Abstract
Grown nucleated particles > 50 nm in diameter are an important source of cloud condensation nuclei (CCN) and when the size is > 100 nm, they can also have direct influence on the climate. In this study, the nucleation and growth of new particles in the atmosphere in Hong Kong were investigated during dry season (monthly averaged RH < 75%). The maximum size of grown nucleated particles was generally less than 40 nm during new particle burst and growth events. The exception, accounting for ∼ 20% of all burst and growth events, was those induced by strong photochemical reactions, in which subsequent particle shrinkage occurred. Temporal particle and gas concentration variability and meteorological conditions support the occurrence of particle shrinkage. The shrinkage rate calculated (∼ 8 nm h–1) was close to the growth rate. The observation of particle shrinkage sheds new light on particle transformation dynamics and it would add to the understanding of particle behavior in the atmosphere.
1. INTRODUCTION
Nucleation and growth of nucleated particles have attracted much interest in the past decade because of potential influence of these particles on the climate. Particles < 30 nm in diameter are conventionally considered to be nucleation mode particles (CitationDal Maso et al. 2005) although nucleated particles have diameters around 1 nm (CitationKulmala et al. 2004; CitationKulmala and Kerminen 2008). The size of grown nucleated particles is crucial to the climate. CitationDusek et al. (2006) found that cloud condensation nuclei (CCN) concentrations are mainly determined by ambient particle size (number) distribution rather than chemical composition and < 50 nm particles are almost incapable of becoming CCN because of the impractical supersaturation needed for activation.
In the past few years, quite a number of researchers have studied particle formation and growth, and measured ultrafine particle (UFP, diameter < 100 nm) size distributions (CitationO'Dowd et al. 1999 2007; McMurry et al. 2000; Shi et al. 2001; McMurry and Woo 2002; Young and Keeler 2004; Dal Maso et al. 2005; Jung et al. 2006; Watson et al. 2006; Wen et al. 2006, Gao et al. 2009). Reviews of the formation and growth rates of nucleated particles can be found in CitationKulmala et al. (2004), CitationHolmes (2007), and CitationKulmala and Kerminen (2008). During most new particle burst events, new particles do not grow larger than 50 nm. Theoretically, there are three possibilities for these < 50 nm grown nucleated particles after new particle burst events: continuous growth due to gas condensation and particle-particle coagulation, no growth, and a reversal of growth due to evaporation of semi-volatile and volatile species. The same are also applicable to the > 50 nm grown nucleated particles.
CitationBarsanti et al. (2008) proposed that low molecular weight volatile/semi-volatile organic salts associated with ammonium and amines can potentially contribute to particle growth. CitationZhang et al. (2004) and CitationJacobson et al. (2005), on the other hand, hypothesized the possibility of a reversal in nucleated particle growth in combustion plumes but this has not been reported for atmospheric particles. CitationGramotnev and Gramotnev (2005) proposed a mechanism for the thermal fragmentation of nanoparticles in vehicular plume induced by the evaporation of volatile molecules in the particle surface. Recently, CitationLee et al. (2008) reported that octanal absorbed by sulfuric acid particles can subsequently evaporate when the particles are exposed to octanal free environment. Thus, it is conceivable when the meteorological and chemical conditions become unfavorable for particle growth, a reversal in growth could occur due to thermal fragmentation and the evaporation of the volatile/semi-volatile components, leading to the shrinking of the grown particles. The shrinkage would lower the probability of the particles becoming CCN which is important to the understanding of atmospheric transformation.
In this paper, we analyzed a dry season dataset obtained from a year-long continuous measurement of 3–100 nm particles at a suburban site in Hong Kong, in search of the reversal in growth of nucleated particles. Shrunken particle sizes and corresponding ambient conditions are discussed, and shrinkage rates are estimated.
2. METHODS
2.1. Sampling Sites and Sampling Methods
A TSI Scanning Mobility Particle Sizer (SMPS) was used to sample UFP from February 2003 to January 2004 on the roof of a 7-storey student residence building at the Hong Kong University of Science and Technology (HKUST) located in eastern Hong Kong (). The sampling site is approximately 600 m from the coastline and approximately 100 meters above sea level. The site is shielded by hills on three sides and facing Port Shelter Bay on the east side. The campus with plenty of trees and vegetation is 5–10 km from the nearest urban areas. This site is thereby considered to be coastal suburban (CitationYao et al. 2002). Potential sources of air pollutants include traffic emissions from three service roads, parking lots, kitchens of student cafeterias and restaurants, and university staff residential units, as well as occasional barbeque fires. The sources usually become active at fixed time slots: 8:30–9:30, 12:00–14:00, 17:00–18:00, and 18:00–22:00. However, emissions from local power plants (20–40 km east of the site) and regionally transported air pollutants occasionally invade this site (CitationLam et al 2001; CitationYao et al. 2006a).
FIG. 1 Map of sampling sites.
The SMPS was equipped with a Nano-Differential Mobility Analyzer (NDMA, TSI Model 3085) having a Kr-85 neutralizer and an Ultrafine Condensation Particle Counter (UCPC, TSI Model 3025A). The SMPS time was corrected to Hong Kong local time. A diffusion dryer was used to remove water vapor upstream of the SMPS and dry particles were measured. The sampling probe was 1 m in length with a residence time of 2 s and each scan took 5 min. The SMPS scans particle size ranging from 3.2 nm to 106 nm in 98 size bins and the bin distribution is 32, 45 and 21 for < 10 nm, 10–50 nm, and 30–106 nm particles, respectively. TSI Aerosol Instrument Manager Software was used to process the particle size distributions. Internal consistency tests were performed to remove those outliners from the raw dataset due to sampling artifacts related to scanning and sampling rates reported by CitationRussell et al. (1995) and CitationYao et al. (2006b). Meteorological data were measured every second. Land-sea breezes were frequent except when strong synoptic winds occurred.
SO2, NO2, and O3 and PM10 concentrations were not measured, instead, hourly data reported by Hong Kong Environmental Protection Department (HKEPD) were used. The reported concentrations followed the U.S. Environmental Protection Agency QA/QC protocol and are ± 10% accurate (CitationYao et al. 2006a; CitationZhang et al. 2007; http://www.epd-asg.gov.hk/english/report/aqr.php). In , the Tap Mun “background” station, located on a remote island in northeast Hong Kong, is used to monitor the marine background and transported air plumes from outside of Hong Kong. Tai Po, Sha Tin, and Kwun Tong, situated in the east, are all urban sites but in different airsheds. HKUST is situated between Sha Tin and Kwun Tong.
Temporal and spatial variations of (NO2+O3) instead of O3 were used to study the photochemical activities because of the titration reaction O3+ NO → NO2, although O3 is more commonly used. Increase of (NO2+O3) concentrations in daytime could be the result of photochemical activities and could also be due to the vertical transport of O3 from aloft to the ground level. The former usually leads to a large spatial variation of (NO2+O3) while the latter results in a spatially-uniform distribution of (NO2+O3) concentration (CitationYao et al. 2006a; CitationZhang et al. 2007). A combination of temporal and spatial variability of (NO2+O3) would isolate the two processes from each other. Photochemically formed H2SO4 gas is the major precursor of UFP (CitationKulmala et al. 2004, CitationHolmes 2007, CitationKulmala and Kerminen 2008), and photochemical secondary organic species could be important in nucleation and particle growth; but evidence is still lacking (CitationKulmala and Kerminen 2008).
2.2. Computational Methods
Apparent formation rates and growth rates are calculated using the method provided by CitationKulmala et al. (2004), Dal Maso et al. (2005), and CitationQian et al. (2007). The apparent formation (J) is expressed as:
The apparent particle growth rate (GR) is expressed as:
The apparent particle shrinkage rate (SR) is calculated using the same equation as GR but the value is negative.
4. RESULTS AND DISCUSSION
3.1. Classification of Long-Term Ultrafine Particle Bursts
In this study, we focused on long-term (> 1 h) UFP bursts in the dry season (October 2003 to January 2004, ). These long-term UFP bursts can be classified into four regimes and the occurrence frequencies are summarized in . Also included in the table are the number of sampling days and effective scans.
TABLE 1 Summary of meteorological data from February 2003 to January 2004 (*January 2004, ** background air pollutant concentrations at the “Tap Mum” station).
TABLE 2 Occurrence of long-term ultrafine particle bursts (detailed definitions are given in the text).
In Regime 1, UFP bursts were characterized by the growth of nucleated particles from < 10 nm to < 40 nm. In Regime 2, the growth events were also observed except they started at < 20 nm and ended sometimes at > 50 nm while other times at < 50 nm. These two regimes have been suggested by Dal Maso et al. (2005) and CitationQian et al. (2007) to be regional nucleation events. In Regime 3, a broad spectrum of particle size (number concentration) was detected at the initial burst. The smaller UFP concentration decreased faster than the larger ones, leading to a continuous increase of the geometric mean mobility diameter of UFP throughout the event. Regime 3 occurred in daytime in association with elevated concentrations of PM10, SO2, and NO2+O3 (not shown in ). Regime 3 probably included both primary UFP emitted from point sources and secondarily formed UFP in pollution plumes. In Regime 4, there was no growth of particles during the bursts and the particle size was constant at 40–50 nm or > 80 nm. Regime 4 occurred in both daytime and nighttime in association with elevated concentrations of PM10; Regime 4 probably reflected aged UFP emitted from point sources.
In this study, we focus on Regimes 1 and 2. Regimes 1 and 2 were not detected in October, but they occurred for 2 and 1 days each in November, 6 and 0 days each in December, and 3 and 1 days each in January. Reversal in growth and the shrinkage of the grown nucleated particles were observed in 1 out of the 11 days for Regime 1, and 2 out of the 2 days for Regime 2. The events with growth reversal accounted for ∼ 20% of the new particle burst events.
3.2. Growth of Nucleated Particles During New Particle Bursts
The characteristics of Regimes 1 and 2 new particle bursts are summarized in . The estimated formation rate of the new particles varied from 2.3 to 11.5 particle cm–3 s–1 with a median of 3.6 particle cm–3 s–1. The rate of coagulation loss was estimated to be 0.2–0.4 particle cm–3 s–1 using the same approach as CitationQian et al. (2007). The formation rates listed in have been corrected for coagulation loss. The particle growth rate was in the range of 2–11.8 nm h–1 with a median of 3.7 nm h–1. The duration of new particle bursts had median of 5 h and a range of 2 to 10 h.
TABLE 3 Characteristics of Regimes1 and 2 new particle bursts.
Particle growths with no observed reversals are examined first to be used as the baseline and the 25 January 2004 dataset is used as an example (, , , , ). On that day, the new particle burst event lasted for 10 h, the longest of the 11 Regime 1 events measured (). The burst started at 10:25 and the minimum size of nucleated particles was ∼ 7 nm (). The total UFP concentration increased from 5*103 particle cm–3 before the burst to a maximum of 2.6*104 particle cm–3 at 12:20.
FIG. 2 (a–e) Concentrations of particles and gases on 25 January 2004.
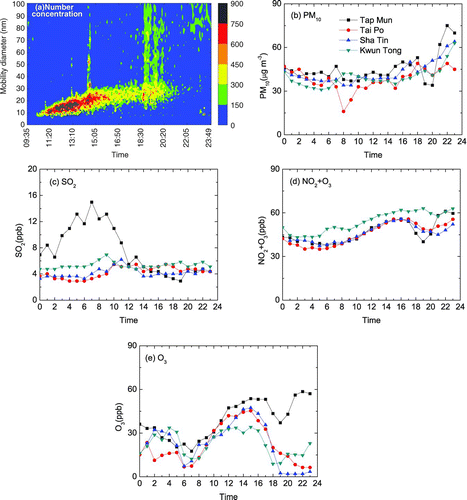
The < 30 nm particle formation rate was estimated to be 3.2 particle cm–3 s–1 (). This is within the range reported in Beijing (3.3–81.4 particle cm–3 s–1, CitationWu et al. (2007)) and St. Louis, Missouri, USA, (1–80 particle cm–3 s–1, CitationKulmala et al. (2004)). A uni-modal size distribution of UFP was exhibited during the entire particle burst (). Like those size distribution spectra of UFP reported in the literature, the degree of smoothness of the curves in was lower than those made by the fast particle sizer and was probably ascribed to the sampling artifact caused a low scanning rate of the SMPS (CitationYao et al. 2005). The calculated growth rate for the initial 5 h was 3.2 nm h–1 and was < 1.0 nm h–1 for the last 5 h ( and ). The growth of particles stopped at 35 nm. The growth rate was apparently too low for the nucleated particles to grow to larger than 50 nm, the CCN size. CitationKulmala et al. (2004) reviewed the formation and growth rates of UFP and the typical growth rate is 1–20 nm h–1 although in some coastal environments it can be as high as 200 nm h–1. They also reported that nucleation events could be induced by a change in wind direction.
FIG. 3 (a–d) Temporal variation of ultrafine particles and size distributions on 25 January 2004 (a: dN< 30nm/dt = 2.8 cm–3 s–1 and the average of Fcoag = 0.4 cm–3 s–1 during the initial 2 h of particle burst, A-10:25-10:40, B-15:20-15:40, C-19:15-19:45; c: GMD, geometric mean diameter; GR = 3.2 nm h–1 during the initial 5 h).
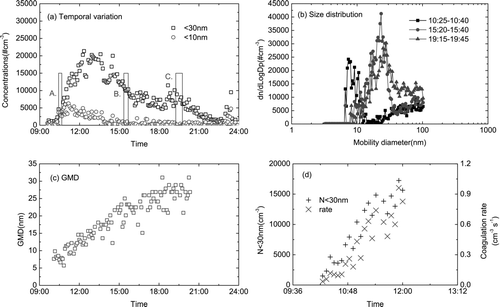
A particle burst occurred on 25 January 2004 when the wind direction changed from north to east (onshore wind) (http://envf.ust.hk/dataview/aws/current/index.py?station=UST). It is apparent that a nucleation event was induced by the change in wind direction. The wind speed was ∼ 5 m s–1 at the onset of the particle burst and gradually decreased to 0.5 m s–1 toward the end. The ambient temperature was 10–12°C and RH 40–45%. The daytime maximum irradiation was 450 W m–2 and the smooth diurnal variation suggested that it was a mostly clear day. CitationWen et al. (2006) reported that nucleation events occurred mostly at 10–13°C at Bodega Bay, California.
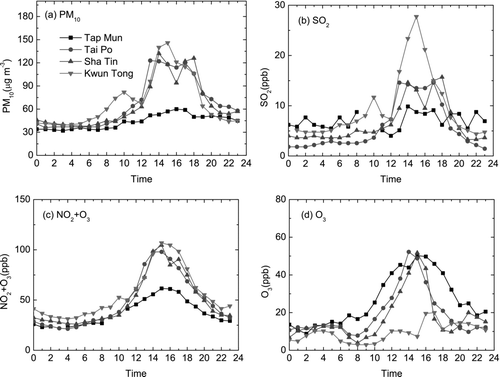
There was no significant increase in PM10 and SO2 concentrations at the three HKEPD urban sites during the particle burst (, , , ) and concentrations of (NO2+O3) at Tai Po and Sha Tin were close to the background site concentration. The high concentration of (NO2+O3) at Kwun Tong during daytime and nighttime was probably due to the primary emission of NO2 rather than photochemical NO2 and O3(CitationYao et al. 2006a). Based on these observations, we can infer that there were no strong photochemical reactions in east Hong Kong, while HKUST is situated in the east between Sha Tin and Kwun Tong. This suggests that a lack of strong photochemical reactions at low ambient temperature probably limited the growth of the nucleated particles to < 50 nm. From 22:00 to 24:00, the wind direction changed from west to north and the wind speed increased from 1 m s–1 to 4 m s–1, making the transport of pollutants from outside of Hong Kong likely, thus increasing the concentrations of PM10 and (NO2+O3) (, , , ).
Regime 1 particle growth was also observed to be induced by a strong northwest wind. During 8:00–12:00 on 21 January 2004, the wind was from the northwest and the wind speed was 7–8 m s–1, the ambient temperature was 7.5–12.5°C and RH was 42–60%. The smooth diurnal variation of irradiation indicated that it was a clear day. A nucleated particle burst started from 7 nm (12:00) and grew to 32 nm (16:00) () when the wind speed was larger than 5 m s–1. The formation rate was 2.7 particle cm–3 s–1 and the growth rate was 3.4 nm h–1. PM10 and gaseous species data (, , , ) suggest that the lack of strong photochemical reactions in east Hong Kong limited the growth of the nucleated particles to < 50 nm in diameter. Lack of strong photochemical reactions is likely the reason for other Regional 1 cases where the grown nucleated particles were < 50 nm in diameter.
FIG. 4 (a–e) Concentrations of particle and gases on 21 January 2004.
The “official” lunch time in Hong Kong is 13:00–14:00 and short-term large particle bursts are commonly observed at ∼ 14:00 (e.g., on 21 and 25 January 2004; and ) which are mere reflections of lunch-time traffic activities.
3.3. Shrunken Particle Sizes and Estimated Shrinkage Rates
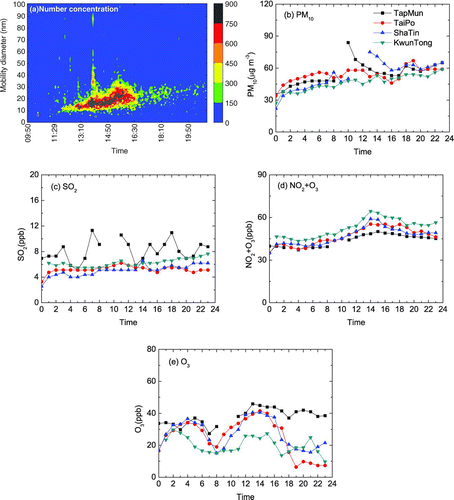
On 12 January 2004, a reversal of particle growth was observed. A nucleation particle burst started at 10:40 and the initial particle mode was ∼ 18 nm (). The total UFP concentration increased from ∼ 7.6*103 particle cm–3 to a maximum of 2.8*104 particle cm–3. Temporal variations of 3–10 nm, 3–30 nm, and UFP in number concentration are shown in . From this data, the formation rate of the < 30 nm particles was estimated to be 4.3 particle cm–3 s–1. shows the size distribution of UFP in number concentration at the initial stage (10:40–11:00), during maximum growth (14:20–15:00), during particle shrinkage (15:40–16:00), and at the end of the shrinkage (18:00–18:20). The calculated growth rate in the initial 4 h was 11.8 nm h–1 and the maximum GMD reached ∼ 60 nm (). The short-term particle bursts at ∼ 14:00 were probably vehicle emissions from the nearby parking lot rather than by transport. CitationYao et al. (2005) reported that aged vehicular particles in tunnels in Hong Kong dominate the 30 to 50 nm size range and CitationZhu et al. (2002) also reported that aged vehicular particles at > 100 m distance from the traffic roads dominate the > 30 nm size range, therefore contribution of aged vehicular particles to the < 30 nm ambient particles should be insignificant.
FIG. 5 (a–c) Particle growth followed by particle shrinkage on 12 January 2004 (GMD, geometric mean diameter).
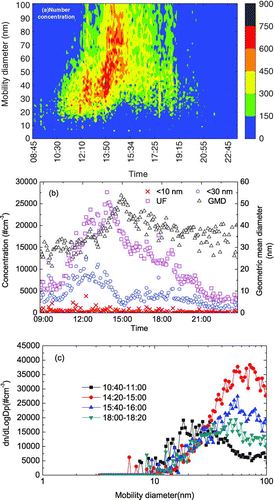
From , it can be seen that after the 14:20 burst, particle size growth was evident and was believed to be an extension of the long-term event started at 10:40. The growth continued until 15:00. A uni-modal size distribution was exhibited after 14:20 () and the uni-modal size distribution suggested that the grown nucleated particles from the long-term event overwhelmed the short-term vehicular UFP. It is important to note that the particle size distribution throughout this period was UNI-MODAL even though there was a shift in the mode (increase of GMD during the period from 14:20 to 15:00). Should mixing be responsible for the “apparent growth or shrinkage” of the particles, a bi-modal or even tri-modal distribution would result. It is also conceivable for the mixed particles to maintain the uni-modal distribution, but given there is a continuous increase of GMD in the uni-modal size distribution, this possibility is not likely.
The calculated shrinkage rate was 8.6 nm h–1 and the particle mode was finally reduced to < 50 nm. From 10:40 to 13:00, new particle formation and particle growth led to an increase in the concentration of < 30 nm particles and an increase in GMD (). From 13:00 to 15:00, the sum of the scavenge rate of the < 30 nm particles and the growth rate of particles from < 30 nm to > 30 nm was probably larger than the new particle formation rate, leading to a decrease in the concentration of < 30 nm particles (). The growth of particles increased the GMD. From 15:00 to 16:00, the shrinkage rate of particles from > 30 nm to < 30 nm was larger than the scavenge rate of < 30 nm particles, leading to an increase in concentration of < 30 nm particles and a decrease in GMD ().
HKEPD air pollution data for the same day showed a simultaneous increase in PM10 and (NO2+O3) concentrations at Tai Po, Sha Tin, and Kwun Tong since 10:00 and the spatial distribution was uniform ( and ). SO2 concentrations also increased but the spatial variation was large (). Maximum concentrations at the three sites occurred at 13:00–16:00 when the average wind at HKUST was 5 m s–1and the wind direction was from the northwest, north and northeast. The similar concentrations at these urban sites suggest the transport of these pollutants from outside of Hong Kong in the northeast, north and northwest directions rather than uniform street-level emission. Maximum PM10 and (NO2+O3) concentrations at these three sites were 2–3 times of the background site at Tap Mun, suggesting the formation of secondary gaseous and particulate species. The particle burst observed at HKUST was caused by the regional transport of air pollutants. After 15:00, PM10 and (NO2+O3) concentrations started to decrease and particle growth reversed while there were no significant changes in temperature and RH.
FIG. 6 (a–d) Spatial distribution of air pollutants on 12 January 2004.
There was a shift in the wind direction during this period; however, the winds were clean marine air from South China Sea. This can be seen from the air quality data at the background station in Tap Mun (TM) upwind of the sampling site in the same (northeast) direction of the winds. Air pollutant concentrations including PM10 at TM on that day were substantially lower than those at the urban sites. The clean marine air would dilute the polluted air mass at sampling site, leading to a decrease of concentrations of the primary and secondary gaseous pollutants (, ). This suggests that the decrease in photochemical activity and concentrations of photochemically formed semi-volatile gases such as HNO3, HCOOH, CH3COOH, etc., could be responsible for the growth reversal. When chemical conditions changed, re-partitioning of the semi-volatile species would occur, thus the thermodynamic equilibrium is re-established.
Summarizing, the above discussion provides evidence that particle growth reversals or shrinkages were observed after 15:00 (a) there was no significant increase of 3–10 nm particles in number concentration suggesting no new particle formation; and (b) the increase in the < 30 nm particles occurred at the expense of a decrease of the > 30 nm particles. The uni-modal size distribution exhibited during particle growth reversals suggested that the grown nucleated particles from long-term events were dominant. Furthermore, the continuous decrease of GMD in the uni-modal size distribution suggested that the possibility of mixing of air masses was not likely; and (c) the polluted air mass diluted by the clean marine air would lead to the re-partitioning of the semi-volatile species and thus particle shrinkage.
On 21 November 2003, particle growth reversal was observed at 10:15-∼ 13:20 for Regime 1 and at ∼ 13:30–16:40 for Regime 2 (, , ). The Regime 1 formation rate was 5.1 particle cm–3 s–1, and the maximum growth of 7.5 nm h–1 occurred at 12:30. From 12:30 to 13:15, the shrinkage rate was 10.7 nm h–1 and the particle mode decreased from 30 to 21 nm (). For Regime 2, maximum growth occurred at 15:10. Because of the overlap of Regimes 1 and 2, the exact start time of Regime 2 could not be isolated. From 15:10 to 16:40, the shrinkage rate was 8.2 nm h–1 and the particle mode decreased from 37 nm to 23 nm (). Using the same arguments for the 12 January dataset, growth reversal most likely occurred.
FIG. 7 (a–c) Particle growth followed by particle shrinkage on 21 November 2003 (GMD, geometric mean diameter).
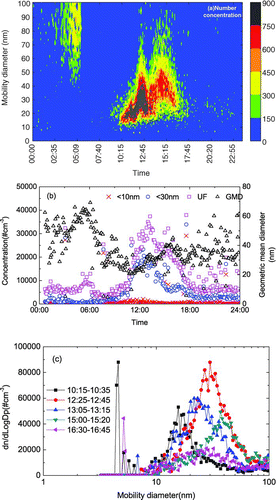
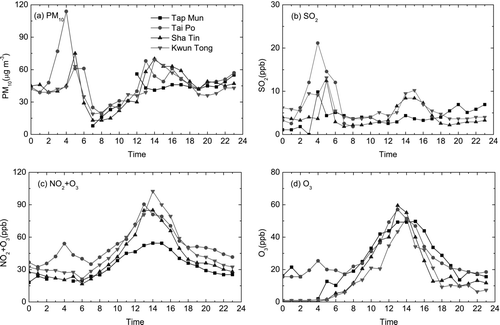
HKEPD air pollution data at 7:00–10:15 on the same day showed PM10 and SO2 concentrations at the three urban sites were < 30 μ g m–3 and 5 ppb, respectively ( and ). Regime 1 was observed in a clean atmosphere although PM10 and SO2 concentrations were high at 4:00 ( and ). There was no significant increase in < 50 nm particle number concentration but there was an increase of > 60 nm particles at 4:00 (); the absence of < 50 nm particles was likely because of no photochemical reactions. After 12:00, there was a marked increase in PM10 and SO2 concentrations and higher SO2 probably induced the occurrence of Regime 2. Maximum solar irradiation was observed at 13:00. The concentration of (NO2+O3) started to decrease at 14:00 as shrinkage of particles started (), probably due to the decrease in photochemical activity. Again, no relationship between the reversal in growth of the nucleated particles with ambient temperature and relative humidity could be found.
FIG. 8 (a–d) Spatial distribution of air pollutants on 21 November 2003.
3.4. Possible Causes of the Reversal in Growth of Nucleated Particles
Three possible processes are responsible for the reversal in growth of the nucleated particles: thermal fragmentation of nanoparticles triggered by water evaporation, evaporation of water, and/or evaporation of volatile/semi-volatile species from grown nucleated particles (CitationZhang et al. 2004; CitationGramotnev and Gramotnev 2005). Thermal fragmentation was used to explain the jump in carbonaceous particle mode in vehicle plumes (CitationGramotnev and Gramotnev 2005). Since there was no significant change in ambient temperature during the transition from particle growth to particle shrinkage, thermal fragmentation is not likely to be the explanation.
Our best efforts found three papers on the chemical components of grown nucleated particles in the atmosphere and one on the density (CitationMäkelä et al. 2001; CitationKannosto et al. 2008; CitationSmith et al. 2005, Citation2008). A decrease in RH would cause morphological changes, leading to size change such as shrinkage. RH increased from 57% to 59% from 14:00 to 18:00 when growth reversal was observed on 12 January 2004; therefore, water evaporation from the particles was not likely to be the cause for the shrinkage of the particles. Mäkelä et al. (2001) reported grown nucleated particles to be acidic and consisted mainly of sulfates such as (NH4)2SO4, NH4HSO4, ((CH3)2NH2)2SO4, (CH3)2NH2HSO4 with minor components such as methanosulfonic salts and dicarboxylic acids in the boreal forest in Finland. Sulfates are stable and do not evaporate. CitationSmith et al. (2005) reported ammonium sulfate to be the major component of grown nucleated particles in Atlanta, and in a later study in Tecamac, Mexico, they reported the major component to be organics (84 ± 5%) including nitrogen-containing organic compounds, organic acids (formate, acetate) and hydroxyl organic acids (CitationSmith et al. 2008). Nitrate and sulfate accounted for 6 ± 2% and 10 ± 1%, respectively. Most of these organics are volatile/semi-volatile and can repartition between the gas and particle phases when conditions change. CitationKannosto et al. (2008) reported the densities of nucleation, Aitken (grown nucleated particles) and accumulation particles to be 1.1, 0.8, and 1.5 g cm–3 in the boreal forest, respectively. The low density of grown nucleated particle is ascribed to the condensation of lighter compounds on nucleated particles during particle growth. In the atmosphere, the density of NH4NO3 and most organics is closer to 0.8 g cm–3 and the growth of nucleated particles was likely related to condensation formation of particulate NH4NO3 and organics.
Comparing the case of no growth reversal to one with growth reversal suggests that strong photochemical formation of secondary gaseous and particulate species is an important factor in governing the reversal of growth of particles, and evaporation of the organics and NH4NO3 was the most probable mechanism of the shrinking of the grown nucleated particles.
CitationJung et al. (2006) simulated a nucleation and growth event with particles containing sulfate, ammonium and non-volatile organics, and no shrinkage of grown nucleated particles was reported. This was probably due to the lack of volatile and semi-volatile species in the simulation. Most nucleation and growth events in Regime 1 in this study were likely caused by the mechanism as simulated by CitationJung et al. (2006). In a simulated evolution of particles in vehicle plumes, CitationZhang et al. (2004) and CitationJacobson et al. (2005) reported a reversal in nucleated particle growth when volatile and semi-volatile species were present. For Regime 2 in this study, strong photochemical activity was present and probably formed a large amount of secondary volatile and semi-volatile organic and inorganic species in the gas phase. CitationHo et al. (2003) reported the concentration of organic carbon in PM2.5 in Hong Kong reached ∼ 10 μ g m–3 in the season same as this study. The grown nucleated particles could consist mainly of secondary volatile and semi-volatile organic and inorganic species as observed in Mexico by CitationSmith et al. (2008). Thus it is not surprising that shrinkage of particles was observed when photochemical activity decreased.
4. CONCLUSION
The reversal of nucleated particle growth resulting in the shrinkage of these particles was discovered in a large dry-season UFP (3–300 nm) dataset (30,663 scans) taken at a suburban site in Hong Kong. The shrinkage occurred after the particles reached maximum diameter and the shrinkage rate was close to the growth rate. The particulates in Hong Kong contain large fractions of volatile/semi-volatile organic and inorganic species, and it is believed that when atmospheric conditions (meteorological parameters and pollutant concentrations) change, a shift in gas/particle partitioning takes place and the evaporation of the volatile/semi-volatile species caused this phenomenon. This phenomenon is worthy of further study because of its potential impact on the climate.
Acknowledgments
This work was supported by the Hong Kong Jockey Club Charities Trust. XHY would like to acknowledge the support of NSFC No. 40775082 and thanks Guangliang Liu and Fan Yang for their help on preparing figures.
Notes
*Formation rate of < 30 nm particles was corrected for particle-particle coagulation effect using the same method by CitationQian et al. (2007)
**Regime 1 and Regime 2 overlapped and the rate cannot be estimated.
Related Research Data
REFERENCES
- Barsanti , K. C. , McMurry , P. H. and Smith , J. N. 2008 . The Potential Contribution of Organic Salts to New Particle Growth . Atmos. Chem. Phys. Discuss. , 8 : 20723 – 20748 .
- Dal Maso , M. , Kulmala , M. , Riipinen , I. , Wagner , R. , Hussein , T. , Aalto , P. P. and Lehtinen , K. E. J. 2005 . Formation and Growth of Fresh Atmospheric Aerosols: Eight Years of Aerosol Size Distribution Data from SMEAR II, Hyytiala, Finland . Boreal Environ. Res. , 10 : 323 – 336 .
- Dusek , U. , Frank , G. P. , Hildebrandt , L. , Curtius , J. , Schneider , J. , Walter , S. , Chand , D. , Drewnick , F. , Hings , S. , Jung , D. , Borrmann , S. and Andreae , M. O. 2006 . Size Matters More Than Chemistry for Cloud-Nucleating Ability of Aerosol Particles . Science , 312 : 1375 – 1378 .
- Gao , J. , Wang , T. , Zhou , X. H. , Wu , W. and Wang , W. X. 2009 . Measurement of Aerosol Number Size Distributions in the Yangtze River Delta in China: Formation and Growth of Particles Under Polluted Conditions . Atmos. Environ , 43 : 829 – 836 .
- Gramotnev , D. K. and Gramotnev , G. 2005 . A New Mechanism of Aerosol Evolution Near a Busy Road: Fragmentation of Nanoparticles . J. Aerosol Sci , 36 : 323 – 340 .
- Jacobson , M. Z. , Kittelson , D. B. and Watts , W. F. 2005 . Enhanced Coagulation Due to Evaporation and its Effect on Nanoparticle Evolution . Environ. Sci. Technol , 39 : 9486 – 9492 .
- Jung , J. G. , Adams , P. J. and Pandis , S. N. 2006 . Simulating the Size Distribution and Chemical Composition of Ultrafine Particles during Nucleation Events . Atmos. Environ , 40 : 2248 – 2259 .
- Ho , K. F. , Lee , S. C. , Chan , C. K. , Yu , J. C. , Chow , J. C. and Yao , X. H. 2003 . Characterization of Chemical Species in PM2.5 and PM10 Aerosols in Hong Kong . Atmos. Environ , 37 : 31 – 39 .
- Holmes , N. S. 2007 . A Review of Particle Formation Events and Growth in the Atmosphere in the Various Environments and Discussion of Mechanistic Implications . Atmos. Environ , 41 : 2183 – 2201 .
- Kannosto , J. , Virtanen , A. , Lemmetty , M. , Mäkelä , J. M. , Keskinen , J. , Junninen , H. , Hussein , T. , Aalto , P. and Kulmala , M. 2008 . Mode Resolved Density of Atmospheric Aerosol Particles . Atmos. Chem. Phys , 8 : 5327 – 5337 .
- Kulmala , M. , Vehkamaki , H. , Petaja , T. , Dal Maso , M. , Lauri , A. , Kerminen , V.-M. , Birmili , W. and McMurry , P. H. 2004 . Formation and Growth Rates of Ultrafine Atmospheric Particles: A Review of Observations . J. Aerosol Sci , 35 : 143 – 176 .
- Kulmala , M. and Kerminen , V.-M. 2008 . On the Formation and Growth of Atmospheric Nanoparticles . Atmos. Res , 90 : 132 – 150 .
- Lam , K. S. , Wang , T. J. , Chan , L. Y. , Wang , T. and Harris , J. 2001 . Flow Patterns Influencing the Seasonal Behavior of Surface Ozone and Carbon Monoxide at A Coastal Site Near Hong Kong . Atmos. Environ. , 35 : 3121 – 3135 .
- Lee , A. K. Y. , Li , Y. J. , Lau , A. P. S. and Chan , C. K. 2008 . A Re-Evaluation on the Atmospheric Significance of Octanal Vapor Uptake by Acidic Particles: Roles of Particle Acidity and Gas-Phase Octanal Concentration . Aerosol Sci. Technol. , 42 : 992 – 1000 .
- Mäkelä , J. M. , Yli-Koivisto , S. , Hiltunen , V. , Seidl , W. , Swietlicki , E. , Teinilä , K. , Sillanpää , M. , Koponen , I. K. , Paatero , J. , Rosman , K. and Hämeri , K. 2001 . Chemical Composition of Aerosol during Particle Formation Events in Boreal Forest . Tellus, Ser. B , 53 : 380 – 393 .
- McMurry , P. H. , Woo , K. S. , Weber , R. , Chen , D.-R. and Pui , D. Y. H. 2000 . Size Distribution of 3-10 nm Atmospheric Particles: Implication for Nucleation Mechanisms . Phil. Trans. Royal Soc. London, A , 358 : 2625 – 2642 .
- McMurry , P. H. and Woo , K. S. 2002 . Size Distribution of 3-100 nm Urban Atlanta Aerosols: Measurement and Observations . J. Aerosol Med. , 15 : 169 – 178 .
- O'Dowd , C. D , McFiggans , G. , Creasey , D. J. , Pirjola , L. , Hoell , C. , Smith , M. H. , Allan , B. J. , Plane , J. M. C. , Heard , D. E. , Lee , J. D. , Pilling , M. J. and Kulmala , M. 1999 . On the Photochemical Production of new Particles in the Coastal Boundary Layer . Geophys. Res. Lett. , 26 : 1707 – 1710 .
- O'Dowd , C. D. , Yoon , Y. J. , Junkerman , W. , Aalto , P. , Kulmala , M. , Lihavainen , H. and Vilsanen , Y. 2007 . Airbone Measurements of Nucleation Mode Particles I: Coastal Nucleation and Growth Rates . Atmos. Chem. Phys. , 7 : 1491 – 1501 .
- Qian , S. , Sakurai , H. and McMurry , P. H. 2007 . Characteristics of Regional Nucleation Events in Urban East St. Louis . Atmos. Environ. , 41 : 4119 – 4127 .
- Russell , L. M. , Flagan , R. C. and Seinfeld , J. H. 1995 . Asymmetric Instrument Response Resulting from Mixing Effects in Accelerated DMA-CPC Measurements . Aerosol Sci. Technol. , 23 : 491 – 509 .
- Shi , J. P. , Evans , D. E. , Khan , A. A. and Harrison , R. M. 2001 . Sources and Concentrations of Nanoparticles (< 10 nm Diameter) in the Urban Atmosphere . Atmos. Environ. , 35 : 1193 – 1202 .
- Smith , J. N. , Moore , K. F. , Eisele , F. L. , Voisin , D. , Ghimire , A. K. , Sakurai , H. and McMurry , P. H. 2005 . Chemical Composition of Atmospheric Nanoparticles During Nucleation Events in Atlanta . J. Geophys. Res. , 110 : D22S03 doi:10.1029/2005JD005912
- Smith , J. N. , Dunn , M. J. , VanReken , T. M. , Iida , K. , Stolzenburg , M. R. , McMurry , P. H. and Huey , L. G. 2008 . Chemical Composition of Atmospheric Nanoparticles Formed from Nucleation in Tecamac, Mexico: Evidence for an Important Role for Organic Species in Nanoparticle Growth . Geophys. Res. Lett , 35 : L04808 doi:10.1029/2007GL032523
- Watson , J. G. , Chow , J. C. , Park , K. and Lowenthal , D. H. 2006 . Nanoparticle and Ultrafine Particle Events at the Fresno Supersite . J. Air & Water Manag. Assoc. , 56 : 417 – 430 .
- Wen , J. , Zhao , Y. and Wexler , A. S. 2006 . Marine Particle Nucleation: Observation at Bodega Bay, California . J. Geophys. Res. , 111 : D08207 doi:10.1029/2005JD006210
- Wu , Z. J. , Hu , M. , Liu , S. , Wehner , B. , Bauer , S. , Maßling , A. , Wiedensohler , A. , Petäjä , T. , Dal Maso , M. and Kulmala , M. 2007 . New Particle Formation in Beijing, China: Statistical Analysis of a 1-Year Data Set . J. Geophys. Res. , 112 : D09209 doi:10.1029/2006JD007406
- Yao , X. H. , Fang , M. and Chan , C. K. 2002 . Size Distributions and Formation of Dicarboxylic Ions in Atmospheric Particles . Atmos. Environ. , 36 : 2099 – 2107 .
- Yao , X. H. , Lau , N. T. , Fang , M. and Chan , C. K. 2005 . Real-Time Observation of the Transformation of Ultrafine Atmospheric Particle Modes . Aerosol Sci. Technol. , 39 : 831 – 841 .
- Yao , X. H. , Lau , N. T. , Fang , M. and Chan , C. K. 2006a . Use of Stationary and Mobile Measurements to Study Power Plant Emissions . J. Air Waste Manage. Assoc. , 54 : 144 – 151 .
- Yao , X. H. , Chan , C. K. , Lau , N. T. , Lau , P. S. and Fang , M. 2006b . Possible Sampling Artifact in Real Time Particle Size Distributions Related to Sampling Rate . Aerosol Sci. Technol. , 40 : 1080 – 1089 .
- Young , L.-H. and Keeler , G. J. 2004 . Characterization of Ultrafine Particle Number Concentration and Size Distribution during a Summer Campaign in Southwest Detroit . J. Air & Waste Manage. Assoc. , 54 : 1079 – 1090 .
- Zhang , K. M. , Wexler , A. S. , Zhu , Y. , Hinds , W. C. and Sioutas , C. 2004 . Evolution of Particle Number Distribution Near Roadways. Part II: The ‘Road-to-Ambient’ Process . Atmos. Environ. , 38 : 6655 – 6665 .
- Zhang , J. , Wang , T. , Chameides , W. L. , Cardelino , C. , Kwok , J. , Blake , D. R. , Ding , A. and So , K. L. 2007 . Ozone Production and Hydrocarbon Reactivity in Hong Kong, Southern China . Atmos. Chem. Phys. , 7 : 557 – 573 .
- Zhu , Y. , Hinds , W. C. , Kim , S. , Shen , S. and Sioutas , C. 2002 . Study of Ultrafine Particles Near a Major Highway with Heavy-Duty Diesel Traffic . Atmos. Environ. , 36 : 4323 – 4335 .