Abstract
Gas-wall partitioning of organic compounds (OC) that included C 8 –C 16 n-alkanes and 1-alkenes and C 8 –C 13 2-alcohols and 2-ketones was investigated in two Teflon FEP chambers whose walls were either untreated, oxidized in sunlight, or previously exposed to secondary organic aerosol (SOA). Partitioning was nearly independent of chamber treatment, reversible, and obeyed Henry's law. The fraction of an OC that partitioned to the walls at equilibrium ranged from 0 to ∼ 65%. Values increased with increasing carbon number within an OC class and for OC with similar vapor pressures increased in the order n-alkanes <1-alkenes <2-alcohols <2-ketones. Estimated time constants for achieving partitioning equilibrium ranged from ∼ 60 min for n -alkanes to ⩽ 8 min for 2-ketones. The observations are consistent with a sorption mechanism in which OC dissolve into the film but are restricted to the near-surface region by a sharp permeability gradient that develops in response to OC-induced stresses in polymer chains. When the results were analyzed using a model analogous to one commonly employed for gas-particle partitioning, it was estimated that the sorption properties of the chamber walls were equivalent to organic aerosol mass concentrations of 2, 4, 10, and 24 mg m– 3 with respect to the partitioning of n -alkanes, 1-alkenes, 2-alcohols, and 2-ketones. These values are up to ∼ 4 orders of magnitude larger than concentrations used in most laboratory studies of SOA, which are typically 1–10 3 μ g m– 3 , meaning that if full partitioning equilibrium is established in the chamber then semi-volatile OC will reside overwhelmingly in the chamber walls. Model simulations of gas-particle-wall partitioning were also carried out using the experimental data, and demonstrate quantitatively the large potential effects of Teflon walls on measured yields of gas-phase OC products and SOA.
INTRODUCTION
Laboratory studies of atmospheric chemistry, including the kinetics, products, and mechanisms of gas and aerosol reactions are often performed in large, Teflon film reactors (commonly referred to as smog, environmental, or reaction chambers) with volumes ranging from about 4 to 270 m3 (CitationMcMurry and Grosjean 1985; CitationCocker et al. 2001; CitationVolkamer et al. 2001; CitationFolkers et al. 2003; CitationCarter et al., 2005; CitationPaulsen et al. 2005; CitationRohrer et al. 2005; CitationWang et al. 2006; CitationPresto and Donahue 2006; CitationIinuma et al. 2007; CitationLi et al. 2007; CitationHallquist et al. 2009; Shilling et al. 2009). These chambers are attractive for experiments because they can be constructed fairly easily and economically using transparent, low-reactivity Teflon film and their low surface area/volume ratios allow reaction times up to ∼10 h or so, comparable to the timescales for many atmospheric chemical processes. Recently, such chambers have been widely used to study secondary organic aerosol (SOA) formation, in large part because of the potential impacts of particles on climate, visibility, and human health. In a typical experiment, particle wall-loss rates are on the order of ∼20% h–1 (CitationMcMurry and Grosjean 1985) and their effect on measurements of SOA yield (the most commonly measured aerosol quantity) can be routinely accounted for using aerosol measurements or modeling approaches. Wall losses of organic compounds (OC) present in the gas phase, especially reaction products, are more difficult to quantify or even identify because a real-time analyzer for semi-volatile oxidized organic gases comparable to the scanning mobility particle sizer (CitationWang and Flagan 1990) is not widely available. Although the proton transfer reaction mass spectrometer might provide this capability, the commercial instrument must be modified to allow heating of internal surfaces that otherwise adsorb semi-volatile OC (CitationMikoviny et al. 2010). Nonetheless, it has been recognized from gas-phase kinetics and product studies that such losses can be substantial (CitationBiermann et al. 1985; CitationWang et al. 2006), and in a few studies of SOA formation (CitationKroll et al. 2007; CitationPathak et al. 2008) reduced SOA yields measured in experiments carried out in the absence of seed particles were attributed at least in part to enhanced wall losses of semi-volatile reaction products. In a recent study of the temperature-dependence of SOA yields from α-pinene ozonolysis, CitationSaarthoff et al. (2009) attempted to correct their two-product yield model for losses of the semi-volatile OC product by assuming irreversible loss from the gas phase to the walls of their aluminum chamber, but to our knowledge this is the first time this has been done.
Although it has been recognized for many years that the loss of gas-phase OC to walls could impact the results of experiments carried out in Teflon chambers, to date only CitationGrosjean (1985) and CitationMcMurry and Grosjean (1985) have carried out systematic, quantitative studies of this phenomena. In a series of experiments, they measured mass accommodation coefficients for deposition of a suite of OC onto the walls of Teflon chambers and determined that the values were sufficiently small that lifetimes with respect to wall loss were >15 h. The OC investigated were all quite volatile, however, and no similar studies have been performed for semi-volatile OC that are important in SOA formation and are more likely to be lost to chamber walls.
In a recent chamber study aimed at quantifying the yields of gas-phase 2-ketones formed from OH radical-initiated reactions of 2-methyl-1-alkenes we noticed that for carbon numbers above ∼10 the yields decreased, indicating the possibility of wall losses for the larger, less volatile 2-ketones. To further explore this phenomenon we carried out an extensive investigation of the gas-wall partitioning of a variety of OC, including C8–C16 n-alkanes and 1-alkenes and C8–C13 2-alcohols and 2-ketones, in an effort to quantify the sorption and to determine the mechanism and its dependence on OC volatility and molecular structure. In this publication we report the results of these experiments and of model simulations of gas-particle-wall partitioning that investigate quantitatively the potential effects of Teflon walls on measurements of the yields of gas-phase OC products and SOA.
EXPERIMENTAL SECTION
Experiments were carried in two Dupont Teflon FEP (fluorinated ethylene propylene) chambers. One is 5.9 m3 and has been used for hundreds of experiments on SOA formation, whereas the other is 1.7 m3 and was newly constructed for these experiments. Chambers were filled with clean, dry air (<5 ppbv hydrocarbons, <1% RH) and operated at ∼298 K and atmospheric pressure. In all but one experiment, an equimolar mixture of either C8–C16 n-alkanes, C8–C16 1-alkenes, C8–C13 2-alcohols, or C8–C13 2-ketones, sufficient to achieve a concentration of 300 ppbv for each OC in the chamber (in the absence of wall losses), was prepared in a 300 cm3 glass bulb using a microliter syringe and known OC densities and molecular weights. In the other experiment, only the C13 2-ketone was used and the target chamber concentrations were 100–500 ppbv in 100 ppbv increments. The purities, vapor pressures, saturation ratios, and sources of all chemicals are given in Table S1 in the online Supplemental Information. The bulb has 6 cm long glass inlet and outlet tubes by which it was connected to the chamber and flushed with a stream of clean air while it was heated with a heat gun over its entire surface in order to evaporate the OC mixture into the chamber. Heating was continued for at least a minute after OC were no longer visible in the bottom of the glass bulb, which remained hot for many minutes after the heat was removed. The 6 cm long glass outlet tube is sufficiently long to extend 3 cm into the chamber but sufficiently short that it is well heated by conduction from the hot bulb. This bulb configuration and experimental protocol prevented condensation of OC in the tube and guaranteed that the entire OC mixture entered the chamber. After adding the OC the chamber was mixed for 1 min (the large chamber with a Teflon-coated fan and the small one by rocking back and forth). To sample OC from the large chamber, chamber air was first drawn through a 12 cm long × 0.375 cm O.D. stainless steel tube for 20 min at 300 cm3 min–1 to allow the walls of the tube to equilibrate with OC in the chamber. Experience shows that sampling much sooner than this can lead to lower measured concentrations due to wall losses in the tube. This is always a concern when sampling semi-volatile OC, regardless of the type of tubing used. The need for passivation might be avoided by heating the sampling line (CitationWilliams et al. 2006), but this is cumbersome and was not done here. Simple calculations (CitationHinds, 1982) show that for our conditions diffusion-limited loss is sufficiently fast that for concentrations >5 ppbv the tube walls will be coated with a monolayer of OC (assuming 5 × 1013 molecules cm–2 (CitationSchwarzenbach et al. 2003)) in 20 min. Without passivation, monolayer adsorption could remove ∼50 ppbv of OC from 500 cm3 of air, which for these experiments would correspond to less than ∼10% of the observed loss to the chamber walls. Significant multilayer adsorption will not occur under the subsaturated conditions used here. After the first sample was collected, chamber air was drawn through the sampling line for the remainder of the experiment so the tube walls remained passivated. The same protocol was used for the small chamber, but all tubing was Teflon. After allowing the sampling line to equilibrate, OC were collected by drawing 500 cm3 of chamber air through a glass tube containing Tenax TA solid absorbent and analyzed by gas chromatography with flame ionization detection (GC-FID) (CitationDocherty et al. 2005). GC-FID calibration curves were generated for all OC using liquid-phase injection of OC solutions prepared as dilution series in ethyl acetate. Aerosol mass concentrations were measured using a scanning mobility particle sizer (CitationWang and Flagan 1990). In no experiment did particles form when OC were added; aerosol mass concentrations stayed at background levels of <1 μg m–3. Since no particles were present, all sampled OC were in the gas phase and there was no need to use filters to analyze gases and particles separately.
FIG. 1 Fractions of 300 ppbv concentrations of C8–C13 2-ketones measured in the gas phase, [OC]g/[OC]T, in the (a) SOA chamber and (b) new and oxidized chambers at different times after adding the OC. Fractions in (c) are averages of values shown in (a) for the SOA chamber (only 25–105 min samples) and in (b) for the new and oxidized chambers.
![FIG. 1 Fractions of 300 ppbv concentrations of C8–C13 2-ketones measured in the gas phase, [OC]g/[OC]T, in the (a) SOA chamber and (b) new and oxidized chambers at different times after adding the OC. Fractions in (c) are averages of values shown in (a) for the SOA chamber (only 25–105 min samples) and in (b) for the new and oxidized chambers.](/cms/asset/e6e01517-a42c-4e6a-8eef-62b5b3979017/uast_a_501044_o_f0001g.gif)
Vapor pressures were measured for the C13 n-alkane, 1-alkene, 2-ketone, and 2-alcohol to verify that the 300 ppbv concentrations used did not exceed saturation values. Approximately 50 mg of compound was deposited on the bottom of a 6 L glass bottle, which was enough to see that liquid was present throughout the experiment. The liquid spread across the bottom surface to achieve areas of tens of cm2 that enhanced evaporation rates and therefore reduced equilibration times. Samples were collected periodically for GC-FID analysis by drawing 100 cm3 of air from a port on the top of the bottle through a glass tube containing Tenax TA solid absorbent while clean air entered through a port in the bottom to replace the air removed. The jar was allowed to sit at room temperature, which was maintained within 1–2 K of the ∼298 K temperature at which the chamber experiments were performed, and multiple samples were taken over periods of hours to days to verify that gas–liquid equilibrium was achieved.
RESULTS AND DISCUSSION
Characterization of Gas-Wall Partitioning
Effects of Wall Conditions
The results of an experiment in which the concentrations of C8–C13 2-ketones were measured in the large 5.9 m3 chamber (hereafter referred to as the “SOA chamber”) from 25–400 min are shown in . Values are plotted as ratios of the concentration of each 2-ketone in the gas phase, [OC]g, to the total concentration of that 2-ketone added to the chamber, [OC]T. It is worth emphasizing that in this study the values of [OC]T are accurately known because of the precautions (described above) that were taken to guarantee that measured amounts of OC were quantitatively transferred into the chamber, and that the values of [OC]g are accurately known because authentic OC solution standards were used to calibrate the GC-FID and past experience has shown that the sampling protocol provides high accuracy and reproducibility. Because there were no particles in the chamber, [OC]T= [OC]g+ [OC]w, where [OC]w is the amount of an OC sorbed to the chamber walls per chamber volume. In this experiment, [OC]T= 300 ppbv for each 2-ketone. Measured [OC]g/[OC]T ratios were nearly constant over the entire sampling period, indicating rapid establishment of gas-wall partitioning equilibrium. Although concentrations decreased slightly between 25–105 min and 300–400 min, this was probably due to dilution by room air slowly leaking into the chamber. Values were close to 1 for C8 and C9, but decreased with increasing carbon number to a minimum of ∼0.5 for C13. These observations show that the smaller 2-ketones were present entirely in the gas phase but that the larger ones partitioned to the chamber walls and did so to an extent that increased with increasing carbon number.
The 300 ppbv concentrations of 2-ketones added to the chamber were all well below saturation concentrations, so the reason these OC partitioned to the walls was not that they reached saturation with respect to the liquid phase. As shown in Table S1, saturation ratios ranged from ∼0.0002–0.08. Partitioning was instead due to sorption to the walls, probably by absorption into the Teflon film. As will be shown below, it is unlikely that sorption occurred by adsorption onto the film surface or by absorption into a layer of deposited SOA.
FIG. 2 Fractions of 300 ppbv concentrations of C8–C13 2-ketones measured in the gas phase, [OC]g/[OC]T, in the new chamber before (Fill 1) and after (Fill 2) evacuating the chamber and refilling with clean air. Values are averages for three samples collected and analyzed 25–105 min after adding the OC (Fill 1) and 25–105 min after the chamber was refilled with clean air (Fill 2).
![FIG. 2 Fractions of 300 ppbv concentrations of C8–C13 2-ketones measured in the gas phase, [OC]g/[OC]T, in the new chamber before (Fill 1) and after (Fill 2) evacuating the chamber and refilling with clean air. Values are averages for three samples collected and analyzed 25–105 min after adding the OC (Fill 1) and 25–105 min after the chamber was refilled with clean air (Fill 2).](/cms/asset/52ce9e28-d467-466c-9b10-95765fd67153/uast_a_501044_o_f0002g.gif)
A number of additional experiments were performed to further investigate the sorption process. These were carried out in a small 1.7 m3 chamber. This chamber was constructed for these studies with new Teflon film of the same type used in the SOA chamber so as to eliminate the possibility that the walls were coated with an absorbing layer of SOA deposited in previous experiments (even though the SOA chamber was routinely cleaned by exposure to light and oxidants). This chamber is hereafter referred to as the “new chamber.” In one set of experiments 300 ppbv of each of the C8–C13 2-ketones were added, the chamber was mixed, and three samples were collected and analyzed after 25–115 min (Fill 1). The chamber was then completely evacuated without flushing, refilled with clean air, and three more samples were collected and analyzed after 25–100 min (Fill 2). The chamber took ∼5 min to evacuate and ∼15 min to refill. After completing these experiments, the effect on gas-wall partitioning of exposing the Teflon film to O3 and OH radicals was evaluated by adding ∼30 ppmv of O3 to the chamber and placing it outside in full sunlight for 7 h. The chamber was then flushed overnight with clean air and the experiments with 2-ketones were repeated, with four samples collected and analyzed after 25–155 min. This cleaning procedure has been recommended for Teflon chambers and should remove most hydrocarbon contaminants (CitationKelly, 1982). This chamber is hereafter referred to as the “oxidized chamber.”
Similar to the results shown for the SOA chamber, [OC]g/[OC]T ratios measured for 2-ketones in the new and oxidized chambers were constant over time within measurement uncertainties (±5%), as shown in . Ratios measured at different times were therefore averaged to obtain values of [OC]g/[OC]T for each OC, and results for the SOA, new, and oxidized chambers are compared in . Trends in gas-wall partitioning with respect to carbon number were the same for the three chambers, but for the new and oxidized chambers the fraction of C12 and C13 2-ketones on the walls was higher ([OC]g/[OC]T was ∼30% lower). Since sorption was lowest in the SOA chamber the amount of residual SOA present on the walls was insufficient to significantly impact partitioning. The observation that sorption was not very sensitive to the condition of the chamber walls offers hope that understanding gas-wall partitioning of OC may not be as difficult as one might think.
FIG. 3 Measured relationship between [OC]g and [OC]T when the C13 2-ketone was added to the oxidized chamber in five sequential 100 ppbv doses. The slope of the linear least-squares fit to the data is 0.33.
![FIG. 3 Measured relationship between [OC]g and [OC]T when the C13 2-ketone was added to the oxidized chamber in five sequential 100 ppbv doses. The slope of the linear least-squares fit to the data is 0.33.](/cms/asset/7cb0beee-585b-4a1f-9bc6-77cf9ba6ae83/uast_a_501044_o_f0003g.gif)
FIG. 4 Fractions of 300 ppbv concentrations of (a) C14–C16
n-alkanes, (b) C14–C16 1-alkenes, (c) C10–C13 2-alcohols, and (d) C10–C13 2-ketones measured in the gas phase, [OC]g/[OC]T, in the SOA chamber at different times after adding the OC. The curves are least-squares fits of the exponential function A
t
= A∞+ (A○ – A∞)e–![]()
![FIG. 4 Fractions of 300 ppbv concentrations of (a) C14–C16 n-alkanes, (b) C14–C16 1-alkenes, (c) C10–C13 2-alcohols, and (d) C10–C13 2-ketones measured in the gas phase, [OC]g/[OC]T, in the SOA chamber at different times after adding the OC. The curves are least-squares fits of the exponential function A t = A∞+ (A○ – A∞)e–Display full size, where A = [OC]g/[OC]T, to the data, with the exception of the 2-ketone curves, which were calculated using this equation and a time constant of 8 min (see text for explanation). Plots for which [OC]g/[OC]T decreased by <5% (the measurement uncertainty) are not shown.](/cms/asset/ac460ce2-5e23-4886-8734-375556ce0b37/uast_a_501044_o_f0004g.gif)
Reversibility and Henry's Law
Regardless of the sorption mechanism, it is reversible, as demonstrated by the observation that 2-ketones desorbed from the walls when the chamber was evacuated without flushing and refilled with clean air. Average [OC]g/[OC]T ratios measured for the C8–C13 2-ketones in the new chamber for the Fill 1 and Fill 2 periods are shown in . Ratios were calculated relative to [OC]T= 300 ppbv for both periods, so the height of a segment of a bar (hatched or solid area) represents the fraction of a particular OC present in the chamber during the Fill 1 (hatched) or Fill 2 (solid) period, relative to the total amount of that OC originally added. The results show that when the chamber air containing 2-ketones was replaced with clean air, 2-ketones desorbed from the walls to re-establish gas-wall partitioning equilibrium. Values of [OC]g/[OC]T measured for the Fill 2 period increased with increasing carbon number because of the greater sorption of the larger OC to the walls in the Fill 1 period, followed by desorption when the chamber was evacuated and refilled with clean air. The fraction of each 2-ketone on the walls relative to the total amount of each that was estimated to be in the chamber after evacuating and refilling (assumed equal to the amount on the walls in Fill 1) was somewhat lower for Fill 2 than for Fill 1, which may be due to OC loss from the chamber during evacuation. Results for the SOA, oxidized, and new chambers were similar, again indicating that SOA coatings played no significant role in wall sorption.
In order to investigate the possible effects of OC concentration and OC mixtures (which might promote co-condensation) on the observed gas-wall partitioning, an experiment was performed in which 500 ppbv of the C13 2-ketone were added to the oxidized chamber in five sequential 100 ppbv doses. The corresponding range of saturation ratios was 0.027–0.14. The results are shown in , where a clear linear relationship is observed between [OC]g and [OC]T, with near-zero intercept. The slope of the line is 0.33, which agrees well with the [OC]g/[OC]T ratios of 0.38 and 0.36 measured when 300 ppbv of this OC were added to the new and oxidized chambers as a component of the C8–C13 2-ketone mixtures (). This behavior is consistent with Henry's law (CitationSchwarzenbach et al. 2003) and shows that the fraction of the OC that partitioned to the walls was independent of its concentration and the presence of other OC.
Effects of Organic Compound Structure and Vapor Pressure
The experiments described above for C8–C13 2-ketones were repeated with C8–C16
n-alkanes, C8–C16 1-alkenes, and C8–C13 2-alcohols in the SOA chamber to investigate the impact of molecular structure and vapor pressure on gas-wall partitioning. The chamber evacuation-refill behavior of these OC was similar to that described above for 2-ketones so only Fill 1 results are discussed. Samples were collected and analyzed after 25 min and then one or two times after 65–120 min. [OC]g/[OC]T ratios were calculated using [OC]T= 300 ppbv and plotted against time, as shown in . The data were fit by a least-squares method to the exponential function A
t
= A∞+ (A0 – A∞)e![]() , where A = [OC]g/[OC]T and A0= 1, to obtain values for A∞ and τgw. OC for which [OC]g/[OC]T ratios decreased by <5% (the measurement uncertainty) are not shown. Values of [OC]g/[OC]T= A∞, the fraction of OC in the gas phase at equilibrium, are plotted in . They show that, within an OC class, partitioning to the walls increased with increasing carbon number, apparently due to decreasing vapor pressure. Also, for a given carbon number, partitioning to the walls increased in the order n-alkanes < 1-alkenes < 2-alcohols ∼ 2-ketones. This was apparently due to the effects of functional groups on OC vapor pressures and activity coefficients in Teflon FEP, as discussed below.
, where A = [OC]g/[OC]T and A0= 1, to obtain values for A∞ and τgw. OC for which [OC]g/[OC]T ratios decreased by <5% (the measurement uncertainty) are not shown. Values of [OC]g/[OC]T= A∞, the fraction of OC in the gas phase at equilibrium, are plotted in . They show that, within an OC class, partitioning to the walls increased with increasing carbon number, apparently due to decreasing vapor pressure. Also, for a given carbon number, partitioning to the walls increased in the order n-alkanes < 1-alkenes < 2-alcohols ∼ 2-ketones. This was apparently due to the effects of functional groups on OC vapor pressures and activity coefficients in Teflon FEP, as discussed below.
FIG. 5 Fractions of 300 ppbv concentrations of n-alkanes, 1-alkenes, 2-alcohols, and 2-ketones present in the gas phase at equilibrium in the SOA chamber, [OC]g/[OC]T= A∞, obtained from the curve fits shown in or, when [OC]g/[OC]T decreased by <5% (the measurement uncertainty), by averaging all values for times ⩾25 min. The curves were drawn to aid the eye.
![FIG. 5 Fractions of 300 ppbv concentrations of n-alkanes, 1-alkenes, 2-alcohols, and 2-ketones present in the gas phase at equilibrium in the SOA chamber, [OC]g/[OC]T= A∞, obtained from the curve fits shown in Figure 4 or, when [OC]g/[OC]T decreased by <5% (the measurement uncertainty), by averaging all values for times ⩾25 min. The curves were drawn to aid the eye.](/cms/asset/84a4db38-6d3a-474a-b87b-222e06d035d2/uast_a_501044_o_f0005g.gif)
Equilibration Timescales
The average values of τgw obtained by fitting exponential curves to the data shown in were 60 ± 20, 16 ± 3, 17 ± 10, and 2 ± 4 min for the n-alkanes, 1-alkenes, 2-alcohols, and 2-ketones. The curves shown for the 2-ketones are not the fitted curves, for which 0.01 min ⩽τgw⩽ 9 min, but were instead calculated using a time constant of 8 min. The values obtained from the fitted curves were not used because most are less than the value of 8 min we think is the minimum value that can be reliably measured with our method. This value was arrived at by assuming that equilibrium was achieved when the measured [OC]g/[OC]T ratios were within 5% (the measurement uncertainty) of A∞. Because for an exponential curve this condition is reached in a time equal to 3τgw, the observation that the 2-ketones were already in gas-wall partitioning equilibrium when the first sample was taken at 25 min means that τgw⩽
25 min/3∼8 min.
The value of τgw∼ 8 min estimated for the 2-ketones is similar to the minimum lifetime of ∼10 min measured for the heterogeneous conversion of 1,4-hydroxycarbonyls to dihydrofurans on the walls of an ∼7 m3 Teflon FEP chamber (CitationAtkinson et al. 2008). And the range of τgw values of about 8–60 min bracket the value of ∼30 min calculated from the inverse of the wall-loss rate constant of 5 × 10–4 s–1 calculated using the equation given by CitationMcMurry and Grosjean (1985) for a turbulently mixed chamber where gas transport to the walls is limited by diffusion through an air boundary layer. This model requires a wall accommodation coefficient greater than ∼10–5, and these OC were assumed to have gas-phase diffusion coefficients of 0.03 cm2 s–1 (CitationDavis et al. 1980). This level of agreement is reasonable considering the uncertainties in the CitationMcMurry and Grosjean (1985) model and measurements (such as particle size-dependent wall loss rates) used to calculate the coefficient of eddy diffusion, as well as potential differences in convection in chambers. The observation that τgw values measured here were not the same for all OC classes, however, indicates that diffusion through the air boundary layer was not the only factor that determined τgw and that molecular structure also played a role. The results are consistent with a gas-wall partitioning process in which diffusion through the air boundary layer limits τgw to values greater than ∼8 min, with larger values being determined by diffusion of OC into the Teflon film after they equilibrated at the air–film interface. It is possible that these time constants were affected by our 1 min use of a small fan to mix the SOA chamber (CitationOkuyama et al. 1986) and by our rocking method for mixing the new and oxidized chambers (although the latter method is much less vigorous), but mixing is also induced whenever chemicals are flushed into a chamber using an air stream as well as by temperature gradients in the room. In the experiments discussed in the Introduction, in which 2-ketones were formed in the SOA chamber from reactions of a series of 2-methyl-1-alkenes with OH radicals, the reactions were initiated by turning on blacklights ∼40 min after the mixing fan was turned off. The time-dependent concentrations of the 2-ketones formed in these 6 min reactions (measured using sampling and analytical methods similar to those used here) indicated that the time constants for equilibration with the walls ranged from ⩽8 min to ∼30 min.
Sorption Mechanism
Numerous studies have shown that amorphous, glassy polymer films absorb gases and that equilibrium sorption isotherms and diffusion in the film can be explained using a dual mode model that consists of two terms: a Langmuir term for adsorption of gas into microvoids, which are fixed in number, and a Henry's law term for dissolution of gas into the amorphous assemblage of polymer chains (CitationBarrer et al. 1958; CitationChan et al. 1978; CitationPace and Daytner 1980). Diffusion of OC through the polymer chains can be explained by the Eyring “hole” theory (CitationPrager and Long 1951; CitationPace and Daytner 1980). The process is initiated by the creation of holes when thermal energy leads to the random breaking of van der Waals bonds that hold chains together. Diffusing molecules occasionally jump into and pass through these holes. They preferentially move lengthwise, since this orientation presents the smallest cross-sectional area to the hole. Longer molecules and those with branching and functional groups diffuse most slowly.
Diffusion of OC in polymers often obeys Fick's laws (CitationFrisch 1980), as has been shown for small n-alkanes (n⩽ 4) in Teflon FEP (CitationYi-Yan et al. 1980) and so is expected for (at least) small OC in chambers. This is not the case for the OC investigated here, however, since Fick's laws cannot explain the observation that OC in the gas phase reach equilibrium with the walls at non-zero concentrations. Instead, according to Fick's laws, OC should diffuse through the walls until their concentrations reach zero, the same as those outside the chamber. Our results can only be explained by non-Fickian diffusion, which has been observed in many polymer systems (CitationFrisch 1980). The mechanism proposed here has some similarities to one suggested by CitationMeares (1958) to explain his observations of transient permeability of allyl chloride in polyvinyl acetate films. The basic premise is that when large OC molecules penetrate the Teflon film they distort the polymer structure, creating holes that enhance local permeability (OC diffusivity). Because the concentration of OC (and so their perturbing effect) decreases with depth into the film, permeability also decreases, eventually becoming so small that OC are effectively trapped in the near-surface region. Rather than continuing to diffuse inward in response to the concentration gradient across the film, OC establish gas-wall partitioning equilibrium. The depth to which OC penetrate the film will depend on OC partial pressure and solubility, film permeability, and the response of polymer chains to stress. Our observation that sorption obeys Henry's law suggests that OC penetrate the film to a depth that is proportional to their partial pressure, which seems plausible. The observed rapid desorption of OC from the walls when the chamber was evacuated and refilled with clean air is also consistent with this model of a thin surface layer of relatively mobile OC that can quickly diffuse to the air–wall interface and evaporate in response to the removal of gas-phase OC. A more detailed discussion of this proposed sorption mechanism is presented in online Supplemental Information.
Readers should also be warned that although one might be tempted to try to explain the observed gas-wall partitioning of OC as occurring by adsorption to the film surface instead of by absorption into the film, our results and everything we have seen in the literature argues against this. In addition to the studies discussed above and in online Supplemental Information, which have demonstrated and quantified absorption of OC into polymer films including Teflon FEP, measurements by CitationLi et al. (1993) indicate that alkane vapors do not adsorb onto fluorocarbon surfaces. Also, for example, the amount of 2-ketones sorbed in our experiments is equivalent to ∼8 monolayers (calculated as the surface area of sorbed molecules divided by the smooth chamber surface area = (0.5 ppmv × 2.5 × 1019 molecules m–3 ppmv–1× 6 m3= 8 × 1019 sorbed molecules)(2 × 10–18 m2 per molecule (CitationSchwarzenbach et al. 2003)/20 m2)), but layers of this thickness cannot exist at the low saturation ratios (≪1) present in the chamber. Furthermore, calculations based on van der Waals interactions and compound vapor pressures (CitationSchwarzenbach et al. 2003) indicate that even for sub-monolayers the fraction of 2-ketones adsorbed on a Teflon surface should be ≪1%.
FIG. 6 Relationship between the measured ratios of n-alkanes, 1-alkenes, 2-alcohols, and 2-ketones on the walls and in gas phase, [OC]w/[OC]g, and RT/P°. Ratios were calculated using the results shown in , measured in separate experiments performed for each compound class in the SOA chamber. The lines are linear least-squares fits to the data with slopes of 9, 20, 50, and 120 μmole m−3 for n-alkanes, 1-alkenes, 2-alcohols, and 2-ketones.
![FIG. 6 Relationship between the measured ratios of n-alkanes, 1-alkenes, 2-alcohols, and 2-ketones on the walls and in gas phase, [OC]w/[OC]g, and RT/P°. Ratios were calculated using the results shown in Figure 5, measured in separate experiments performed for each compound class in the SOA chamber. The lines are linear least-squares fits to the data with slopes of 9, 20, 50, and 120 μmole m−3 for n-alkanes, 1-alkenes, 2-alcohols, and 2-ketones.](/cms/asset/97285c81-7ab6-433a-bb92-bc23403f992a/uast_a_501044_o_f0006g.gif)
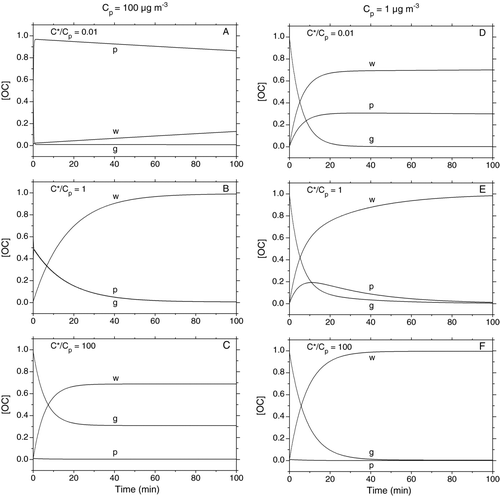
FIG. 7 Model simulations of the time evolution of the concentrations of OC in the gas (g) and particle (p) phases and on the walls (w) in the SOA chamber, assuming gas-wall partitioning behavior similar to 2-ketones. Calculations were performed using the following pairs of values of Cp (μg m−3), C*/Cp: (A) 100, 0.01; (B) 100, 1; (C) 100, 100; (D) 1, 0.01; (E) 1, 1; (F) 1, 100.
Effects of Gas-Wall Partitioning on SOA Formation
The results shown in can be used with OC vapor pressures to gain more quantitative insights into the possible effects of gas-wall partitioning on studies carried out in Teflon chambers. To do this, it is convenient to use gas-particle partitioning theory (CitationPankow, 1994) to treat the walls as if they behave like an equivalent or effective mass concentration of organic aerosol particles (assumed to be liquid), Cw, into which OC partition. The fraction of OC in the gas phase is then given by the equation
This equation can be used to determine the important quantity, Cw/Mwγw, which is the slope of the line obtained by plotting [OC]w/[OC]g (equal to [OC]T/[OC]g – 1) vs. RT/P°. shows plots made using the data in , which give values of Cw/Mwγw of 9, 20, 50, and 120 μmole m–3 for n-alkanes, 1-alkenes, 2-alcohols, and 2-ketones. Values of Cw/Mwγw can be used to model the effects of gas-wall partitioning in chamber studies, as is shown below.
Although we cannot determine individual values of Cw, Mw, and γw from the data available, it is useful to consider two scenarios in order to gain additional physical insights into gas-wall partitioning. If, for example, Cw/Mw is treated as a property of the Teflon film independent of the OC, the observation that the slopes are not all the same indicates that sorption into the Teflon film is non-ideal and depends on compound activity coefficients as well as vapor pressures. The activity coefficients decrease with the addition of C=C double bonds, carbonyl, and hydroxyl groups to the carbon chain, indicating a greater affinity of the Teflon film for compounds with these functional groups. Alternatively, because gas-particle partitioning can also be modeled using Equations (Equation1) and (Equation2) [with w replaced by p (for particles)], it can be instructive to assign values for γw and Mw that are similar to the values of γp and Mp typically used to model gas-particle partitioning of OC in chambers. When this is done (even though the values of γ and M are certainly not similar for the Teflon film and particles), the partitioning properties of the walls and particles can be directly compared using the values of Cp, the organic aerosol particle mass concentration, and Cw, the equivalent or effective organic mass concentration of the walls (in units of mass per chamber volume). This is convenient, since aerosol scientists are used to thinking in terms of particle mass concentrations. Therefore, taking γw= 1 (an ideal solution) and Mw= 200 g mole–1 (CitationSeinfeld et al. 2001), the values of Cw for n-alkanes, 1-alkenes, 2-alcohols, and 2-ketones are 2, 4, 10, and 24 mg m–3. These values are up to ∼4 orders of magnitude larger than the organic aerosol particle mass concentrations present in most laboratory SOA studies, which typically range from ∼1–103μg m–3, and means that if full partitioning equilibrium is established in the chamber then OC will reside overwhelmingly in the chamber walls. It is important to note, however, that although it is valid to treat gas-wall partitioning using an equivalent organic aerosol particle mass concentration, the timescales for gas-particle and gas-wall partitioning are different and must be accounted for in a model.
The simple model employed here uses four first-order rate equations (all of the form d[OC]x/dt = k[OC] x ) with appropriate rate constants given below to describe the following processes: (1) partitioning of the gas-phase OC to the particle phase, (2) partitioning of the particle-phase OC to the gas phase, (3) partitioning of the gas-phase OC to the walls, and (4) partitioning of the OC from the walls to the gas phase.
The equation for k1 is appropriate for gas-phase, diffusion-limited mass transfer (CitationFriedlander, 2000) and depends on the diffusion coefficient of the OC in the gas phase, Dg, the particle surface area concentration, Sp (equal to 6Cp/ρpdp), the particle density, ρp, the mean particle diameter, dp, and Cp. The equations for k2 and k4 were obtained from the partitioning equilibrium equations k2/k1= 1/KpCp and k4/k3= 1/KwCw, where Kp is the gas-particle partitioning coefficient and the other quantities are defined above. For the purposes of this discussion it is convenient to describe the OC volatility in terms of a saturation mass concentration, C*, which is equal to 1/Kp= MpγpP°/RT (CitationDonahue et al. 2006), instead of using OC vapor pressure. Substituting Kp= 1/C* into Equation (Equation4) and Kw= RT/MwγwP° and C*= MpγpP°/RT into Equation (Equation6) gives the final forms used here. It was assumed the particles are spherical and that dp= 0.2 μm, ρp= 1 g cm–3, Dg= 0.03 cm2 s–1 (CitationDavis et al. 1980), γp= 1, Mp= 200 g mole–1, Cw/Mwγw= 120 μmole m–3, and k3= 2 × 10–3 s–1 (the inverse of τgw∼ 8 min). The latter two values are those measured for 2-ketones in the SOA chamber and were chosen so as to estimate upper limits for OC wall losses. In the calculations performed here the OC was assumed to be present initially in the gas phase, which is the case for most products formed in chamber reactions. The time evolution of its concentration in the gas phase, [OC]g, particle phase, [OC]p, and in the walls, [OC]w, was then calculated using the model written in FACSIMILE 4.0 software for Windows (MCPA Software Ltd) kinetics modeling software.
It will be helpful to note that the time constants for reaching gas-particle and gas-wall partitioning equilibrium, τgp and τgw, respectively, can be calculated from each pair of kinetic equations as the inverse of the sum of the rate constants (CitationSchwarzenbach et al. 2003) and so are equal to
The approximate equations give time constants in seconds for mass concentrations in μg m–3.
From partitioning theory and the time constant equations given above, the behavior of the system will depend on C*, Cp, and C*/Cp. In general, OC wall losses should decrease as C* and Cp increase and C*/Cp decreases. This is because (1) the timescale for reaching gas-particle partitioning equilibrium [proportional to 1/(C*+ Cp)] decreases so less time is available for gas-phase OC transport to the walls, (2) the fraction of OC in the gas phase decreases as C*/Cp (equal to [OC]g/[OC]p at equilibrium, as seen by replacing “w” with “p” in Equation (Equation1) or (2) and rearranging) decreases thereby reducing the rate of gas-phase OC transport to the walls, and (3) the fraction of OC in the gas and particle phases at equilibrium will increase relative to the walls.
FIG. 8 Model simulations of the effects of gas-wall partitioning on the concentrations of OC in the gas (g) and particle (p) phases in the SOA chamber at 20 and 100 min, assuming gas-wall partitioning behavior similar to 2-ketones. Values of [OC] (walls)/[OC] (no walls) are ratios of OC concentrations in the gas or particle phase calculated with and without gas-wall partitioning included in the model. Calculations were performed using all combinations of Cp (μg m−3)= 1, 10, 102, 103, and C* (μg m−3)= 0.01, 0.1, 1, 10, 102, 103 for each of the following phases, times (min): (a) gas, 20; (b) gas, 100; (c) particle, 20; (d) particle, 100. The solid and dashed curves were drawn to aid the eye. The dashed curves connect points of equal C*/Cp. Starting on the upper left of each figure the dashed curves correspond to C*/Cp= 0.001, 0.01, 0.1, and 1. The curves connecting C*/Cp= 10 points are essentially the same as the solid curves connecting the high C* points for Cp= 1 μg m−3 at the bottom of each figure and so were not drawn.
![FIG. 8 Model simulations of the effects of gas-wall partitioning on the concentrations of OC in the gas (g) and particle (p) phases in the SOA chamber at 20 and 100 min, assuming gas-wall partitioning behavior similar to 2-ketones. Values of [OC] (walls)/[OC] (no walls) are ratios of OC concentrations in the gas or particle phase calculated with and without gas-wall partitioning included in the model. Calculations were performed using all combinations of Cp (μg m−3)= 1, 10, 102, 103, and C* (μg m−3)= 0.01, 0.1, 1, 10, 102, 103 for each of the following phases, times (min): (a) gas, 20; (b) gas, 100; (c) particle, 20; (d) particle, 100. The solid and dashed curves were drawn to aid the eye. The dashed curves connect points of equal C*/Cp. Starting on the upper left of each figure the dashed curves correspond to C*/Cp= 0.001, 0.01, 0.1, and 1. The curves connecting C*/Cp= 10 points are essentially the same as the solid curves connecting the high C* points for Cp= 1 μg m−3 at the bottom of each figure and so were not drawn.](/cms/asset/16309df0-dd38-4d6b-8d81-c1098204e8d3/uast_a_501044_o_f0008g.gif)
Results of calculations carried out using values of Cp= 1 and 100 μg m–3, and C*/Cp= 0.01, 1, and 100 are shown in for a period of 100 min. The values of these quantities and the time period are typical of many chamber studies of SOA formation (CitationOdum et al. 1997; CitationGriffin et al. 1999). The results are consistent with expectations. In Figures , where Cp= 100 μg m–3 and τgp= 0.1–10 s is much less than τgw= 350–500 s, gas-particle partitioning equilibrium is rapidly established and maintained so that [OC]g/[OC]p= C*/Cp= 0.01, 1, and 100 throughout the 100 min period. For C*/Cp= 0.01 the fractions of OC in the gas phase, particle phase, and in the walls after 100 min are 0.01, 0.86, and 0.13, far from equilibrium values of 0.00, 0.00, and 1.00. Conversely, for C*/Cp= 1 the fractions are 0.00, 0.00, and 1.00, and for C*/Cp= 100 they are 0.29, 0.00, and 0.71, essentially equilibrium values. In Figures , where Cp= 1 μg m–3, τgp= 10–1000 s and τgw= 500 s so the timescale for achieving gas-particle partitioning equilibrium ranges from being much smaller than, to being comparable to, that for gas-wall partitioning. Nonetheless, the fractions of OC in the gas phase, particle phase, and in the walls after 100 min are all very close to equilibrium values. This is because in all cases the fraction of OC in the gas phase is sufficiently large to allow gas-phase OC transport between the particles and walls to nearly establish equilibrium in 100 min, a period of time that is ∼6–12 times larger than the timescales for reaching gas-particle and gas-wall partitioning equilibrium.
As an alternative way of viewing the extent to which sorption to the walls can impact OC, the model was used to calculate OC concentrations in the gas and particle phases in the presence and absence of sorbing walls. The results are shown in as ratios of OC concentrations in each phase calculated with and without wall sorption at 20 and 100 min. As expected from the earlier discussion, for a given C*, losses of both gas-phase and particle-phase OC to the walls increases with decreasing Cp, and for a given Cp, losses increase with increasing C*. For C*⩽ 1 μg m–3 and Cp⩾ 102μg m–3, OC losses after 20 min are relatively independent of C* and <5% for the both particle and gas phases because τgp< 10 s and C*/Cp⩽ 0.01, resulting in very little time and gas-phase OC being available for transport to the walls. As Cp decreases below 102μg m–3 both the timescale for reaching gas-particle partitioning equilibrium and the fraction of OC in the gas phase increase significantly, enhancing wall losses to ∼60% for the particle phase and ∼90% for the gas phase for Cp= 1 μg m–3. Losses are slightly larger after 100 min, but since τgp< 1000 s and C*/Cp⩽ 1 (for C*⩽ 1 μg m–3 and Cp⩾ 1 μg m–3) a large fraction of the OC originally in the gas phase partitions to the particles in 20 min, significantly reducing the rate of additional loss. For C*> 1 μg m–3, wall losses increase more rapidly with increasing C* and decreasing Cp, but for Cp= 1 μg m–3 they are relatively independent of C*. The latter observation is probably due to compensating effects of gas-particle partitioning timescales, which decrease with increasing C*, and equilibria, which shift OC to the gas phase with increasing C*. It is noteworthy that, for Cp= 10–100 μg m–3, which are typical values for many SOA yield experiments, the average loss of OC from the particle and gas phases (the values are similar) to the walls after 100 min are approximately 15%, 20%, 70%, and >99% for C*/Cp= 0.001, 0.01, 0.1, and 1. These values of C*/Cp correspond to OC fractions in the gas phase at gas-particle partitioning equilibrium (in the absence of walls) of 0.1%–50%, thus including not only obviously semi-volatile compounds but also compounds that are on the border of being considered non-volatile (CitationDonahue et al. 2009). For Cp= 1 μg m–3, losses range from ∼70% to >99% for C*⩾ 10–2μg m–3.
It is worth noting that if these calculations were performed assuming that OC behaved like n-alkanes instead of 2-ketones with regard to gas-wall partitioning, so that k3= 3 × 10–4 s–1 instead of 2 × 10–3 s–1 in Equations (Equation5), (6), and (8), and Cw= 2 mg m–3instead of 24 mg m–3in Equations (Equation6) and (Equation8), then for all values of C* and Cp the values of [OC]g/[OC]w and [OC]p/[OC]w at equilibrium (where [OC]g/[OC]w= C*/Cw and [OC]p/[OC]w= Cp/Cw) would be smaller and τgw would be larger (600–3600 s compared to 350–500 s). Thus less OC would be sorbed in the walls at equilibrium and gas-wall partitioning equilibrium would be reached more slowly. The [OC]w curves in would therefore be shifted downwards and to longer times, whereas the [OC]p and [OC]g curves would be shifted upwards but relatively little with respect to time. Likewise, the [OC]p and [OC]g curves in would be shifted upwards at 20 and 100 min.
CONCLUSIONS AND IMPLICATIONS
It appears from the results presented here that losses of OC to the walls of a Teflon chamber can be large for semi-volatile compounds and could have a major impact on measurements of OC in both the gas and particle phases. This could lead to significant underestimates of OC product yields and SOA yields. In the latter case, not only are OC lost from particles to the walls, thereby directly reducing the SOA yield; loss of gas-phase OC reduces the possibility of further oxidation or particle-phase oligomer formation, both of which can lead to SOA-forming products. A current trend in SOA yield measurements is toward the use of SOA mass concentrations on the order of ∼10 μg m–3 or less in an attempt to better simulate atmospheric conditions (CitationPresto and Donahue 2006; CitationShilling et al. 2008). Values measured in such experiments are usually higher than those obtained by extrapolating yield curves measured at higher mass concentrations downwards. Considering that OC wall losses are enhanced under these conditions, however, this approach may also underestimate yields. One possibility might be to carry out experiments at much higher concentrations, where SOA yields are near a plateau in the yield curve (CitationOdum et al. 1996), and then use the SOA volatility distribution measured with a thermal denuder or by dilution (CitationFaulhaber et al. 2009; CitationGrieshop et al. 2009) to extrapolate results to atmospheric conditions. But there are also problems with this approach, such as the difficulty in extrapolating results to low mass concentrations, and the potential for high mass concentrations to promote oligomer formation via enhanced sorption of OC. It is not possible to quantify the effects of gas-wall partitioning on previously measured SOA yields, but yield curve parameters determined from reactions of a large number of biogenic (CitationGriffin et al. 1999) and aromatic compounds (CitationOdum et al. 1997) correspond to C* values of about 1–103μg m–3 and the range of SOA mass concentrations in those experiments was similar, indicating that the measured SOA yields probably underestimate true values. This may be a contributing factor to the disagreement often observed between measured and modeled atmospheric SOA mass concentrations (deGouw and Jimenez 2009), since SOA yields measured in the laboratory are used in most regional and global models to predict atmospheric SOA formation (CitationHallquist et al. 2009). In the light of the results presented here and their potential importance for measurements and modeling of atmospheric chemistry and SOA formation, it is recommended that future studies further investigate the mechanism, magnitude, and timescale for OC sorption into Teflon films and the effects of factors such as OC size, structure, concentration, and mixture composition; oxidation conditions; humidity, temperature, and light; and chamber volume (CitationMcMurry and Grosjean 1985) observed no difference between 4 and 60 m3 chambers), wall material, and mixing; in both static and dynamic flow chambers. It appears that, as with gas-particle partitioning, which is now known to be fundamental to understanding the formation and processing of both primary (CitationRobinson et al. 2007) and secondary (CitationOdum et al. 1996) organic aerosol, gas-wall partitioning must be considered when studying these systems in Teflon chambers.
UAST_A_501044_SUP_0001.zip
Download Zip (157.8 KB)Acknowledgments
This material is based on work supported by the National Science Foundation under Grant ATM-0650061. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation (NSF). We thank Roger Atkinson for helpful discussions.
[Supplementary materials are available for this article. Go to the publisher's online edition of Aerosol Science and Technology to view the free supplementary files.]
Notes
‡Also in the Department of Environmental Sciences and Department of Chemistry.
Related Research Data
REFERENCES
- Atkinson , R. , Arey , J. and Aschmann , S. M. 2008 . Atmospheric Chemistry of Alkanes: Review and Recent Developments . Atmos. Environ. , 42 : 5859 – 5871 .
- Barrer , R. M. , Barrie , J. A. and Slater , J. 1958 . Sorption and Diffusion in Ethyl Cellulose: Part III. Comparison Between Ethyl Cellulose and Rubber . J. Polym. Sci , 27 : 177 – 197 .
- Biermann , H. W. , Mac Leod , H. , Atkinson , R. , Winer , A. M. and Pitts , J. N. Jr. 1985 . Kinetics of the Gas-Phase Reactions of the Hydroxyl Radical with Napthalene, Phenanthrene, and Anthracene . Environ. Sci. Technol , 19 : 244 – 248 .
- Carter , W. P. L. , Cocker , D. R. III , Fitz , D. R. , Malkina , I. L. , Bumiller , K. , Sauer , C. G. , Pisano , J. T. , Bufalino , C. and Song , C. 2005 . A New Environmental Chamber for Evaluation of Gas-Phase Chemical Mechanisms and Secondary Aerosol Formation . Atmos. Environ. , 39 : 7768 – 7788 .
- Chan , A. H. , Koros , W. J. and Paul , D. R. 1978 . Analysis of Hydrocarbon Gas Sorption and Transport in Ethyl Cellulose Using the Dual Sorption/Partial Immobilization Models . J. Membr. Sci. , 3 : 117 – 130 .
- Cocker , D. R. III , Flagan , R. C. and Seinfeld , J. H. 2001 . State-of-the-Art Chamber Facility for Studying Atmospheric Aerosol Chemistry . Environ. Sci. Technol. , 35 : 2594 – 2601 .
- Davis , E. J. , Ravindran , P. and Ray , A. K. 1980 . A Review of Theory and Experiments on Diffusion from Submicroscopic Particles . Chem. Eng. Commun. , 5 : 251 – 268 .
- de Gouw , J. and Jimenez , J. L. 2009 . Organic Aerosols in the Earth's Atmosphere . Environ. Sci. Technol. , 43 : 7614 – 7618 .
- Docherty , K. S. , Wu , W. , Lim , Y. B. and Ziemann , P. J. 2005 . Contributions of Organic Peroxides to Secondary Aerosol Formed from Reactions of Monoterpenes with O3 . Environ. Sci. Technol. , 39 : 4049 – 4059 .
- Donahue , N. M. , Robinson , A. L. , Stanier , C. O. and Pandis , S. N. 2006 . Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics . Environ. Sci. Technol. , 40 : 2635 – 2643 .
- Donahue , N. M. , Robinson , A. L. and Pandis , S. N. 2009 . Atmospheric Organic Particulate Matter: From Smoke to Secondary Organic Aerosol . Atmos. Environ. , 43 : 97 – 109 .
- Faulhaber , A. E. , Thomas , B. M. , Jimenez , J. L. , Jayne , J. T. , Worsnop , D. R. and Ziemann , P. J. 2009 . Characterization of a Thermodenuder-Particle Beam Mass Spectrometer System for the Study of Organic Aerosol Volatility and Composition . Atmos. Meas. Tech. , 2 : 15 – 31 .
- Folkers , M. , Mentel , T. F. and Wahner , A. 2003 . Influence of an Organic Coating on the Reactivity of Aqueous Aerosols Probed by the Heterogeneous Hydrolysis of N2O5 . Geophys. Res. Lett , 30 : 1644 doi:10.1029/ 2003GL017168
- Friedlander , S. K. 2000 . Smoke, Dust, and Haze: Fundamentals of Aerosol Dynamics, , 2nd ed , 283 New York : Oxford University Press, Inc. .
- Frisch , H. L. 1980 . Sorption and Transport in Glassy Polymers: A Review . Polym. Eng. Sci. , 20 : 2 – 13 .
- Grieshop , A. P. , Miracolo , M. A. , Donahue , N. M. and Robinson , A. L. 2009 . Constraining the Volatility Distribution and Gas-Particle Partitioning of Combustion Aerosols Using Isothermal Dilution and Thermodenuder Measurements . Environ. Sci. Technol. , 43 : 4750 – 4756 .
- Griffin , R. J. , Cocker , D. R. III , Flagan , R. C. and Seinfeld , J. H. 1999 . Organic Aerosol Formation from the Oxidation of Biogenic Hydrocarbons . J. Geophys. Res. , 104 : 3555 – 3567 .
- Grosjean , D. 1985 . Wall Loss of Gaseous Pollutants in Outdoor Teflon Chambers . Environ. Sci. Technol. , 19 : 1059 – 1065 .
- Hinds , W. C. 1982 . Aerosol Technology: Properties, Behaviour, and Measurement of Airborne Particles , 146 New York, NY : John Wiley & Sons .
- Hallquist , M. , Wenger , J. C. , Baltensperger , U. , Rudich , Y. , Simpson , D. , Claeys , M. , Dommen , J. , Donahue , N. M. , George , C. , Goldstein , A. H. , Hamilton , J. F. , Herrmann , H. , Hoffmann , T. , Iinuma , Y. , Jang , M. , Jenkin , M. E. , Jimenez , J. L. , Kiendler-Scharr , A. , Maenhaut , W. , McFiggans , G. , Mentel , Th. F. , Monod , A. , Prévôt , A. H. S. , Seinfeld , J. H. , Surratt , J. D. , Szmigielski , R. and Wildt , J. 2009 . The Formation, Properties, and Impact of Secondary Organic Aerosol: Current and Emerging Issues . Atmos. Chem. Phys. , 9 : 5155 – 5236 .
- Iinuma , Y. , Müller , C. , Berndt , T. , Böge , O. , Claeys , M. and Herrmann , H. 2007 . Evidence for the Existence of Organosulfates from Beta-Pinene Ozonolysis in Ambient Secondary Organic Aerosol . Environ. Sci. Technol. , 41 : 6678 – 6683 .
- Kelly , N. A. 1982 . Characterization of Fluorocarbon-Film Bags as Smog Chambers . Environ. Sci. Technol. , 16 : 763 – 770 .
- Kroll , J. H. , Chan , A. W. H. , Ng , N. L. , Flagan , R. C. and Seinfeld , J. H. 2007 . Reactions of Semivolatile Organics and Their Effects on Secondary Organic Aerosol Formation . Environ. Sci. Technol. , 41 : 3545 – 3550 .
- Li , D. , Xie , M. and Neumann , A. W. 1993 . Vapour Adsorption and Contact Angles on Hydrophobic Solid Surfaces . Colloid Polym. Sci. , 217 : 573 – 580 .
- Li , Q. , Hu , D. , Leungsakul , S. and Kamens , R. M. 2007 . Large Outdoor Chamber Experiments and Computer Simulations: (I) Secondary Organic Aerosol Formation from the Oxidation of a Mixture of d-Limonene and α -Pinene . Atmos. Environ. , 41 : 9341 – 9352 .
- McMurry , P. H. and Grosjean , D. 1985 . Gas and Aerosol Wall Losses in Teflon Film Smog Chambers . Environ. Sci. Technol. , 19 : 1176 – 1182 .
- Meares , P. 1958 . Diffusion of Allyl Chloride in Polyvinyl Acetate. Part II: The Transient State of Permeation . J. Polym. Sci. , 27 : 405 – 418 .
- Mikoviny , T. , Kaser , L. and Wisthaler , A. 2010 . Development and Characterization of a High Temperature Proton-Transfer-Reaction Mass Spectrometer (HT-PTR-MS). Atmos. Meas. Tech . Discuss. , 3 : 185 – 202 .
- Odum , J. R. , Hoffmann , T. , Bowman , F. , Collins , D. , Flagan , R. C. and Seinfeld , J. H. 1996 . Gas/Particle Partitioning and Secondary Organic Aerosol Yields . Environ. Sci. Technol. , 30 : 2580 – 2585 .
- Odum , J. R. , Jungkamp , T. P. W. , Griffin , R. J. , Flagan , R. C. and Seinfeld , J. H. 1997 . Aromatics, Reformulated Gasoline, and Atmospheric Organic Aerosol Formation . Environ. Sci. Technol. , 31 : 1890 – 1897 .
- Okuyama , K. , Kousaka , Y. , Yamamoto , S. and Hosokawa , T. 1986 . Particle Loss of Aerosols with Particle Diameters between 6 and 2000 nm in Stirred Tank . J. Colloid Interface Sci. , 110 : 214 – 223 .
- Pace , R. J. and Daytner , A. 1980 . Model of Sorption of Simple Molecules in Polymers . J. Polym. Sci. , 18 : 1103 – 1124 .
- Pankow , J. F. 1994 . An Absorption Model of Gas/Particle Partitioning of Organic Compounds in the Atmosphere . Atmos. Environ. , 28 : 185 – 188 .
- Pathak , R. , Donahue , N. M. and Pandis , S. N. 2008 . Ozonolysis of β -Pinene: Temperature Dependence of Secondary Organic Aerosol Mass Fraction . Environ. Sci. Technol. , 42 : 5081 – 5086 .
- Paulsen , D. , Dommen , J. , Kalberer , M. , Prevot , A. S. H. , Richter , R. , Sax , M. , Steinbacher , M. , Weingartner , E. and Baltensperger , U. 2005 . Secondary Organic Aerosol Formation by Irradiation of 1,3,5-Trimethylbenzene-NO x -H2O in a New Reaction Chamber for Atmospheric Chemistry and Physics . Environ. Sci. Technol. , 39 : 2668 – 2678 .
- Prager , S. and Long , F. A. 1951 . Diffusion of Hydrocarbons in Polyisobutylene . J. Amer. Chem. Soc. , 73 : 4072 – 4075 .
- Presto , A. A. and Donahue , N. M. 2006 . Investigation of α -Pinene + Ozone Secondary Organic Aerosol Formation at Low Total Aerosol Mass . Environ. Sci. Technol. , 40 : 3536 – 3543 .
- Robinson , A. L. , Donahue , N. M. , Shrivastava , M. K. , Weitkamp , E. A. , Sage , A. M. , Grieshop , A. P. , Lane , T. E. , Pierce , J. R. and Pandis , S. N. 2007 . Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging . Science. , 315 : 1259 – 1262 .
- Rohrer , F. , Bohn , B. , Brauers , T. , Brüning , D. , Johnen , F.-J. , Wahner , A. and Kleffmann , J. 2005 . Characterisation of the Photolytic HONO-Source in the Atmosphere Simulation Chamber SAPHIR . Atmos. Chem. Phys. , 5 : 2189 – 2201 .
- Saarthoff , H. , Naumann , K.-H. , Möhler , O. , Jonsson , Å. M. , Hallquist , M. , Kiendler-Scharr , A. , Mentel , Th. F. , Tillmann , R. and Schurath , U. 2009 . Temperature Dependence of Yields of Secondary Organic Aerosols from the Ozonolysis of α-Pinene and Limonene . Atmos. Chem. Phys , 9 : 1551 – 1577 .
- Seinfeld , J. H. , Erdakos , G. B. , Asher , W. E. and Pankow , J. F. 2001 . Modeling the Formation of Secondary Organic Aerosol (SOA). 2. The Predicted Effects of Relative Humidity of Aerosol Formation in the α -Pinene, Δ3-Carene, and Cyclohexene-Ozone Systems . Environ. Sci. Technol. , 35 : 1806 – 1817 .
- Schwarzenbach , R. P. , Gschwend , P. M. and Imboden , D. M. 2003 . Environmental Organic Chemistry , Hoboken, NJ : John Wiley & Sons, Inc. .
- Shilling , J. E. , Chen , Q. , King , S. M. , Rosenoern , T. , Kroll , J. H. , Worsnop , D. R. , McKinney , K. A. and Martin , S. T. 2008 . Particle Mass Yield in Secondary Organic Aerosol Formed by the Dark Ozonolysis of α-Pinene . Atmos. Chem. Phys. , 8 : 2073 – 2088 .
- Volkamer , R. , Platt , U. and Wirtz , K. 2001 . Primary and Secondary Glyoxal Formation from Aromatics: Experimental Evidence for the Bicycloalkyl−Radical Pathway from Benzene, Toluene, and p-Xylene . J. Phys. Chem. A , 105 : 7865 – 7874 .
- Wang , L. , Arey , J. and Atkinson , R. 2006 . Kinetics and Products of the Photolysis and Reaction with OH Radicals of a Series of Aromatic Carbonyl Compounds . Environ. Sci. Technol. , 40 : 5465 – 5471 .
- Wang , S. C. and Flagan , R. C. 1990 . Scanning Electrical Mobility Analyzer . Aerosol Sci. Technol. , 13 : 230 – 240 .
- Williams , B. J. , Goldstein , A. H. , Kreisberg , N. M. and Hering , S. V. 2006 . An In-Situ Instrument for Speciated Organic Composition of Atmospheric Aerosols: Thermal Desorption Aerosol GC/MS-FID (TAG) . Aerosol Sci. Technol. , 40 : 627 – 638 .
- Yi-Yan , N. , Felder , R. M. and Koros , W. J. 1980 . Selective Permeation of Hydrocarbon Gases in Poly(tetrafluoroethylene) and Poly(fluoroethylene-Propylene) Copolymer . J. Appl. Polym. Sci. , 25 : 1755 – 1774 .