Abstract
We compared measurements of organic molecular markers made using a novel Thermal Desorption Aerosol Gas Chromatograph/Mass Spectrometer (TAG) with two offline filter-based methods: solvent extraction GC/MS (SE-GC/MS) and thermal desorption GC/MS (TD-GC/MS). TAG is designed for automated, high time-resolved measurements of organic aerosol speciation. Laboratory and field measurements were performed to compare TAG and filter performance for n-alkanes, n-alkanoic acids, cholesterol, PAHs, and hopanes. Laboratory measurements of model organic aerosol mixtures of known composition were made in the Carnegie Mellon University smog chamber, and field measurements were made in downtown Pittsburgh. There was excellent agreement between techniques for hopanes and several PAHs, which are important markers for motor vehicle emissions. Agreement was also strong for moderately polar and nonpolar species in the high-concentration smog chamber experiments. Poorer agreement between filter and TAG observations was obtained for n-alkanes in ambient measurements. To further investigate the differences in n-alkane performance between these methods, potential matrix effects and internal consistency within the TAG and filter ambient air data sets were examined. We spiked a subset of ambient TAG samples with deuterated internal standards to examine potential matrix effects. Under typical conditions, there was little bias in recovery of deuterated standards. At extreme ambient aerosol levels, however, there were large biases in relative recovery, indicating matrix effects may be important under those conditions. Similar results were observed in laboratory experiments with engine lubricating oil. Applying internal standards in the TAG system would help track and correct for matrix effects influencing compound recovery.
1. INTRODUCTION
Significant advances in instrumentation for measuring atmospheric aerosols have been made over the last decade (CitationSolomon and Sioutas 2008; CitationWexler and Johnston 2008). These advances provide higher total mass recovery, more detailed chemical speciation (e.g., PM2.5 ions, carbon fractions, organic molecules), and better time resolution, providing new insight into the complexity of ambient aerosols. Detailed chemical speciation data are critical for source apportionment—particularly for organic aerosols, where multiple contributing sources potentially confound interpretation of bulk organic carbon (OC) data. OC sources include vehicles, biomass burning, cooking, industrial activity, and secondary organic aerosol (SOA) formation (CitationSchauer et al. 1996). High time resolution also greatly aids source apportionment by capturing dynamic processes related to source activity, such as rush-hour traffic or emissions plumes from point sources. The ideal instrument would combine detailed chemical speciation with high mass recovery and high time resolution.
Traditionally, molecular marker measurements for source apportionment of OC have relied on manual collection of integrated 12 or 24 h samples followed by solvent extraction and offline gas chromatography/mass spectrometry (SE-GC/MS) analysis. This method allows for quantification of source-specific organic marker compounds at the molecular level, and has been used to apportion sources of atmospheric aerosols in many areas of the country (CitationZheng et al. 2002; CitationOndov et al. 2006; CitationShrivastava et al. 2007; CitationJaekels et al. 2007). However, the solvent extraction approach requires large samples, which sacrifices time resolution. In addition, sample collection and extraction from filters is also extremely time- and labor-intensive, which limits the number of samples and frequency of measurements. The potential for positive and negative artifacts from quartz filters collection is well documented (CitationTurpin et al. 1994; CitationSubramanian et al. 2004), and extensive handling of filters samples also introduces potential artifacts.
Recently, a novel in situ Thermal Desorption Aerosol GC/MS (TAG) was developed to provide automated, high time-resolved measurements of organic molecular markers (CitationWilliams et al. 2006). The high time resolution afforded by TAG is a significant advantage over longer integrated samples, and molecular-level speciation complements other high time-resolved techniques such as the Aerodyne AMS (CitationJayne et al. 2000). TAG has been deployed in field campaigns where it has been used to interpret concurrent bulk measurements from other instruments (CitationWilliams et al. 2007; CitationGoldstein et al. 2008; CitationLambe et al. 2009a).
In this article we compare marker concentrations derived from TAG measurements with those obtained by traditional filter-based techniques. Comparisons are made for chamber studies and for 3 weeks of ambient sampling in Pittsburgh, PA. We present results for low-polarity organics as a function of volatility, examine internal consistency within the dataset, and examine the potential influence of aerosol loadings on individual marker recoveries.
2. METHODS
2.1. TAG Operation
The TAG is an in situ instrument designed for the automated, high time-resolved collection and GC/MS analysis of mid-range volatility organic compounds in airborne particles. As described by Williams et al. (Citation2006, Citation2007), it has two modes of operation: (1) sampling with concurrent GC/MS analysis of the previous sample, and (2) thermal desorption of the sample just transfered onto the GC column. During sampling, air is pulled through a PM2.5 cyclone, through a custom collection inlet, and into a collection and thermal desorption (CTD) cell where airborne particles are collected by impaction. The inlet, fabricated by Aerosol Dynamics, Inc. (Berkeley, CA), humidifies the particles at a relative humidity of 75 to 95% to minimize bounce in the collection cell. The air sampling rate is 8.5 lpm, and collection duration varies. For the experiments conducted here the CTD cell is maintained at 30°C during sampling. Following sample collection, the CTD cell is isolated from the sample line and heated to 50°C to purge water and volatile compounds with 50 sccm helium carrier gas. The cell is then ramped from 50°C to 300°C at approximately 25°C min−1 and held for 3 min, after which a 6-port valve switches the cell to the inject position. Helium carrier gas then flows through the cell, transfer lines, and a 6-port valve at 1 sccm to transfer the sample to the GC column (maintained at 45°C). During desorption all transfer lines are maintained at 300°C to minimize losses. The valve body and transfer lines in the sample transfer path were chemically passivated using an Inertium coating (AMCX; Bellefonte, PA).
During this study we performed online TAG GC/MS analysis using an Agilent 5890 GC coupled to an Agilent 5971 MSD. Chromatographic separation was achieved using a Restek Rtx-5MS fused capillary column (30 m × 0.25 mm × 0.25 μ m) with 1 sccm flow in helium. The GC method took 1 hour to complete and used the following temperature protocol: initial temperature 80°C, ramp 50°C min−1 to 45°C, hold for injection of thermally desorbed sample; ramp 8.6°C min−1 to 310°C, hold 10 min; ramp 70°C min−1 to 80°C final temperature for the start of the next run. The MSD was operated in Selected Ion Monitoring (SIM) mode to improve measurement signalto-noise.
TAG is calibrated by direct introduction of liquid standards into the CTD cell by means of a specifically designed injection port (CitationKreisberg et al. 2009). These standards are analyzed using the same protocol as for aerosol samples. For ambient sampling, a single tracking standard at a fixed level is introduced once each day. At the outset and at the conclusion of the field campaign multilevel calibrations were done with this same standard mix. For the field campaign, the tracking standard was prepared by Carnegie Mellon University and combined stock solutions of n-alkanes and PAHs (Accustandard DRH-008S-R1 and H-QME-01; Chemservice 1007S, 1052S, 1047S), hopanes (Chiron 0615,27; 1321,29; 0613,30; 1339,31), cholesterol and β -sitosterol (Sigma-Aldrich), and custom-prepared solutions of n-alkanoic acids (Ultra Scientific), palmitoleic acid (Ultra Scientific), oleic acid, phthalic acid, 9-fluorenone, 9,10-anthracenedione, and 6,10,14-trimethyl-2-pentadecanone (Sigma-Aldrich). For the chamber study, calibration standards were created by diluting the standard mixtures used to generate chamber aerosols. Multipoint calibrations for each mixture were by combining the standards and pure solvent at several in-needle dilution levels.
2.2. Filter Measurements
For both chamber and ambient measurements, filter samples were collected onto pre-fired quartz filter (47 mm, Pall Corp). For the chamber studies these were solvent-extracted using an established method (CitationHuff Hartz et al. 2007) and analyzed with an Agilent 6890 GC/5975 MSD. This filter SE-GC/MS method uses BSTFA for silylation of polar functional groups. A multipoint calibration for target analytes (n-alkanes, n-alkanoic acids, alkenoic acids, cholesterol) was performed using appropriate internal standards (deuterated n-alkanes, n-alkanoic acids, cholesterol) to account for extraction and derivitization efficiencies.
For ambient measurements, filter samples were analyzed by thermal extraction using a Gerstel Thermal Desorption System (TDS3) coupled to a Gerstel Cooled Injection System (CIS4) with helium carrier gas. One or two 1.2 cm2 punches were taken from each filter, placed in an empty glass sample tube, spiked with 5 μ L of an internal standard, and thermally extracted in the TDS3 oven chamber. During desorption, the TDS3 was ramped from an initial temperature of 30°C to a final hold temperature of 320°C (5 min) at 100°C min−1. The transfer line connecting the TDS3 to the CIS4 was maintained at 300°C during extraction. The CIS4 used a glass wool inlet liner and was held at an initial temperature of –120°C during sample transfer from the TDS3 to the CIS4. Following transfer, the CIS4 was ramped at 720°C min−1 to a final hold temperature of 320°C min−1 (3 min). A 10:1 split ratio was implemented at the CIS4 during transfer to the GC column. The sample was then analyzed using an Agilent 6890 GC coupled to a 5975 MSD. The 6890 GC method used a column flow of 1.2 sccm and a temperature protocol consisting of: initial hold temperature 60°C (3 min); ramp 5°C min−1 to final hold temp 320°C (6 min). The 5975 MSD was operated in combined SIM/scan mode. A multipoint calibration for target analytes (n-alkanes, PAHs, hopanes) was performed by taking blank filter punches, placing them in empty sample tubes, spiking different amounts of calibration standards and 5 μ L of internal standards onto the filters, and thermally extracting them in the TDS3 oven chamber. The internal standard was comprised of deuterated n-alkanes (C16, C20, C24, C30, C36) and two alkanoic acids (C16 and C18). Each analyte was matched to one of the internal standards based on volatility, e.g., tetracosane-d50 was used as an internal standard for C23–C27 n-alkanes.
2.3. Smog Chamber Studies
Simultaneous TAG and filter samples were collected of aerosols created by flash vaporizing mixtures of standards in the Carnegie Mellon University smog chamber. One set of experiments used a mixture of polar and nonpolar analytes: cholesterol (Sigma-Aldrich), palmitoleic acid (Sigma-Aldrich), oleic acid (Sigma-Aldrich), palmitic acid (Sigma-Aldrich), stearic acid (Sigma-Aldrich), pentacosane (C25, Sigma-Aldrich), hexacosane (C26, Sigma-Aldrich), and octacosane (C28, Fluka) in 2-butanol (Sigma-Aldrich). A second set considered a mixture of nonpolar species: eicosane (C20, Alfa Aesar), C25, C26, C28, dotriacontane (C32, Sigma-Aldrich), hexatriacontane (C36, Sigma-Aldrich), phenanthrene (TCI America), fluoranthene (TCI America), chrysene (Ultra Scientific), benzo(e)pyrene (Ultra Scientific), and coronene (Acros Organics) in toluene (Fisher Scientific). TAG and filter collection times were synchronized and set from 5–60 min depending on the suspended aerosol mass concentration in the chamber. The TAG sample flow of 8.5 lpm was diluted by a factor of 8–30 depending on the suspended aerosol mass concentration. Filter samples were collected onto pre-fired quartz filter (47 mm, Pall Corp) at 50 lpm, without dilution, and without a size precut.
2.4. Field Measurements
Co-located TAG and filter samples were collected at a field site in downtown Pittsburgh from 5/2/09–5/27/09. Measurements were taken out the window of a 4th-floor office suite in the Diamond Building, located at the intersection of Fifth and Liberty Avenues (CitationLambe et al. 2009a). The site is less than a mile from three major highways (I-279, I-579, and I-376) and is close to many city bus routes. Complementary instrumentation was also deployed at the site; in this article we use results from a multi-wavelength Aethalometer (Magee Scientific) (CitationHansen et al. 1984). Sampling protocols and source apportionment results from this study are described in separate manuscripts (CitationLambe et al. 2009a; CitationLogue et al. 2009). The TAG sampling protocol for these ambient measurements used a 24 h cycle consisting of three 4 h daytime samples (6:30 AM–10:00 AM, 11:30 AM–3:00 PM, 3:30 PM–7:00 PM, one 10 h overnight sample (8:30 PM–6:30 AM), and one 1 h blank sample, and 1 tracking standard introduction following the morning and evening daytime samples. Filter samples were collected at 20 lpm onto pre-fired 47 mm filters (Pall Corp) without precut. A total of ten 24 h, two 48 h, two 72 h, and two handling blank samples were collected. Blank levels were low, less than 5% of analyte levels in ambient filters. After collection, filters were stored in petri dishes (BD-Fisher) at –20°C prior to offline TD-GC/MS analysis. Since integrated filters were collected over 24, 48, or 72 h periods, TAG data were averaged over filter collection periods for intercomparison.
2.5. Influence of Particle Matrix
A liquid standard of deuterated organics was used to compare TAG response for liquid standards introduced into a blank cell to that observed when analyzed in the presence of collected aerosol. This standard was prepared from custom solutions of hexadecane-d34, eicosane-d42, tetracosane-d50, triacontane-d62, hexatriacontane-d74, palmitic acid-d31, and stearic acid-d35, and was also used as the internal standard for ambient filter analyses. During field measurements, a subset of samples (48 of 433) were spiked with 5 μ L of this solution before analysis, usually once per measurement day. During offline calibrations the deuterated standard was also co-injected with the tracking standard at several dilution levels to provide a basis for calculating the recovery in the absence of ambient PM matrix effects. The results from the ambient samples spiked with the deuterated standard were also analyzed to estimate the TAG thermal extraction efficiencies, presented here as a “relative recovery.” Relative recoveries were calculated by normalizing the MSD response in spiked ambient samples to the average response recorded in all of the spiked samples after correcting for MSD decay.
3. RESULTS
3.1. Intercomparison for Smog Chamber Measurements
The first set of chamber experiments studies ozonolysis of an organic mixture containing reactive unsaturated compounds was introduced into the chamber and exposed to ozone. shows results at the outset of one experiment, prior to chemical transformation. Here we compare the mass fraction of cholesterol, palmitoleic and oleic acids, palmitic and stearic acids, C25, C26, and C28 n-alkanes measured by TAG and by the filters to the mass fraction of these compounds in the model mixture injected into the chamber. To obtain sufficient sample for filter measurements, these chamber studies were done at relatively high concentrations, with the result that even with dilution of the TAG sample the analyte mass collected in the TAG CTD cell was about ten times higher than in typical ambient TAG samples (15–30 ng C25, C26, C28; 50–200 ng palmitic and stearic acid; 15–200 ng cholesterol). TAG mass fractions for target analytes were in good agreement with the original solution composition. TAG performance for polar compounds (cholesterol, alkanoic, and alkenoic acids) in the mixture was good, even though TAG does not derivitize these compounds.
FIG. 1 (a) Comparison of TAG and filter recoveries with composition of model mixture containing. cholesterol (Chol), palmitoleic acid (POleic), oleic acid (Oleic), palmitic acid (PAcid), stearic acid (SAcid), and C25, C26, and C28 n-alkanes. (b) Cholesterol, C25, C26, C28, palmitic acid, and stearic acid mass loadings across TAG and filter samples. Samples were collected from smog chamber as aerosols and calibrated with liquid calibration standards injected into TAG collection cell. Filter mass loadings are higher due to their greater sampling rate.
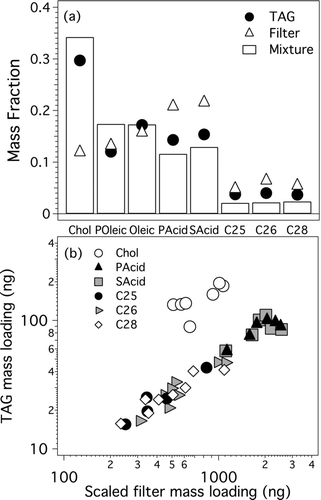
compares TAG and filter measurements for subsequent measurements during two ozonolysis experiments, in which primary organic matter was transformed through oxidation. Shown are mass loadings for cholesterol, n-alkanes and n-alkanoic acids, where we plot the mass of each compound in the TAG CTD cell as a function of the mass collected on the filter scaled by the dilution factor for the TAG sample (which varied from 8 to 30 over the course of these experiments). Due to fast ozone-oxidation kinetics (CitationHuff Hartz et al. 2007; CitationWeitkamp et al. 2008a), palmitoleic and oleic acids were below the TAG detection limits after the first sample. Good correlation was observed between TAG and SE-GC/MS for all mixture compounds (r2 = 0.94–0.99), demonstrating excellent analytical precision of the TAG under these experimental conditions. Absolute concentrations (ng m−3) were in good agreement for cholesterol (m = 1.2 ± 0.1), but there was a factor of 3 discrepancy for C25, C26, and C28 n-alkanes, palmitic acid, and stearic acid with TAG reporting consistently lower concentrations (m = 0.23–0.32). The cause of this discrepancy is not known, and was not seen in some of the other chamber experiments. It may be due to an unknown systematic calibration bias, sampling artifacts on the filter samples, or something associated with the dilution of the TAG air sample stream. The key performance metric for kinetics measurements is analytical precision, which is demonstrated in the intercomparison measurements in .
shows results for a chamber experiment in which a mix of nonpolar organic compounds was introduced into the chamber without chemical reactions. To investigate effects of gas-particle partitioning, this mixture spans a wide range of volatility including intermediate-volatility organic compounds (IVOC: C20, phenanthrene, saturation concentrations C* = 103–106 μg m−3), semivolatile organic compounds (SVOC: fluoranthene, C25, C26, C28, C32, chrysene, benzo(e)pyrene, C * = 10−1–103 μg m−3) and low-volatility organic compounds (LVOC: C36, coronene, C* < 10−1 μg m−3) (CitationRobinson et al. 2007). Calculated mass fractions are normalized by C25 on the filter to illustrate how collection efficiency changes with volatility. In order to collect enough sample for SE-GC/MS, the initial organic aerosol concentration (COA) was about 300 μ g m−3.
FIG. 2 Comparison of TAG relative recoveries with composition of model nonpolar mixture containing C20, C25, C26, C28, C32, C36 n-alkanes, phenanthrene (Phe), fluoranthene (Flu), chrysene (Chr), benzo(e)pyrene (B[e]p), and coronene (Cor). Samples were collected from smog chamber as aerosols and diluted upstream of collection cell. Calibrations were performed with liquid calibration standards injected into TAG collection cell. Analyte masses are normalized by pentacosane (C25) mass. C32, C36, chrysene, benzo(e)pyrene and coronene were below SE-GC/MS detection limits in these samples.
![FIG. 2 Comparison of TAG relative recoveries with composition of model nonpolar mixture containing C20, C25, C26, C28, C32, C36 n-alkanes, phenanthrene (Phe), fluoranthene (Flu), chrysene (Chr), benzo(e)pyrene (B[e]p), and coronene (Cor). Samples were collected from smog chamber as aerosols and diluted upstream of collection cell. Calibrations were performed with liquid calibration standards injected into TAG collection cell. Analyte masses are normalized by pentacosane (C25) mass. C32, C36, chrysene, benzo(e)pyrene and coronene were below SE-GC/MS detection limits in these samples.](/cms/asset/0fe285b6-8fd0-4772-a907-ffc8cea3f385/uast_a_445081_o_f0002g.gif)
In this experiment, the agreement in the absolute chamber concentrations (ng m−3) measured by TAG and by the filters was significantly better than the polar mixture, with TAG yielding concentrations 8% higher than the filter for C25. For compounds that exist primarily in the vapor phase (C20, phenanthrene, and fluoranthene), shows that TAG collected very little, indicative of a relatively low artifact for vapor collection. In the midrange, TAG values are in agreement with those measured by the filter, and the relative distribution among compounds from C25 to C32 agrees with what was introduced into the chamber. For the least volatile species in the nonpolar mixture, TAG recoveries are less than in the injected mixture. This trend may be due to incomplete vaporization of these compounds during aerosol generation in the chamber. Filter data for these compounds were below detection limits.
3.2. Intercomparison of Ambient Measurements
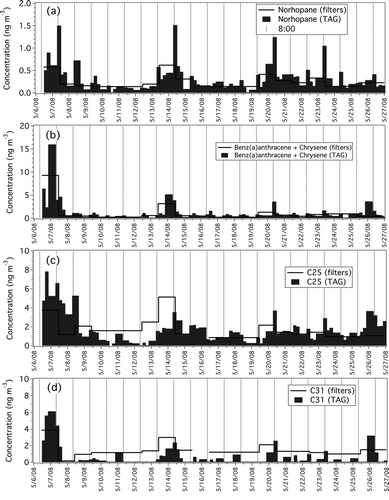
shows time series for the ambient concentrations of selected organic marker compounds measured by TAG and by filter collection for the 3 week field campaign in downtown Pittsburgh. Norhopane () and other hopanes are frequently used in receptor models as molecular markers for motor vehicle emissions (CitationSubramanian et al. 2006). For these compounds the TAG data show midmorning concentration peaks with much lower concentrations midday and overnight, consistent with expected changes in motor vehicle activity and atmospheric stability. A different temporal profile is observed for PAH228 (those compounds with m/z = 228 including chrysene and benz(a)anthracene), and C31. These compounds show several high-concentration overnight samples that were likely an effect of suppressed vertical mixing. The large variability in marker concentrations indicates the strong influence of local sources and the insight gained by high time-resolved TAG measurements. None of this variability is resolved by the long-duration filter samples, underscoring the value of high-time-resolved TAG samples for source apportionment.
FIG. 3 Time series of selected marker concentrations (ng m− 3) measured by TAG (dark grey bars) and filters (black trace). (a) Norhopane, (b) Benz(a)anthracene + Chrysene, (c) C25, (d) C31. TAG data are are averaged over filter collection periods. Vertical bars placed at 8:00 AM for each day of TAG and filter sample collection.
presents scatter plots of the TAG and filter-based measurement of ambient concentrations of 16 different compounds measured during the 3 week Pittsburgh field study. Regression lines were computed using ordinary least squares fitting, and r2 values are the square of the Pearson correlation coefficient. Hopanes (, , , , , ) are in good agreement, with regression slopes ranging from 1.0 ± 0.2 (17a(H)-22,29,30-trisnorhopane; r2 = 0.64) to 1.7 ± 0.2 (22R-17a(H),21b(H)-homohopane; r2 = 0.90). Precision was better for higher molecular weight hopanes (norhopane, hopane, homohopanes; r2 = 0.86–0.95) than smaller hopanes (trisnorhopanes; r2 = 0.55–0.64). The larger scatter in the lower molecular weight hopanes may be due to collection issues for semivolatile species—either sampling artifacts on the filters and/or evaporation in the TAG during the helium purge. This trend was observed for all compound classes.
FIG. 4 Comparison of (a–f) hopane, (g–h) PAH and (i–p) alkane concentrations measured using TAG and offline TD-GC/MS analysis of quartz filters. Dashed line is the one-to-one line indicating perfect agreement. Solid line indicates a linear regression of the data (r2 = Pearson correlation coefficient squared). Regression slope (m) and intercept (b) parameters shown in figure captions.
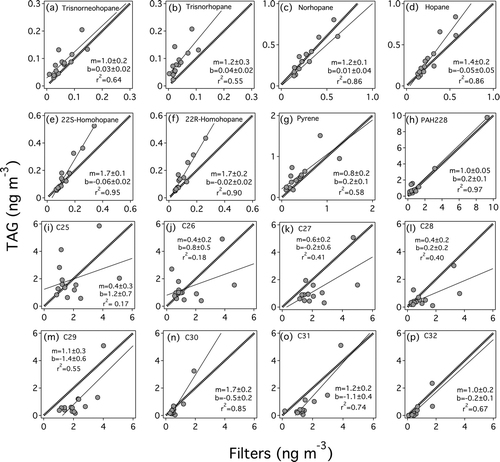
, show intercomparison results for two PAHs. For these PAHs, there was excellent agreement between TAG and TD-GC/MS analysis of filter samples, with regression slopes of 1.0 ± 0.05 (benz(a)anthracene + chrysene, PAH228) and 0.8 ± 0.2 (pyrene). The best correlation (r2 = 0.97) was observed for PAH228; dropping the highest-concentration point from the PAH228 regression modestly reduces the correlation (m = 0.97 ± 0.14, r2 = 0.83). Pyrene, which is partitioned appreciably into both the vapor and condensed phases, was less well correlated between methods (r2 = 0.58).
, , , , , show scatter plots for C27–C32 n-alkanes. Compared to the PAHs and hopanes, there was somewhat poorer agreement (m = 0.4–1.7) and correlation (r2 = 0.17–0.85) between TAG and filter concentrations for n-alkanes. For the smaller, more volatile n-alkanes there was more bias between the two techniques, and more scatter compared to the larger n-alkanes. It is possible that a newer MSD than the one used in this study (Agilent 5971) may improve TAG performance, as signal-to-noise for n-alkanes was about a factor of ten lower than for hopanes and PAHs. Another possible explanation for the greater scatter in the n-alkane data might be interference from other hydrocarbons with similar fragmentation patterns in the MS. For TAG, n-alkanes were quantified using m/z = 57 (C4H9 +), which is formed during electron-impact ionization of many hydrocarbons. This type of non-specificity in n-alkane quantitation ions was suggested as an explanation for poor intercomparison in an 8-laboratory study (CitationSchantz et al. 2005). For the same set of n-alkanes (C25-C32), RSD across labs for individual n-alkanes ranged from 28–47% (SRM1648), 35–59% (Baltimore2PM), and 58–124% (RM8785). The last sample, RM8785, is particularly relevant to this discussion because physical filters were distributed between labs for analysis and reporting rather than bulk dust samples.
shows the correlation among individual n-alkanes within the set of ambient TAG measurements (upper left hand side) and within the set of ambient filter measurements (lower right hand side). For TAG, the correlation coefficient systematically decreases as the n-alkanes become more different (e.g., C25 and C26 are highly correlated (r2 > 0.9) while C25 and C32 have lower correlation (r2 < 0.7)). For the filter measurements, there is similarly high correlation among low carbon number n-alkanes, but poorer correlation between similar high carbon number n-alkanes, with r2∼ 0.7 for comparison of C29 and C31. When comparing the TAG to the filters, the TAG data are better correlated for the larger n-alkanes. It the n-alkanes are derived from the same sources, these data would indicate greater internal consistency in the TAG data.
FIG. 5 Correlation (r2 = Pearson correlation coefficient squared) among individual hopanes, PAHs, and C25-C32 n-alkanes within ambient TAG (upper left hand side) and ambient filter measurements (lower right hand side). TAG data are averaged over filter collection periods.
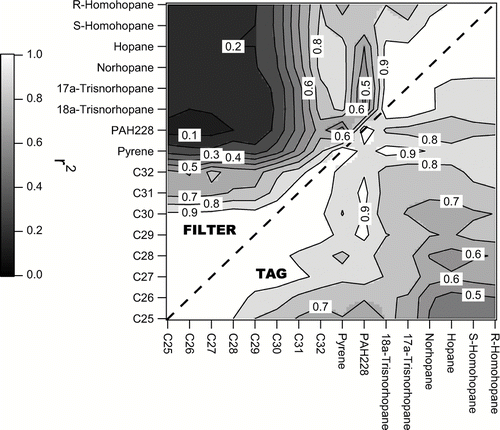
Since the filter and TAG data agree well for hopanes, further insight about the n-alkane measurements may be gained by comparing the internal consistency in each dataset for the n-alkanes versus the hopanes. Correlations between n-alkanes and hopanes in the TAG data were consistently much higher than in the filter data. The r2 of correlations between C25–C30 n-alkanes with 6 different hopanes was consistently between 0.4 to 0.8 for TAG and always < 0.4 for filters. Higher correlations were observed between C31-C32 n-alkanes and hopanes for both methods, but again correlations were higher for TAG than filter data. To the extent that n-alkanes and hopanes should covary due to similar source categories and meteorological conditions, the correlations demonstrate better internal consistency in the TAG data and suggest TAG measured n-alkanes with higher precision.
3.3. Influence of the Particle Matrix
For TAG, ambient marker concentrations are derived from the MSD response to liquid tracking standards injected into a clean collection cell. To assess whether this response is representative of that when particulate matter is present, experiments were conducted by manually spiking a subset of ambient samples with a deuterated standard mixture. Results shown in compare the response of hexadecane-d34, eicosane-d42, tetracosane-d50, and triacontane-d62 spiked onto ambient particulate samples to the response obtained in offline injections. Offline injections with the deuterated standard were done into a blank cell at 4 levels, while the spike onto the ambient samples was only done at the highest level.
FIG. 6 Recovery of deuterated C16, C20, C24, and C30 n-alkanes in offline calibrations (open markers) and ambient (filled markers) samples. In offline calibrations, deuterated n-alkanes were coinjected with a tracking standard into the TAG collection cell, except for the highest calibration point at which only 5uL of deuterated standard was injected. Linear response observed over the range of spiked masses (approx. 0.6−6 ng). In ambient collection, 5 uL of deuterated standard was injected into the TAG collection cell and thermally desorbed with the ambient sample; markers shown are offset for readability. Markers represent the average ambient recovery for each deuterated n-alkane.
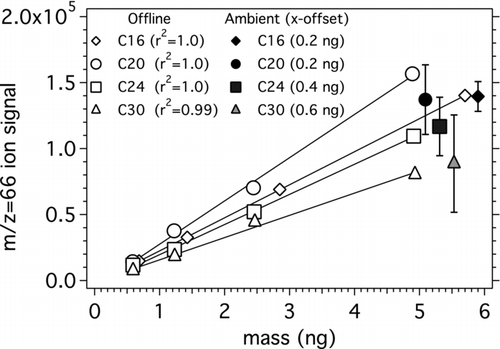
Two important points are shown in this figure. First, the data show that recovery of deuterated standards is the same when co-introduced with ambient particulate samples as when introduced into the blank cell. Second, the response to the offline injections is linear over the range of recoveries measured in ambient samples. This indicates no significant bias from the use of in situ offline calibrations for quantifying ambient TAG data. There is some variability in the recovery of the deuterated standard in the ambient samples, especially for the high molecular weight compounds. Over several weeks of field measurements, the relative standard deviation in recovery of deuterated C16, C20, and C24 n-alkanes was 13%, 20%, and 27%, while that for triacontane (C30) is 60%. This variability was not observed when similarly spiking ten TAG samples of diesel exhaust from two smog chamber experiments. In those experiments, the variability in recovery was 5% to 13% for the deuterated n-alkanes, and 11% to 14% for the deuterated n-alkanoic acids.
and plot TAG relative recovery for deuterated C16 and C30 as a function of black carbon (BC) mass collected in the spiked samples. BC is typically co-emitted with primary organic aerosol and BC levels are high downtown compared to other areas in Pittsburgh due to the high density of mobile sources (CitationLambe et al. 2009a). BC mass was estimated using Aethalometer data averaged over TAG sample collection periods. shows that the relative recovery for C30 is flat in the midrange, when BC ranged from 1 to 10 μ g. The only points falling outside the 95% confidence level for relative recovery = 1 are at the lowest and highest BC levels.
FIG. 7 TAG relative recovery for (a) deuterated C16 and (b) deuterated C30 as a function of BC mass collected in TAG samples. Data points are binned averages of 3–5 spiked samples. Solid lines are power law regressions to guide the eye. (c) TAG relative recovery for 4.93 ng triacontane-d62 coinjected with a liquid motor oil standard. Solid line is power law regression to guide the eye; dashed lines are uncertainty estimates (95% confidence levels).
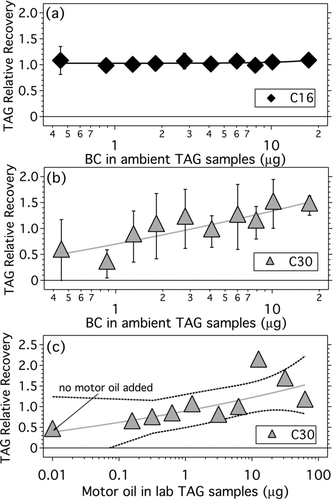
To further investigate whether TAG recovery is influenced by the matrix of material deposited in the CTD cell, experiments were conducted in which we co-injected the deuterated standard with different amounts of liquid motor oil. Engine lubricating oil is thought to be a dominant component of organic matter emitted by motor vehicles (CitationSchauer et al. 1999). presents the results of this experiment for triacontane-d62 co-injected with 0–60 μ g of motor oil. This level of motor oil spans the range of total organic loadings in ambient TAG samples. is suggestive of an upward trend in TAG recovery with increasing amounts of co-injected organic matter, but uncertainties are large. It is possible that co-injected organic matter extracted with target analytes competes for active adsorption sites in the TAG sample transfer lines or 6-port valve (unique to TAG), resulting in better recovery at higher total organic levels. It is also possible that, as suggested in previous studies, transfer issues are more significant for n-alkanes than for other types of compounds, with preferential adsorptive loss of n-alkanes (C22–C36) through heated Silcosteel- and Siltek-treated stainless steel, Silcosteel-treated stainless steel coated with 100% polydimethylsiloxane, and deactivated fused silica capillary transfer lines (CitationModey and Doskey, 2006).
4. DISCUSSION AND CONCLUSIONS
In this paper, we have compared organic aerosol speciation data measured with TAG to traditional filter-based techniques. There was good to excellent agreement between these two techniques for 4-ring PAHs and hopanes. Given that hopanes are key molecular markers for source apportionment of motor vehicles (CitationSchauer et al. 1996; CitationSubramanian et al. 2006), this is an important result. Strong correlations were also observed for moderately polar and nonpolar species (cholesterol, n-alkanes and n-alkanoic acids) in high-concentration smog chamber experiments. Prior work (CitationKreisberg et al. 2009) has also shown good agreement between TAG and other techniques for six more EPA criteria PAHs not covered in this work. There was worse agreement between TAG and the filter measurements for n-alkanes. The underlying cause of these differences is not known: they could be due to problems with one or both of the methods. Similar levels of disagreement have been observed among filter-based GC/MS methods (NIST intercomparison, CitationSchantz et al. 2005). Further development of TAG, such as the incorporation of an internal standard, or improvement in the transfer lines, may provide greater precision.
Several other factors must be considered when evaluating these intercomparison results. TAG uses a very different sample collection methodology (inertial impaction) than filters, and there are well-known problems/shortcomings with both approaches. Particles below the TAG impactor cutpoint of 80 nm are not collected (CitationWilliams et al. 2006), which may be important in urban environments with fresh hydrophobic emissions. On the other hand, filters have significant problems with gas-phase artifacts (CitationTurpin et al. 1994; CitationSubramanian et al. 2004; CitationSihabut et al. 2005). TAG has been designed to exclude vapors; concentrated inertial impaction of particles results in small surface area for retaining vapors, and the collection cell is purged with helium to remove volatile compounds prior to thermal desorption. For the chamber experiments uncertainties may have been introduced by the need to add dilution flow to the TAG samples. Finally, although molecular speciation of filter-based samples is widely performed, intercomparison studies show significant lab-to-lab variability (CitationSchantz et al. 2005).
To date the primary application of TAG has been for source apportionment using the Positive Matrix Factorization (PMF) receptor model (CitationPaatero and Tapper, 1994). Our intercomparison results indicate important molecular markers such as hopanes and PAHs (e.g., benz(a)anthracene and chrysene) are accurately monitored with TAG. With PMF, isolation of source factors only relies on temporal correlations among fitting species; therefore, precision and signal-to-noise are the critical performance metrics. This potentially allows the use of more TAG markers as PMF fitting species, including novel molecular markers with unknown sources, if precision is high and markers are not vulnerable to matrix effects.
The intercomparison results also indicate that TAG is suitable for smog-chamber applications, such as measuring oxidation kinetics of molecular markers in complex aerosols (CitationWeitkamp et al. 2008b). In smog-chamber experiments, concentrations are higher and the matrix effects appear to be either minimal or much easier to isolate. Since TAG requires smaller samples than SE-GC/MS, atmospherically relevant aerosol mass concentrations can be used, which is important for better understanding and characterization of atmospheric aerosols (CitationDonahue et al. 2006; CitationLambe et al. 2009b).
Acknowledgments
We thank Brent Williams and David Worton for contributions from previous work. This work is supported by a grant from the Allegheny County Health Department and the U.S. Environmental Protection Agency and DOE grant DE-FG02-05ER86235. This article has not been subject to EPA's required peer and policy review, and therefore does not necessarily reflect the views of the Agency. No official endorsement should be inferred.
Related Research Data
REFERENCES
- Donahue , N. M. , Robinson , A. L. , Stanier , C. O. and Pandis , S. N. 2006 . Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics . Environ. Sci. Technol. , 40 : 2635 – 2643 .
- Goldstein , A. H. , Worton , D. R. , Williams , B. J. , Hering , S. V. , Kreisberg , N. M. , Panic , O. and Gorecki , T. 2008 . Thermal Desorption Comprehensive Two-Dimensional Gas Chromatography for In-Situ Measurements of Organic Aerosols . J. Chrom. A , 118 : 340 – 347 .
- Hansen , A. D. A. , Rosen , H. and Novakov , T. 1984 . The Aethalometer—An Instrument for the Real-Time Measurement of Optical Absorption by Aerosol Particles . The Science of the Total Environment , 36 : 191 – 196 .
- Huff Hartz , K. E. , Weitkamp , E. A. , Sage , A. M. , Donahue , N. M. and Robinson , A. L. 2007 . Laboratory Measurements of the Oxidation Kinetics of Organic Aerosol Mixtures Using a Relative Rate Constants Approach . J. Geophys. Res. , 112 : D04204
- Jaekels , J. M. , Bae , M.-S. and Schauer , J. J. 2007 . Positive Matrix Factorization Analysis of Molecular Marker Measurements to Quantify the Sources of Organic Aerosols . Environ. Sci. Technol. , 41 : 5763 – 5769 .
- Jayne , J. T. , Leard , D. C. , Zhang , X. , Davidovits , P. , Smith , K. A. , Kolb , C. E. and Worsnop , D. R. 2000 . Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles . Aerosol Sci. Technol. , 33 : 49 – 70 .
- Kreisberg , N. M. , Hering , S. V. , Williams , B. J. , Worton , D. R. and Goldstein , A. H. 2009 . Quantification of Hourly Speciated Organic Compounds in Atmospheric Aerosols, Measured by an In-Situ Thermal Desorption Aerosol Gas Chromatograph (Tag) . Aerosol Sci. Technol. , 43 ( 1 ) : 38 – 52 .
- Lambe , A. T. , Logue , J. M. , Kreisberg , N. M. , Hering , S. V. , Worton , D. R. , Goldstein , A. H. , Donahue , N. M. and Robinson , A. L. 2009a . Apportioning Black Carbon to Sources Using Highly Time-Resolved Ambient Measurements of Organic Molecular Markers in Pittsburgh . Atmos. Environ. , 43 ( 25 ) : 3941 – 3950 .
- Lambe , A. T. , Miracolo , M. M. , Hennigan , C. J. , Robinson , A. L. and Donahue , N. M. 2009b . Effective Rate Constants and Uptake Coefficients for the Reactions of Organic Molecular Markers (n-Alkanes, Hopanes and Steranes) in Motor Oil and Diesel Primary Organic Aerosols With Hydroxyl Radicals . Environ. Sci. Technol. , 43 ( 23 ) : 8794 – 8800 .
- Logue , J. M. , Hartz , K. E. H. , Lambe , A. T. , Donahue , N. M. and Robinson , A. L. 2009 . High Time Resolved Measurements of Organic Air Toxics in Multiple Exposure Regimes . Atmospheric Environment , 43 ( 39 ) : 6205 – 6217 .
- Modey , W. K. and Doskey , P. V. 2006 . Evaluation of a Valveless Thermal Desorption System for Organic Aerosols and Vapors: Transfer Lines and Preconcentration Module . J. Chrom. A , 1121 ( 1 ) : 16 – 22 .
- Ondov , J. M. , Buckley , T. J. , Hopke , P. K. , Ogulei , D. , Parlange , M. B. , Rogge , W. F. , Squibb , K. S. , Johnston , M. V. and Wexler , A. S. 2006 . Baltimore Supersite: Highly Time- and Size-Resolved Concentrations of Urban Pm2.5 and Its Constituents for Resolution of Sources and Immune Responses . Atmos. Environ. , 40 : S224 – S237 .
- Paatero , P. and Tapper , U. 1994 . Positive Matrix Factorization: A Non-Negative Factor Model with Optimal Utilization of Error Estimates of Data Values . Environmetrics , 5 : 111 – 126 .
- Robinson , A. L. , Donahue , N. M. , Shrivastava , M. K. , Weitkamp , E. A. , Sage , A. M. , Grieshop , A. P. , Lane , T. E. , Pierce , J. R. and Pandis , S. N. 2007 . Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging . Science , 315 ( 5816 ) : 1259 – 1262 .
- Schantz , M. M. , Poster , D. L. , Kucklick , J. R. , Wise , S. A. , McDow , S. and Lewtas , J. 2005 . Intercomparison Program for Organic Speciation in Pm2.5 Air Particulate Matter: Description and Results for Trial III. Technical Report NISTIR 7303 , National Institute of Standards and Technology and Environmental Protection Agency .
- Schauer , J. J. , Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. and Cass , G. R. 1996 . Source Apportionment of Airborne Particulate Matter Using Organic Compounds as Tracers . Atmos. Environ. , 30 ( 22 ) : 3837 – 3855 .
- Schauer , J. J. , Kleeman , M. J. , Cass , G. R. and Simoneit , B. R. T. 1999 . Measurement of Emissions From Air Pollution Sources. 2. C1 through C30 Organic Compounds from Medium Duty Diesel Trucks . Environ. Sci. Technol. , 33 : 1578 – 1587 .
- Shrivastava , M. K. , Subramanian , R. , Rogge , W. F. and Robinson , A. L. 2007 . Sources of Organic Aerosol: Positive Matrix Factorization of Molecular Marker Data and Comparison of Results from Different Source Apportionment Models . Atmos. Environ. , 41 : 9353 – 9369 .
- Sihabut , T. , Ray , J. , Northcross , A. and McDow , S. R. 2005 . Sampling Artifact Estimates for Alkanes, Hopanes, and Aliphatic Carboxylic Acids . Atmos. Environ. , 39 : 6945 – 6956 .
- Solomon , P. A. and Sioutas , C. 2008 . Continuous and Semicontinous Monitoring Techniques for Particulate Matter Mass and Chemical Components: A Synthesis of Findings from Epa'S Particulate Matter Supersites Program and Related Studies . J. Air & Waste Manage. Assoc. , 58 ( 2 ) : 164 – 195 .
- Subramanian , R. , Khlystov , A. Y. , Cabada , J. C. and Robinson , A. L. 2004 . Positive and Negative Artifacts in Particulate Organic Carbon Measurements With Denuded and Undenuded Sampler Configurations . Aerosol Sci. Technol. , 38 ( Suppl. 1 ) : 28 – 47 .
- Subramanian , R. , Donahue , N. M. , Bernando-Bricker , A. , Rogge , W. F. and Robinson , A. L. 2006 . Contribution of Motor Vehicle Emissions to Organic Carbon and Fine Particle Mass in Pittsburgh, Pennsylvania: Effects of Varying Source Profiles and Seasonal Trends in Ambient Marker Concentrations . Atmos. Environ. , 40 : 8002 – 8019 .
- Turpin , B. J. , Huntzicker , J. J. and Hering , S. V. 1994 . Investigation of Organic Aerosol Sampling Artifacts in the Los-Angeles Basin . Atmos. Environ. , 26 ( 19 ) : 3061 – 3071 .
- Weitkamp , E. A. , Hartz , K. E. H. , Sage , A. M. , Donahue , N. M. and Robinson , A. L. 2008a . Laboratory Measurements of the Heterogeneous Oxidation of Condensed-Phase Organic Molecular Markers for Meat Cooking Emissions . Environ. Sci. Technol. , 42 ( 14 ) : 5177 – 5182 .
- Weitkamp , E. A. , Lambe , A. T. , Donahue , N. M. and Robinson , A. L. 2008b . Laboratory Measurements of the Heterogeneous Oxidation of Condensed-Phase Organic Molecular Markers for Motor Vehicle Exhaust . Environ. Sci. Technol. , 42 ( 21 ) : 7950 – 7956 .
- Wexler , A. S. and Johnston , M. V. 2008 . What Have We Learned from Highly Time-Resolved Measurements During Epa'S Supersites Program and Related Studies? . J. Air & Waste Manage. Assoc. , 58 ( 2 ) : 303 – 319 .
- Williams , B. J. , Goldstein , A. H. , Kreisberg , N. M. and Hering , S. V. 2006 . An In-Situ Instrument for Speciated Organic Composition of Atmospheric Aerosols: Thermal Desorption Aerosol Gc/Ms-Fid (Tag) . Aerosol Sci. Technol. , 40 : 627 – 638 .
- Williams , B. J. , Goldstein , A. H. , Millet , D. B. , Holzinger , R. , Kreisberg , N. M. , Hering , S. V. , White , A. B. , Worsnop , D. R. , Allan , J. D. and Jimenez , J. L. 2007 . Chemical Speciation of Organic Aerosol During the International Consortium for Atmospheric Research on Transport and Transformation 2004: Results from In Situ Measurements . J. Geophys. Res. , 112 : D10S26
- Zheng , M. , Cass , G. R. , Schauer , J. J. and Edgerton , E. S. 2002 . Source Apportionment of Pm2.5 in the Southeastern United States Using Solvent-Extractable Organic Compounds as Tracers . Environ. Sci. Technol. , 36 : 2361 – 2371 .