSPHERIC IMPLICATIONS
In a future publication, we will present vapor pressures estimated from Tdes values of products formed from reactions of a large variety of alkanes and alkenes. For the purposes of the present discussion, it is sufficient to note that from calibrations carried out with a series of alkanes having known vapor pressures at 25°C (P25) we obtained the following equation:
When applied to the Tdes of ∼ 65–73°C measured here for DOS seed particles, the estimated P25 values are 2 × 10−6–4 × 10−7 Pa, in reasonable agreement with the literature value of 3 × 10−6 Pa (CitationRader et al. 1987). Calculated P25 values are probably accurate to within about an order of magnitude. Although this equation could be used to estimate the vapor pressures of all the products observed here, values calculated for very high Tdes should be viewed with caution. As noted above, these compounds may exist as oligomers, in which case the vapor pressures are matrix dependent. For the range of Tdes values observed in this study, ∼ 12–185°C, corresponding P25 are ∼ 10−2–10−16 Pa. This is an enormously large range in volatility, corresponding to gas-particle partitioning behavior ranging from semi-volatile (distributed between the gas and particle phases) to low volatility (essentially entirely in the particle phase).
We can use Equation (Equation1) with gas-particle partitioning theory (CitationPankow 1994) to gain further insight into SOA formation from the oxidation of alkanes in the atmosphere. For a typical atmospheric organic aerosol mass concentration of 5 μ g m–3 (CitationZhang et al. 2007), compounds with molecular weights of 300 g mol–1 and vapor pressures of 4 × 10–5 Pa will be distributed equally between the gas and particle phases. Products that desorb at temperatures above ∼ 50°C in the present experiments have vapor pressures less than this value, and should therefore exist predominantly in the particle phase (for 60°C the fraction would be ∼ 90%). This temperature falls slightly above Tdes measured for 1,4-HN formed from C15 linear, branched, and cyclic alkanes, the least volatile first-generation products, indicating that for alkanes smaller than ∼ C15 contributions to SOA should consist mostly of multi-generation, more oxidized products. For alkanes larger than C15, each additional carbon number should decrease product vapor pressures by a factor of ∼ 3 (CitationLuxenhofer et al. 1996; CitationPankow and Asher 2008). From Equation (Equation1), the vapor pressures of C15 AN, the most volatile SOA products observed here, are estimated to be ∼ 3 × 10−2 Pa, in good agreement with a value of ∼ 2 × 10−2 Pa obtained by interpolating between literature values for 2-alkyl nitrates (CitationLuxenhofer et al. 1996). Reactions of alkanes larger than ∼ C21 should therefore form AN, 1,4HC (those formed from the HD reaction in the presence of NH3 had essentially the same Tdes as AN), and 1,4HN with vapor pressures less than 4 × 10−5 Pa, which should exist predominantly in the particle phase. The subsequent fate of 1,4HC depends on particle acidity. If the particles are non-acidic, 1,4HC should remain in the particles, whereas if the particles are acidic they should isomerize to CHA that then dehydrate to volatile DHF. In the atmosphere, DHF will react with O3 within a few minutes (and perhaps with NO3 radicals at night) (CitationMartin et al. 2002). The products of the O3 reactions have not been thoroughly investigated (CitationMartin et al. 2002), but are likely similar to the multifunctional compounds formed from reactions of simple cyclic alkenes and monoterpenes (CitationYu et al. 1999; CitationGao et al. 2004). Many of these compounds should exist predominantly in the particle phase, certainly for carbon numbers greater than ∼ C21, and to some extent for smaller carbon numbers as well (especially compounds such as dicarboxylic acids).
As discussed above, AN, 1,4HC, and 1,4HN comprise all the major known and expected (based on current understanding of reaction mechanisms) first-generation products formed from OH radical-initiated reactions of linear alkanes. The alkoxy radicals formed in these reactions do not decompose, so no fragmentation products are formed. Reactions of C6 and C15 cyclic alkanes, whose alkoxy radicals also do not decompose (CitationLim and Ziemann 2009b), form cyclic AN, 1,4HC, and 1,4HN. Alkoxy radicals formed from reactions of cyclic alkanes with carbon numbers between C6 and C15 will decompose to some extent, leading to the formation of ring-opened products. These should be similar, however, to those formed from reactions of the corresponding linear alkanes, but with an additional aldehyde group. Only reactions of branched alkanes are expected to form first-generation products via alkoxy radical decomposition, these being carbonyl fragmentation products [3, 4] that have much higher vapor pressures than AN products. In the atmosphere, the gas-phase oxidation of linear, cyclic, and slightly branched alkanes larger than ∼ C21 is therefore expected to form SOA in high yields (approaching ∼ 1 or higher on a mass basis), with the composition dominated by first-generation AN, 1,4HC (and/or DHF + O3 reaction products), and 1,4HN products. This SOA should be less oxidized than that formed from smaller alkanes, which would contain a greater fraction of multi-generation products. The vapor pressures of alkanes (CitationLemmon and Goodwin 2000) larger than ∼ C30 are sufficiently low that they should exist predominantly in the particle phase, where they would undergo much slower heterogeneous oxidation by OH radicals (CitationBertram et al. 2001).
CONCLUSIONS
Identifying SOA products formed though a complex series of reactions is always a challenge, due in large part to the difficulties inherent in analyzing multifunctional, low volatility compounds, and the lack of authentic standards that can be used for mass spectral matching. Nonetheless, by combining the information available in TDPBMS thermal desorption profiles and time profiles with an understanding of electron ionization mass spectra and a proposed reaction mechanism based on well-established chemistry, it was possible to develop a self-consistent set of reaction product assignments.
The products and mechanisms of SOA formation from OH radical-initiated reactions of linear, branched, and cyclic alkanes in the presence of NOx are similar in many respects, but also differ due to effects of molecular structure. The major first-generation SOA products were alkyl nitrates, 1,4-hydroxycarbonyls, and 1,4-hydroxynitrates, consistent with previous studies of gas-phase reaction products. When 1,4-hydroxycarbonyls partition to the particle phase, they can isomerize to cyclic hemiacetals that then dehydrate to volatile, reactive dihydrofurans. This particle-phase process was monitored with the TDPBMS by following the time profiles of ions that are characteristic of cyclic hemiacetals. Under normal reaction conditions, conversion was rapid, but in the presence of NH3 it slowed dramatically, indicating that HNO3 that forms via OH + NO2 reactions catalyzes the conversion. In addition to first-generation products, more functionalized multi-generation products were also formed through further gas-phase reactions with OH radicals. The final product mixture consisted of compounds with acyclic and cyclic structures containing various combinations of hydroxy, carbonyl, nitrate, ether, and ester groups.
Alkane structure primarily impacts the formation of SOA products through its effect on alkoxy radicals, in particular, the relative rates of isomerization and decomposition of these reactive intermediates. For linear alkanes, isomerization dominates, leading to more functionalized, lower volatility products. Branching and ring strain increases decomposition. For branched alkanes, this leads to fragmentation and the formation of more volatile products, whereas for cyclic alkanes ring-opened products are formed. These ring-opened products contain an aldehyde group that apparently allows them to form low volatility oligomers, possibly hemiacetals. One can therefore expect that not only SOA composition, but also yields, will be impacted by alkane structure. Elsewhere, we report the results of SOA yield measurements made on a large variety of linear, branched, and cyclic alkanes (CitationLim and Ziemann 2009b), and show that this is most definitely the case.
In the atmosphere, the yields of SOA formed from OH radical-initiated reactions of ∼ C21–C30 alkanes in high NOx regions are expected to be on the order of 1, with the composition dominated by first-generation alkyl nitrate, 1,4-hydroxycarbonyl (and/or dihydrofuran + O3 reaction products), and 1,4-hydroxynitrate products. This SOA should be less oxidized than that formed from smaller alkanes, which would contain a greater fraction of multi-generation reaction products. SOA formed from reactions of alkanes smaller than ∼ C15 should be essentially all multi-generation products. The C/O ratio, a quantity that can now be measured routinely to characterize the degree of oxidation of organic aerosol and which is typically ∼ 0.5–1 for atmospheric SOA (CitationAiken et al. 2008), should be ∼ 0.1–0.15 for SOA formed from the oxidation of C21-C30 alkanes. Ratios could approach ∼ 0.5 and perhaps higher values for much smaller alkanes whose oxidation forms SOA comprised mostly of multi-generation products, especially those formed from the reaction of O3 with dihydrofurans.
Acknowledgments
This material is based on work supported by the National Science Foundation under Grants ATM-0234586 and ATM-0650061. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author and do not necessarily reflect the views of the National Science Foundation (NSF). We also thank Roger Atkinson for helpful discussions.
Notes
a Numbers refer to profiles shown in and are predicted from product assignment and observed.
b Not observed.
c Observed for HD reaction in the presence of NH3.
d ND = Not determined because other compounds dominated signal in the profile.
1Also in the Department of Chemistry. Present address: Department of Environmental Sciences, Rutgers University, New Brunswick, NJ
2Also in the Department of Environmental Sciences and Department of Chemistry.
Related Research Data
REFERENCES
- Aiken , A. C. , DeCarlo , P. F. , Kroll , J. H. , Worsnop , D. R. , Huffman , J. A. , Docherty , K. , Ulbrich , I. M. , Mohr , C. , Kimmel , J. R. , Sueper , D. , Zhang , Q. , Sun , Y. , Trimborn , A. , Northway , M. , Ziemann , P. J. , Canagaratna , M. R. , Onasch , T. B. , Alfarra , R. , Prevot , A. S. H. , Dommen , J. , Duplissy , J. , Metzger , A. , Baltensperger , U. and Jimenez , J. L. 2008 . O/C and OM/OC Ratios of Primary, Secondary, and Ambient Organic Aerosols with High Resolution Time-of-Flight Aerosol Mass Spectrometry . Environ. Sci. Technol. , 42 : 4478 – 4485 .
- Andreae , M. O. and Crutzen , P. J. 1997 . Atmospheric Aerosols: Biogeochemical Sources and Role in Atmospheric Chemistry . Science , 276 : 1052 – 1058 .
- Arey , J. , Aschmann , S. M. , Kwok , E. S. C. and Atkinson , R. 2001 . Alkyl Nitrate, Hydroxyalkyl Nitrate, and Hydroxycarbonyl Formation from the NOx-Air Photooxidations of C5-C8 n-Alkanes . J. Phys. Chem. A , 105 : 1020 – 1027 .
- Atkinson , R. 1997 . Gas-Phase Tropospheric Chemistry of Volatile Organic Compounds: 1. Alkanes and Alkenes . J. Phys. Chem. Ref. Data , 26 : 215 – 290 .
- Atkinson , R. 2007 . Rate Constants for the Atmospheric Reactions of Alkoxy Radicals: An Updated Estimation Method . Atmos. Environ. , 41 : 8468 – 8485 .
- Atkinson , R. , Carter , W. P. L. , Winer , A. M. and Pitts , J. N. Jr. 1981 . An Experimental Protocol for the Determination of OH Radical Rate Constants with Organics Using Methyl Nitrite Photolysis as an OH Radical Source . Air Pollut. Control Assoc. , 31 : 1090 – 1092 .
- Atkinson , R. and Arey , J. 2003 . Atmospheric Degradation of Volatile Organic Compounds . Chem. Rev. , 103 : 4605 – 4638 .
- Atkinson , R. , Arey , J. and Aschmann , S. M. 2008 . Atmospheric Chemistry of Alkanes: Review and Recent Developments . Atmos. Environ. , 42 : 5859 – 5871 .
- Bertram , A. K. , Ivanov , A. V. , Hunter , M. , Molina , L. T. and Molina , M. J. 2001 . The Reaction Probability of OH on Organic Surfaces of Tropospheric Interest . J. Phys. Chem. A , 105 : 9415 – 9421 .
- Bruckner , R. 2002 . Advanced Organic Chemistry , San Diego : Harcourt/Academic Press .
- Calvert , J. G. , Atkinson , R. , Kerr , J. A. , Madronich , S. , Moortgat , G. K. , Wallington , T. J. and Yarwood , G. 2000 . The Mechanisms of Atmospheric Oxidation of the Alkenes, , Oxford : Oxford University Press .
- Chattopadhyay , S. and Ziemann , P. J. 2005 . Vapor Pressures of Substituted Monocarboxylic and Dicarboxylic Acids Measured Using an Improved Thermal Desorption Particle Beam Mass Spectrometry Method . Aerosol Sci. Technol. , 39 : 1085 – 1100 .
- Crable , G. F. and Coggeshall , N. D. 1958 . Application of Total Ionization Principles to Mass Spectrometric Analysis . Anal. Chem. , 30 : 310 – 313 .
- Docherty , K. S. and Ziemann , P. J. 2006 . Reaction of Oleic Acid Particles with NO3 Radicals: Products, Mechanism, and Implications for Radical-Initiated Organic Aerosol Oxidation . J. Phys. Chem. A , 110 : 3567 – 3577 .
- Englert , N. 2004 . Fine Particles and Human Health—A Review of Epidemiological Studies . Toxicol. Lett. , 149 : 235 – 242 .
- Gao , S. , Keywood , M. , Ng , N. L. , Surratt , J. , Varutbangkul , V. , Bahreini , R. , Flagan , R. C. and Seinfeld , J. H. 2004 . Low-Molecular Weight and Oligomeric Components in Secondary Organic Aerosol from the Ozonolysis of Cycloalkenes and α -Pinene . J. Phys. Chem. A , 108 : 10147 – 10164 .
- Gong , H. , Matsunaga , A. and Ziemann , P. J. 2005 . Products and Mechanism of Secondary Organic Aerosol Formation from Reactions of Linear Alkenes with NO3 Radicals . J. Phys. Chem. A , 109 : 4312 – 4324 .
- Guenther , A. , Hewitt , C. N. , Erickson , D. , Fall , R. , Geron , C. , Graedel , T. , Harley , P. , Klinger , L. , Lerdau , M. , McKay , W. A. , Pierce , T. , Scholes , B. , Steinbrecher , R. , Tallamraju , R. , Taylor , J. and Zimmermann , P. A. 1995 . Global Model of Natural Volatile Organic Compound Emissions . J. Geophys. Res. , 100 : 8873 – 8892 .
- Handley , S. R. , Clifford , D. and Donaldson , D. J. 2007 . Photochemical Loss of Nitric Acid on Organic Films: A Possible Recycling Mechanism for NOx . Environ. Sci. Technol. , 41 : 3898 – 3903 .
- Holt , T. , Arey , J. and Atkinson , R. 2005 . Effect of Water Vapor Concentration on the Conversion of a Series of 1,4-Hydroxycarbonyls to Dihydrofurans . J. Photochem. Photobiol. A: Chem. , 176 : 231 – 237 .
- Jordan , C. E. , Ziemann , P. J. , Griffin , R. J. , Lim , Y. B. , Atkinson , R. and Arey , J. 2008 . Modeling SOA Formation from OH Reactions with C8-C17 n-Alkanes . Atmos. Environ. , 42 : 8015 – 8026 .
- Kanakidou , M. , Seinfeld , J. H. , Pandis , S. N. , Barnes , I. , Dentener , F. J. , Facchini , M. C. , Van Dingenen , R. , Ervens , B. , Nenes , A. , Nielsen , C. J. , Swietlicki , E. , Putaud , J. P. , Balkanski , Y. , Fuzzi , S. , Horth , J. , Moortgat , G. K. , Winterhalter , R. , Myhre , C. E. L. , Tsigaridis , K. , Vignati , E. , Stephanou , E. G. and Wilson , J. 2005 . Organic Aerosol and Global Climate Modelling: A Review . Atmos. Chem. Phys. , 5 : 1053 – 1123 .
- Kroll , J. H. and Seinfeld , J. H. 2008 . Chemistry of Secondary Organic Aerosol: Formation and Evolution of Low-Volatility Organics in the Atmosphere . Atmos. Environ. , 42 : 3593 – 3624 .
- Lemmon , E. W. and Goodwin , A. R. H. 2000 . Critical Properties and Vapor Pressure Equation for Alkanes C n H2n + 2: Normal Alkanes with n ≤ 36 and Isomers for n = 4 through n = 9 . J. Chem. Phys. Ref. Data , 29 : 1 – 39 .
- Lim , Y. B. and Ziemann , P. J. 2005 . Products and Mechanism of Secondary Organic Aerosol Formation from Reactions of n-Alkanes with OH Radicals in the Presence of NOx . Environ. Sci. Technol. , 39 : 9229 – 9236 .
- Lim , Y. B. and Ziemann , P. J. 2009a . Kinetics of the Heterogeneous Conversion of 1,4-Hydroxycarbonyls to Cyclic Hemiacetals and Dihydrofurans on Organic Aerosol Particles . Phys. Chem. Chem. Phys , In preparation
- Lim , Y. B. and Ziemann , P. J. 2009b . Effects of Molecular Structure on Yields of Secondary Organic Aerosol Formed from OH Radical-Initiated Reactions of Linear, Branched, and Cyclic Alkanes in the Presence of NOx . Environ. Sci. Technol. , In press
- Luxenhofer , O. and Ballschmiter , K. 1996 . Semivolatile Long Chain C6–C17 Alkyl Nitrates as Trace Compounds in Air . Chemosphere , 33 : 393 – 404 .
- Martin , P. , Tuazon , E. C. , Aschmann , S. M. , Arey , J. and Atkinson , R. 2002 . Formation and Atmospheric Reactions of 4,5-Dihydro-2-methylfuran . J. Phys. Chem. A , 106 : 11492 – 11501 .
- Matsunaga , A. and Ziemann , P. J. 2009 . Yields of β -Hydroxynitrates and Dihydroxynitrates in Aerosol Formed from OH Radical-Initiated Reactions of Linear Alkenes in the Presence of NOx . J. Phys. Chem. A , 113 : 599 – 606 .
- McLafferty , F. W. and Turecek , F. 1993 . Interpretation of Mass Spectra , Sausalito, CA : Fourth Edition, University Science Books .
- Pankow , J. F. 1994 . An Absorption Model of the Gas/Aerosol Partitioning Involved in the Formation of Secondary Organic Aerosol . Atmos. Environ. , 28 : 189 – 193 .
- Pankow , J. F. and Asher , W. E. 2008 . SIMPOL.1: A Simple Group Contribution Method for Predicting Vapor Pressures and Enthalpies of Vaporization of Multifunctional Organic Compounds . Atmos. Chem. Phys. , 8 : 2773 – 2796 .
- Rader , D. J. , McMurry , P. H. and Smith , S. 1987 . Evaporation Rates of Monodisperse Organic Aerosols in the 0.02 to 0.2–μ m Diameter Range . Aerosol Sci. Technol. , 6 : 247 – 260 .
- Reisen , F. , Aschmann , S. M. , Atkinson , R. and Arey , J. 2005 . 1,4-Hydroxycarbonyl Products of the OH Radical Initiated Reactions of C5-C8 n-Alkanes in the Presence of NO . Environ. Sci. Technol. , 39 : 4447 – 4453 .
- Robinson , A. L. , Donahue , N. M. , Shrivastava , M. K. , Weitkamp , E. A. , Sage , A. M. , Grieshop , A. P. , Lane , T. E. , Pierce , J. R. and Pandis , S. N. 2007 . Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging . Science , 315 : 1259 – 1262 .
- Seinfeld , J. H. and Pandis , S. N. 1998 . Atmospheric Chemistry and Physics, , First Edition , New York : Wiley-Interscience .
- Taylor , W. D. , Allston , T. D. , Moscato , M. J. , Fazekas , G. B. , Kozlowski , R. and Takacs , G. A. 1980 . Atmospheric Photodissociation Lifetimes for Nitromethane, Methyl Nitrite, and Methyl Nitrate . Int. J. Chem. Kinet. , 12 : 231 – 240 .
- Tobias , H. J. and Ziemann , P. J. 1999 . Compound Identification in Organic Aerosols Using Temperature-Programmed Thermal Desorption Particle Beam Mass Spectrometry . Anal. Chem. , 71 : 3428 – 3435 .
- Tobias , H. J. , Kooiman , P. M. , Docherty , K. S. and Ziemann , P. J. 2000 . Real-Time Chemical Analysis of Organic Aerosols Using a Thermal Desorption Particle Beam Mass Spectrometer . Aerosol Sci. Technol. , 33 : 170 – 190 .
- Yu , J. , Cocker , D. R. III , Griffin , R. J. , Flagan , R. C. and Seinfeld , J. H. 1999 . Gas-Phase Ozone Oxidation of Monoterpenes: Gaseous and Particulate Products . J. Atmos. Chem. , 34 : 207 – 258 .
- Zhang , Q. , Jimenez , J. L. , Canagaratna , M. R. , Allan , J. D. , Coe , H. , Ulbrich , I. , Alfarra , M. R. , Takami , A. , Middlebrook , A. M. , Sun , Y. L. , Dzepina , K. , Dunlea , E. , Docherty , K. , DeCarlo , P. F. , Salcedo , D. , Onasch , T. , Jayne , J. T. , Miyoshi , T. , Shimono , A. , Hatakeyama , S. , Takegawa , N. , Kondo , Y. , Schneider , J. , Drewnick , F. , Weimer , S. , Demerjian , K. , Williams , P. , Bower , K. , Bahreini , R. , Cottrell , L. , Griffin , R. J. , Rautiainen , J. and Worsnop , D. R. 2007 . Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenically-Influenced Northern Hemisphere Mid-Latitudes . Geophys. Res. Lett. , 34 ( L13801 ) doi:10.1029/2007GL029979