Abstract
Total 360 samples (of 8 h each) of PM2.5 were collected from six sampling sites for summer and winter seasons in Kanpur city, India. The collected PM2.5 mass was subjected to chemical speciation for: (Equation1) ionic species (NH+ 4, SO2– 4, NO– 3, and Cl–), (Equation2) carbon contents (EC and OC), and (Equation3) elemental contents (Ca, Mg, Na, K, Al, Si, Fe, Ti, Mn, V, Cr, Ni, Zn, Cd, Pb, Cu, As, and Se). Primary and secondary components of PM2.5 were assessed from speciation results. The influence of marine source to PM2.5 was negligible, whereas the contribution of crustal dust was significant (10% in summer and 7% in winter). A mass reconstruction approach for PM2.5 could distinctly establish primary and secondary components of measured PM2.5 as: (Equation1) Primary component (27% in summer and 24% in winter): crustal, elemental carbon, and organic mass, (Equation2) Secondary component (45% in summer and 50% in winter): inorganic and organic mass, and (Equation3) others: unidentified mass (27% in summer and 26% in winter). The secondary inorganic component was about 34% in summer (NH+ 4: 9%; SO2– 4: 16%; NO– 3: 9%) and 32% in winter (NH+ 4: 8%; SO2+ 4: 13%; NO– 3: 11%). The secondary organic component was 12% in summer and 18% in winter. In conclusion, secondary aerosol formation (inorganic and organic) accounted for significant mass of PM 2.5 (about 50%) and any particulate control strategy should also include control of primary precursor gases.
1. Introduction
Particulate matter (PM) levels have been shown to be very high in Indian cities (PM10: 50–600 μgm–3; PM2.5: 25–200 μgm–3; Sharma and Maloo 2005). For developing PM control policies, knowledge of chemical characterization of PM is essential to provide a cause-effect relationship between ambient PM (especially PM2.5) concentrations and emitting sources. Recent studies in developing countries like India have focused on chemical characterization of PM10 (e.g., CitationShukla and Sharma 2008; CitationDubey and Pervez 2008; CitationKarar and Gupta 2006) and very little is known about concentration levels and chemical composition of PM2.5. Although PM2.5 is a subset of PM10, its sources, characteristics, health effects and behavior in the atmosphere could be very different (CitationChow et al. 1993; CitationLevy et al. 2000; CitationVallius et al. 2000). Typical PM2.5 sources are secondary nitrate, sulfate, and organic compounds (formed in the atmosphere), combustion processes including biomass burning, and soil and road dust (CitationHildemann et al. 1991; CitationKim Oanh et al. 2010; CitationHeo et al. 2009).
Earlier studies (CitationHeisler and Friedlander 1977; CitationLee et al. 2004; CitationCabada et al. 2002) have shown that secondary organic aerosol (SOA) is an important contributor to PM2.5. SOA contributes between 15 and 40% of total organic carbon (OC) (CitationHildemann et al. 1993; CitationCabada et al. 2002). Polar nature of SOA poses challenges in their detection and analysis (CitationPlaza et al. 2006). Several studies have estimated contribution of SOA to PM2.5 by employing elemental carbon (EC) tracer method, where OC/EC ratio (of emitting sources) is utilized (CitationStrader et al. 1999; CitationCabada et al. 2002, Citation2004; CitationRussell et al. 2004; CitationNa et al. 2004; CitationPlaza et al. 2006). These studies partially reconstructed PM2.5 mass contribution from primary sources and secondary formations.
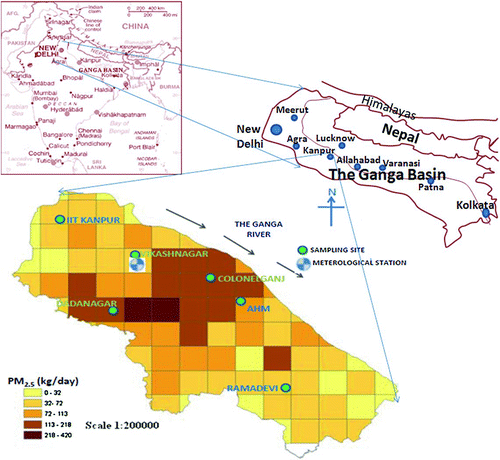
PM emission sources and atmospheric chemistry and properties (e.g., wind speed, temperature, relative humidity, mixing height, and rainfall) vary significantly from one season to another, resulting in variable particulate concentration and composition (CitationSharma et al. 2007). The current study presents a complete reconstruction of PM2.5 composition at six sampling sites in Kanpur, India (latitude 26° 26′ N and longitude 88° 22′ E) for two seasons (summer and winter). The objective of this study is to assess PM2.5 mass and its chemical composition in terms of ions, elements, and EC and OC to reconstruct PM2.5 through its chemical constituents and discern the influence of soil and marine salt, secondary inorganic aerosols (SIA) and SOA in PM2.5 formulation. The study area, Kanpur city represents a typical urban agglomeration in highly polluted Ganga basin (CitationSingh et al. 2004; ). The Ganga Basin (east longitudes 73° 30′ to 89° 0’ and north latitudes 22° 30′ to 31° 30′) is the largest river basin in India, supporting more than 40% of India's population and accounts for 26% of the Indian landmass (). Findings of the present study could also be useful for other cities in the basin.
FIG. 1 The Ganga basin, emission inventory of PM2.5 (grid of 2 km × 2 km) and Air quality sampling sites.
2. Methodology
2.1. Study Area
Kanpur is a large industrial city (population about 4.0 million) having cotton, jute, leather, and wool industries with a total area of about 270 square km. The source activities of air pollution in the city can be broadly classified as: transport, commercial, industrial, domestic, institutional, and fugitive emissions. Sources responsible for NH3 emission are quite significant including livestock, poor sewerage system (open drains), waste collection, and disposal practices and fertilizer application on agricultural fields.
2.2. Materials and Methods
Air quality monitoring was undertaken at six sampling sites (representing different land-use patterns) in summer and winter seasons (; ). The approximate distance between any two consecutive sites was more than 6.0 km except for distance between CG and AHM (∼3.0 km). The distance between the two farthest sites, i.e., IIT and RD is 22 km. The duration of sampling was from 8 April 2007 to 30 June 2007 for summer and 1 December 2007 to 31 January 2008 for winter. The frequency of sampling at each site was a minimum of 9 days (at a stretch) in each season. Filter paper exposure for each sample was 8 h. The sampling height was about 10.0 m at all sites (except for kerb site, CG, where height was about 2.0 m). Reporting and interpretation of results in this article have been done on the basis of a 24-h average data.
TABLE 1 Description of air quality sampling sites
Partisol® Model 2300 4-Channel Speciation Samplers, (Thermo Fisher Scientific Inc., USA) were used to collect air through an inlet (at a flow rate of 10.0 LPM) that removes particles with aerodynamic diameters greater than 2.5 μm; the remaining particles were collected on the filter. Three channels of the sampler were used to capture the PM2.5, one on quartz fiber filter (Whatman grade QM-A quartz filters of 47 mm diameter) and two on Teflon filters (Whatman grade PTFE Filters of 47 mm diameter). Out of these two PTFE filters, one was utilized for ion analysis and the other was used for elemental analysis. The quartz filter was utilized for the analysis of EC and OC. During this monitoring program, meteorological parameters (temperature, relative humidity, wind speed, and wind direction) were recorded using a wind monitor (WM251 Envirotech, New Delhi).
2.3. Gravimetric and Chemical Analyses
PM2.5 mass concentration was determined gravimetrically by weighing the PTFE filters before and after sampling using a digital microbalance (Mettler-Toledo MX-5, USA; sensitivity of 0.001 mg). Prior to weighing, filters were equilibrated in a controlled desiccator (20 ± 5°C and relative humidity 40 ± 2%) for at least 24-h before and after the sampling. Prior to sample collection, quartz filters were baked at 600°C for a minimum of 3-h to remove residual carbon from untreated filters. Before and after particle collection, filters were stored in Petri dishes (lined with aluminum foil), sealed with Teflon tape and were refrigerated. Field blanks (1 in 10 filters) were collected from each site and were analyzed in parallel to the exposed filter papers as a part of QA/QC as per USEPA (1998).
Water soluble ions, NH+ 4, SO2– 4, NO– 3, and Cl– were extracted from one set of PTFE filters using ultra-pure Milli-Q water following the reference method of USEPA (Compendium Method IO-4.2, EPA/625/R-96/010a 1999). Chemical analyses of the water-soluble ions were carried out using Ion Chromatography (Metrohm 761 compact IC, Switzerland). The second set of PTFE filter meant for elemental analysis were digested in hydrochloric/nitric acid solution using a laboratory hot acid extraction apparatus (Compendium Method IO 3.1, EPA/625/R-96/010a 1999). The digested samples were filtered and diluted to 25 ml with deionized (ultra pure) water. The digested samples were analyzed for elemental components (using GBC Avanta ∑ Atomic Absorption Spectrophotometer (AAS)). The elements analyzed by AAS were: Ca, Mg, Na, K, Al, Fe, V, Cr, Ni, Cd, Pb, and Cu. Prior to digestion of second set PTFE filter, elemental components (Si, Ti, Mn, Zn, As, and Se) were analyzed by energy-dispersive X-ray fluorescence (EDXRF). Elemental recovery efficiencies were determined by spiking known quantity of element mass and reproducibility tests were performed by replicate analysis of one out of every 10 samples. Recovery efficiencies varied between 95% and 105%, and reproducibility tests had acceptable results within ±10% for all the elemental species analyzed in this study. OC and EC were analyzed by thermal optical transmittance (DRI Model 2001 Thermal/Optical Carbon Analyzer) using the NIOSH based method. The concentrations reported in this study were blank subtracted using the average filter blank concentration for each site.
3. Results and Discussion
3.1. Ambient PM2.5 Levels and Micrometeorology
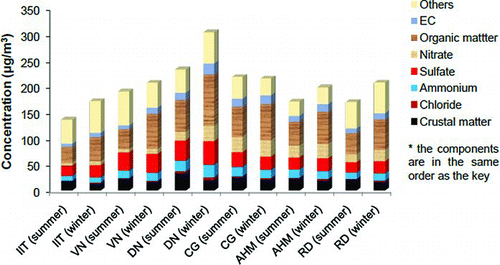
PM2.5 mass concentrations are presented in . The 24-h average concentrations of PM2.5 at six sampling sites varied from 136 to 232 μgm–3 in summer and 172 to 304 μgm–3 in winter. DN was the most polluted site (peak concentration 388 μgm–3 in summer and 477 μgm–3 in winter) followed by CG (peak concentration 322 μgm–3 in summer and 306 μgm–3 in winter). DN was an industrial site with many industries having stack heights below 20 meters and area is characterized by movement of heavy traffic (mostly trucks) engaged in movement of products and raw materials.
TABLE 2 Summary of chemical speciation of PM2.5 (μgm−3) on 24-h average basis*
The combustion related mass in PM2.5, EC, and OC are also quite high in the study (i.e., average of all sites as: 40 μgm–3 of OC and 12 μgm–3 of EC). It is seen that Kanpur is highly polluted in comparison to some Asian cities (). The PM2.5 levels on all days of sampling and at all sites exceed the Indian National Air Quality Standard of 60 μgm–3 (24-h average). The levels are generally higher in winter at all sites except at CG. The possible reason for similar levels at CG in two seasons could be due to the fact that the CG site received direct emissions from vehicles before any dispersion could take place because of low sampling height (2.0 m). It is evident that the city is distressed under high PM2.5 pollution and massive efforts are required to reduce PM2.5 levels. One needs to know the composition of PM2.5 and relative strength of contributing sources to plan any control actions. In addition, understanding the contribution of secondary aerosols, which cannot be estimated by traditional emission inventory or simple model, is important for PM2.5 control.
TABLE 3 Pollution status in some Asian cities
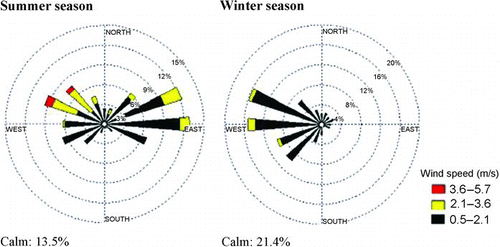
shows wind roses during summer and winter seasons. Prevailing wind direction was WNW-NW and E-ENE for summer and W-WNW for winter. The average ambient temperature was 35.4 ± 5.1°C in summer and 13.4 ± 4.6°C in winter and the corresponding relative humidity was 48.0 ± 18.5% (summer) and 67.2 ± 18.4% (winter). Wind speed was 1.1 ± 0.7 ms–1 in summer and 0.6 ± 0.5 ms–1 in winter. Mixing height in summer varies from 500 m during midnight to 3500 m during midday and for winter it varies between 200 m (midnight) and 1600 m (midday). The atmospheric stability remains unstable, moderately unstable and slightly unstable during daytime and neutral, slightly stable and stable during nighttime for summer. Winter atmosphere is more stable than the summer one (CitationSantosh 2001). Meteorology suggests that winter season could be critical from dispersion point of view.
FIG. 2 Seasonal wind roses during the monitoring program 2007–08.
3.2. Chemical Compositions of PM2.5 and Secondary Aerosol
The levels of water-soluble ions, elements, and OC–EC are presented in . Generally for all species, levels in winter were higher than those in summer in a statistical sense (at 5% level), except for elements of crustal origin (Ca, Mg, Si, Fe, Ti, and Al), which showed higher concentrations in summer. Further, Zn at VN, Cd, Ni at DN, EC, Mn at CG, Pb at AHM, and Cu; Se at RD did not show statistical difference in two seasons. The possible reason for higher levels in winter could be due to poor dispersion and more primary emissions. However, in addition to dispersion, the atmospheric chemistry and physics also play an important role in seasonal variability of pollutant levels and this aspect is examined below.
Based on earlier studies (CitationZhang et al. 2002; CitationHo et al. 2003), we have categorized the elements present in PM2.5 into two major groups: (Equation1) earth crustal elements; Ca, Mg, Na, K, Al, Si, Fe, Ti, Mn and (Equation2) anthropogenic tracer; V, Cr, Ni, Zn, Cd, Pb, Cu. The sum of the mass concentrations of crustal elements was more in summer than in winter (i.e., 16 μgm–3 (summer) and 14 μgm–3 (winter)). The reason for high level of crustal elements in summer could be due to high wind speeds and dry condition that favors re-suspension of road and soil particles (CitationHo et al. 2003). Anthropogenic tracer elements were observed to be higher in winter than in summer (1.87 μgm–3 (summer) and 2.62 μgm–3 (winter)) probably due to more anthropogenic activities and poor atmospheric dispersion.
3.2.1. Secondary Inorganic Aerosol
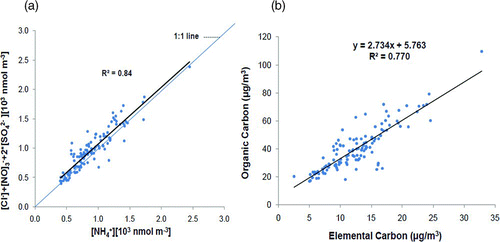
The ionic composition of PM2.5 has also been evaluated with regard to the charge balance between the major anions (Cl–, NO– 3, and SO2– 4) and NH+ 4(). NH+ 4 correlated well with the sum of Cl–, NO– 3, and SO2– 4 (r= 0.91). It can be concluded that for most of the samples, ammonium quantity was generally sufficient to balance the negative charges of nitrate, sulfate and chloride, as most of the data points are above or on 1:1 line in . The ammonium salts, largely of secondary formation, result from the following processes: ammonia is first depleted by reaction with H2SO4 to form (NH4)2SO4 and the remaining free ammonia is depleted by reaction with HNO3 and HCl to form NH4NO3 and NH4Cl, respectively (CitationBaek et al. 2004). The secondary aerosols mass also show seasonal variability. While SO2– 4 formation is more in summer (due to high solar radiation and OH radical), the net SO2– 4 concentration was higher in winter due to poor dispersion. Overall increase in SO2– 4 in winter is 8%. The formation of nitrate is enhanced in the winter. The lower temperatures and higher humidity in winter would shift the gas phase of reaction between NH3 and HNO3 to form particle phase NH4NO3 (CitationSeinfeld and Pandis 1998).
FIG. 3 (a) Charge balance between Cl–, NO– 3, SO2– 4 and NH+ 4; and (b) correlation between OC and EC (all sampling sites).
3.2.2. Secondary Organic Aerosol
The average summer and winter OC concentrations (from six sites) were 32.76 and 47.85 μgm–3, and for EC, the corresponding concentrations were 10.60 and 14.10 μgm–3 (). The high loadings of carbonaceous aerosol could be due to: (Equation1) heavy traffic flow in the city (EC from diesel vehicles and most of the OC from gasoline combustion and fuel evaporation); (Equation2) industrial emission sources; (Equation3) use of coal as domestic cooking fuel by economically poor residents (mostly bituminous and anthracite); and (Equation4) use of ground-based diesel generator sets at the time of power failure in the commercial areas (NILU 2008).
presents the linear regression between EC and OC. It is observed that in both seasons, the OC is about 2.8 times higher than EC at all sites. These EC and OC measurements can assist in identifying the presence of SOA in the study area based on OC/EC ratio from primary sources. If one assumes EC is largely contributed by the urban sources (vehicles and industry), the background OC level of 5.7 μgm–3 (intercept in ) can be taken as an indicative of the OC contribution from primary sources of non-combustion origin (i.e., biogenic, soil and road re-suspended, long-range transport, and evaporation of fuel and solvents).
3.3. Enrichment Factors for Chemical Components of PM2.5
Enrichment factors (EF) can be calculated to show the degree of enrichment of a given element compared to the relative abundance of that element in crustal and sea salt components. These were used as a first step to evaluate the influence of crustal and sea salt sources on the components of PM2.5 (CitationZhang et al. 2002). Aluminum and sodium are reference elements for crustal and sea salt particles in EF calculations (CitationZhang et al. 2002). The non-crustal EFc was estimated using the following expression:
Where, “(Cx/Al)aerosol” represents the concentration ratio of element x (Cx) (in aerosol) to Al in aerosol, and “(Cx/Al)crust” represents corresponding element (Cx) to Al ratio in crustal matter. Similarly, the contribution of non-sea salt (EFs) can be estimated by the following expression:
Where, “(Cx/Na)aerosol” is the element x (Cx) to Na ratio in aerosol, and “(Cx/Na)sea salt” is the concentration ratio of element x (Cx) to Na in sea salt. By convention (CitationZhang et al. 2002), if EF ⩽ 10 it is considered to show that element in aerosols has a significant crust and/or marine contribution, and hence termed as the non-enriched element. The EF > 10 indicates that element has an important proportion of non-crustal and/or non-marine sources and hence termed as the enriched element (CitationZhang et al. 2002). In this study, we have used average seawater and earth crustal compositions as references in calculating EFc and EFs.
Cd and Se exhibit the highest enrichment factor (EFc ∼10000) followed by Zn, Pb, Cu, and As (EFc >100). Na, V, and Cr are moderately enriched (10 < EFc < 100) (). The enrichment of these elements suggests that the dominant sources for these elements were non-crustal and a variety of emission sources might have contributed to their loading in the ambient air. In the study area the enrichment of these elements could be attributed to coal (200 MW power plant and fly ash disposal are in operation since 1970) and other combustion processes. On the contrary, Ca, Mg, K, Si, Fe, Ti, Mn, and Ni have EFc less than 10, which suggests negligible contribution of anthropogenic sources.
FIG. 4 (a) Non-crustal Enrichment factors (EFc), (b) Non-sea salt Enrichment factors (EFs).
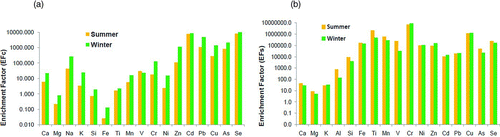
It can be observed from that Cr exhibits the highest enrichment factor (EFs > 106). Fe, Ti, Mn, Zn, Cu, and Se show high EFs (105< EFs < 106) and V, Ni, Cd, Pb, As, and Si show a moderately high EFs (>103). Al, Ca, and K are slightly enriched (EFs > 10). The high enrichment of these elements (Cr, Fe, Ti, Mn, Zn, Cu, Se, V, Ni, Cd, Pb, As, Si, Al, Ca, and K) suggests that the dominant sources for these elements were non-marine. It appears that marine source had no influence on the chemical components of the fine aerosols in the lower atmosphere of Kanpur city which is more than 1200 km from the nearest sea coast.
3.4. Reconstruction of Chemical Components in PM2.5
Components of PM2.5 were classified into seven major types including: crustal matter, chloride, ammonium, sulfate, nitrate, organic matter, and EC. The generic “crustal matter” component of PM2.5 is estimated from concentrations of oxides of Al, Si, Ca, Fe, K, and Ti (CitationEldred et al. 1987; CitationMarcazzan et al. 2001):
Where Fe and K indicate the part of iron and potassium concentration considered of natural origin. The factor 1.15 was used for compensating Na and Mg oxides (CitationEldred et al. 1987). The chemical composition of the organic fraction of PM2.5 is largely unknown. A conversion factor between 1.2 and 1.4 is generally used for converting OC to organic mass (OM) to account for hydrogen, oxygen, and nitrogen present in OM (CitationTurpin and Lim 2001; CitationHo et al. 2003). In absence of any reported value for Indian cities to estimate OM, OC concentrations were multiplied by a factor of 1.4 based on a study in an Asian city (CitationHo et al. 2003).
The reconstructed chemical compositions (based on the average concentration) of PM2.5 mass for summer and winter seasons are presented in . The sum of seven major PM2.5 components accounts for 64–85% mass of PM2.5. Overall, the major components are organic matter (>20% mass) and sulfate (>11% mass). Unidentified mass estimated from mass closure analysis could be oxides of unaccounted elements and water. The sequence for percentage of contribution of these components to PM2.5 mass is: OM > sulfate > crustal matter > nitrate > ammonium > EC > chloride. It is noteworthy that OM followed by sulfate is a matter of concern and these need further break-up analysis in terms of primary and secondary components.
FIG. 5 Mass reconstruction of chemical composition in PM2.5 in Kanpur.
3.5. Primary and Secondary Components of PM2.5
EC is normally emitted from combustion sources and its chemical transformations are limited. OC can be emitted from primary emission sources (combustion and evaporation of fuel and solvents) and can be formed through chemical reactions among primary gaseous OC in the atmosphere (CitationTurpin et al. 1991; CitationHo et al. 2003). The lack of direct chemical analysis method for the determination of either primary or secondary organic aerosol led to development of different indirect approaches including EC tracer method. The EC tracer method is widely used for its simplicity and it does not require any information on composition of OM (CitationTurpin et al. 1991; CitationStrader et al. 1999; CitationYuan et al. 2005). The underlying hypothesis is, in a particular area, if SOA is not formed, the representative ratio of OC (primary) to EC emissions should remain unchanged in ambient PM2.5. If the measured ratio of OC to EC (in ambient air) exceeds the representative ratio of primary OC/EC, then the additional OC is considered to be of secondary origin. Primary ratios of OC to EC of emitting sources present in the study area can be derived from the emissions inventory of PM2.5, EC, and OC (CitationCabada et al. 2002, Citation2004). presents the average ambient air OC to EC ratio in PM2.5 (i.e., 2.73 to 4.33) and the OC to EC ratio in the primary sources near the sampling sites of this study. The higher ratio in ambient air than at sources suggests the presence of SOA.
TABLE 4 Ratios between OC and EC
Primary OC concentration can be expressed as the sum of OC from non combustion sources and OC from combustion sources:
-
a represents OC from non combustion sources (i.e., biogenic, soil and road re-suspended, long-range transport, etc.); and
-
bECmeasured represents OC from combustion sources (e.g., traffic, heating).
OC from combustion sources can be estimated from measured EC concentrations (in ambient air) and by assuming that at each combustion source, the emission ratio of primary OC and primary EC is relatively constant over space and time, implying OCcombustion= ((OC/EC)pri) × ECmeasured (CitationSaylor et al. 2006). In other words, b is the ratio of OC to EC at an emitting source (i.e., (OC/EC)pri). The value of a can be estimated as the intercept of a straight line fit between OCmeasured (y-axis) and ECmeasured(x-axis) (e.g., ).
OC from secondary formation (OCsec) in carbon mass can be estimated as the difference between the measured OC (OCmeasured) and primary OC (OCpri) calculated from Equation (Equation4):
The SOA mass can be estimated from OCsec as:
A conversion factor of 1.4 is used to compensate for the mass of hydrogen, oxygen, and nitrogen present in OCsec (for details see section 3.4. We have estimated the intercept a in Equation (Equation4) (as background concentration of OC) from the best fit linear plots (e.g., intercept in ) between OC and EC for summer and winter seasons ().
TABLE 5 OC-EC regression parameters for the sampling period
These value of (OC/EC)pri was taken from primary emission inventory completed under a joint program between Norwegian Institute for Air Research (NILU) and IIT-Kanpur (NILU 2008). In this study, primary sources were identified through a door-to-door survey in each of the seven grids (2 km × 2 km), surrounding the sampling site. The sources considered for emission inventory were: domestic fuel, area and point source industries, transport sector, garbage burning, agricultural waste burning, hotel and restaurants, DG sets, and medical waste incinerators. Emission factors of PM2.5, OC, and EC suitable for our primary source activities were taken from literatures (CitationTurpin and Huntzicker 1995; CitationHildemann et al. 1991; CitationCabada et al. 2002). Vehicles running on Indian roads in year 2007 are equivalent to EURO-III and industrial processes are same as reported in USEPA AP42. Emissions from garbage burning (a specific activity in Kanpur) could be at variance compared to international literature; the contribution of this source is only about 5%. Therefore, emission factors from international literature have been used to estimate EC and OC emissions.
presents the overall emission inventory for the city from different emission sectors. Estimated contributions of major sources to primary OC were: industries (38%), domestic fuel (31%), and transport (16%). Similarly estimated EC emissions were: industries (37%), transport (28%), and domestic fuel (22%).
TABLE 6 Overall emission inventory for the study area (kg/day)
The overall (OC/EC)pri ratio of 1.5 was utilized in Equation (Equation4) to calculate OCpri for both summer and winter seasons. SOA was first estimated site-wise for the two seasons. The average of SOA at all sites is presented in . A near uniform distribution of SOA concentration was observed; variation in SOA concentration was less than 20% from one site to another.
FIG. 6 Relative composition of sources in PM2.5 for summer and winter.
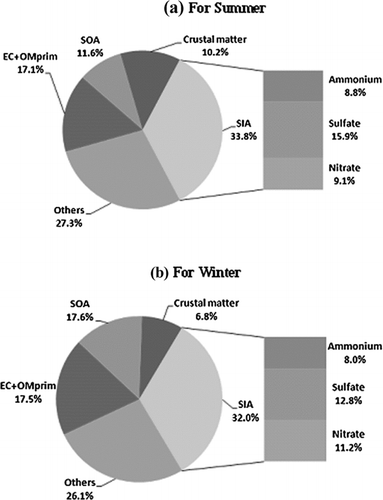
shows the relative composition of PM2.5 for summer and winter based on averages of six sampling sites. The components of PM2.5 are: (Equation1) EC and OMpri (derived from OCpri), (Equation2) SOA, (Equation3) SIA, (NH+ 4, SO2– 4 and NO– 3), and (Equation4) crustal matter. It has been estimated that for PM2.5 composition, SIA was the predominant contributing component (32–40%) followed by EC and OMpri (17–18%) and SOA (12–18%). The contributions of sulfate, nitrate and ammonium in PM2.5 are also presented in . Higher percent contribution of sulfate in PM2.5 for summer can be attributed to high temperature, high solar radiation and presence of significant amount of OH radical that readily converts SO2 into sulfate (CitationSharma et al. 2007). Estimated ammonium contribution was more in summer which could be due to large evaporative losses of ammonia from several sources (e.g., open sewerage system, live stocks) (CitationHoek et al. 1996). Nitrate contribution to PM2.5 was more in winter than in the summer. This might be due to the meteorological conditions that favor particulate nitrate formation due to low temperature and high relative humidity in winter (CitationSeinfeld and Pandis 1998). The crustal matter contribution to PM2.5 was higher in summer (10%) than in winter (7%), primarily due to the fact that high wind speeds in summer can make the loose and dry soil airborne (CitationHo et al. 2003).
The contribution of primary carbon (EC + OCpri) to PM2.5 is slightly higher in winter (18%) than in summer (17%). This could be due to poor dispersion in and more biomass burning in winter. However, contribution of SOA to PM2.5 is much higher in winter (18%) than in summer (12%). The reasons for higher SOA in winter could be due to enhanced condensation (due to low temperature) of organic vapors on particles, which are present in abundance and other processes like oxidation, coagulation and aging. These processes enhance conversion of primary organic vapors to fine aerosols (Ho et al. 2006). It can, therefore, be concluded that PM2.5 has significant contribution from secondary formations, both for inorganic and organic components.
4. Conclusions
Based on PM2.5 bulk mass reconstruction exercise, it has been estimated that crustal matter, chloride, ammonium, sulfate, nitrate, organic matter and EC accounted for 64–85% of the PM2.5 mass in Kanpur city (Ganga basin, India). Overall, the major PM2.5 components in Kanpur were organic matter (>20%) and sulfate (>11%). The sequence for percentage of contribution of these components to PM2.5 mass was: organic matter > sulfate > crustal matter > nitrate > ammonium > EC > chloride.
In summer, secondary species (SIA and SOA) accounted for about 47% of the PM2.5 bulk mass, with SIA contribution of 33%. Overall primary components accounted for about 26% of mass, including primary contribution of carbons (EC + OM) at 17%. For winter, secondary species (SIA and SOA) account for about 50% of the PM2.5 with SIA contribution of 32%. Primary components account for about 25%, with primary contribution of carbons (EC+OM) at 18% in winter. The significant contribution of the secondary component to PM2.5 mass suggests that control actions must be aimed not only to control the primary PM emissions but also the precursor gases, (NO x , SO2, NH3, and VOCs), which all play an important role in the secondary aerosol formation.
We are thankful to anonymous reviewers for their critical comments which have substantially improved the quality of this article.
Related Research Data
REFERENCES
- Baek , B. H. , Aneja , V. P. and Tong , Q. 2004 . Chemical Coupling Between Ammonia, Acid Gases, and Fine Particles . Environ. Pollut. , 129 : 89 – 98 .
- Cabada , J. C. , Pandis , S. N. and Robinson , A. L. 2002 . Sources of Atmospheric Carbonaceous Particulate Matter in Pittsburgh, Pennsylvania . J. Air and Waste Manag. Assoc. , 52 : 732 – 41 .
- Cabada , J. C. , Pandis , S. N. , Subramanian , R. , Robinson , A. L. , Polidori , A. and Turpin , B. 2004 . Estimating the Secondary Organic Aerosol Contribution to PM2.5 Using the EC Tracer Method. . Aerosol Sci. Technol. , 38 ( S1 ) : 140 – 155 .
- Chow , J. C. , Watson , J. G. , Lowenthal , D. H. , Solomon , P. A. , Magliano , K. L. , Ziman , S. D. and Richards , L. W. 1993 . PM10 and PM2.5 Compositions in California's San Joaquin Valley . Aerosol Sci. Technol. , 18 : 105 – 128 .
- Dubey , N. and Pervez , S. 2008 . Investigation of Variation in Ambient PM10 Levels within an Urban Industrial Environment . Aerosol and Air Qual. Res. , 8 : 54 – 64 .
- Eldred , R. A. , Cahill , T. A. and Feeney , P. J. 1987 . Particulate Monitoring at US National Parks Using PIXE . Nuclear Instruments and Methods in Physics Research , B22 : 289 – 295 .
- Heisler , S. L. and Friedlander , S. K. 1977 . Gas-to-Particle Conversion in Photochemical Smog: Aerosol Growth Laws and Mechanisms for Organics . Atmos. Environ. , 11 : 157 – 168 .
- Heo , J.-B. , Hopke , P. K. and Yi , S.-M. 2009 . Source Apportionment of PM2.5 in Seoul, Korea . Atmos. Chem. Phys. , 9 : 4957 – 4971 .
- Hildemann , L. M. , Cass , G. R. , Mazurek , M. A. and Simoneit , B. R. T. 1993 . Mathematical Modeling of Urban Organic Aerosol: Properties Measured by High–Resolution Gas Chromatography. . Environ. Sci. Technol. , 27 : 2045 – 2055 .
- Hildemann , L. M. , Markowski , G. R. and Cass , G. R. 1991 . Chemical Composition of Emissions from Urban Sources of Organic Aerosol . Environ. Sci. Technol. , 25 : 744 – 759 .
- Ho , K. F. , Lee , S. C. , Chan , C. K. , Yu , J. C. , Chow , J. C. and Yao , X. H. 2003 . Characterization of Chemical Species in PM2.5 and PM10 Aerosols in Hong Kong . Atmos. Environ. , 37 : 31 – 39 .
- Hoek , G. , Mennen , M. G. , Allen , G. A. , Hofschreuder , P. and Meulen , T. V. D. 1996 . Concentrations of Acidic Air Pollutants in the Netherlands . Atmos. Environ. , 30 : 3141 – 3150 .
- Karar , K. and Gupta , A. K. 2006 . Seasonal Variations and Chemical Characterization of Ambient PM10 at Residential and Industrial Sites of an Urban Region of Kolkata (Calcutta), India . Atmos. Res. , 81 : 36 – 53 .
- Kim Oanh , N. T. , Thiansathit , W. , Bond , T. C. , Subramanian , R. , Winijkul , E. and Paw-armart , I. 2010 . Compositional Characterization of PM2.5 Emitted from in-Use Diesel Vehicles . Atmos. Environ. , 44 : 15 – 22 .
- Kim Oanh , N. T. , Upadhyay , N. , Zhuang , Y.-H. , Hao , Z.-P. , Murthy , D. V. S. , Lestari , P. , Villarin , J. T. , Chengchua , K. , Co , H. X. , Dung , N. T. and Lindgren , E. S. 2006 . Particulate Air Pollution in Six Asian Cities: Spatial and Temporal Distributions, and Associated Sources. . Atmos. Environ. , 40 : 3367 – 3380 .
- Lee , H. S. and Kang , B.-W. 2001 . Chemical Characteristics of Principal PM2.5 Species in Chongju, South Korea . Atmos. Environ. , 35 : 739 – 746 .
- Lee , S. , Jang , M. and Kamens , R. M. 2004 . SOA Formation From the Photo Oxidation of α–Pinene in the Presence of Freshly Emitted Diesel Soot Exhaust . Atmos. Environ. , 38 : 2597 – 2605 .
- Levy , J. I. , Hammittf , J. K. and Spengler , J. D. 2000 . Estimating the Mortality Impacts of Particulate Matter: What Can be Learned from Between-Study Variability . Environ. Health Perspect. , 108 : 109 – 117 .
- Marcazzan , G. M. , Vaccaro , S. , Valli , G. and Vecchi , R. 2001 . Characterization of PM10 and PM2.5 Particulate Matter in the Ambient Air of Milan, Italy . Atmos. Environ. , 35 : 4639 – 4650 .
- Na , K. , Sawant , A. A. , Song , C. and Cocker , D. R. III . 2004 . Primary and Secondary Carbonaceous Species in the Atmosphere of Western Riverside County, California . Atmos. Environ. , 38 : 1345 – 1355 .
- Norwegian Institute for Air Research (NILU) . 2008 . Environmental Health Assessment: Respiratory Disease in relation to Air Pollution in Kanpur, Uttar Pradesh, Final Report. Project no. O–106082, ref.nr. IND3025 05/51.
- Plaza , J. , Go’mez-Moreno , F. J. , Núñez , L. , Pujadas , M. and Artíñano , B. 2006 . Estimation of Secondary Organic Aerosol Formation from Semi-Continuous OC–EC Measurements in a Madrid Suburban Area . Atmos. Environ. , 40 : 1134 – 1147 .
- Russell , M. , Allen , D. , Collins , D. and Fraser , M. 2004 . Daily, Seasonal, and Spatial Trends in PM2.5 Mass and Composition in Southeast Texas. . Aerosol Sci. Technol. , 38 ( S1 ) : 14 – 26 .
- Santosh , B. C. J. 2001 . “ A Methodology to Determine Minimum Stack Height for Thermal Power Plants Based on Dispersion Capacity ” . M. Tech. Thesis, Indian Institute of Technology Kanpur .
- Saylor , R. D. , Edgerton , E. S. and Hartsell , B. E. 2006 . Linear Regression Techniques for Use in the EC Tracer Method of Secondary Organic Aerosol Estimation . Atmos. Environ. , 40 : 7546 – 7556 .
- Seinfeld , J. H. and Pandis , S. N. 1998 . Atmospheric Chemistry and Physics , New York : Wiley Interscience .
- Sharma , M. , Kishore , S. , Tripathi , S. N. and Behera , S. N. 2007 . Role of Atmospheric Ammonia in the Formation of Inorganic Secondary Particulate Matter: A Study at Kanpur, India . J. Atmos. Chem. , 58 : 1 – 17 .
- Sharma , M. and Maloo , S. 2005 . Assessment and Characterization of Ambient Air PM10 and PM2.5 in the City of Kanpur, India . Atmos. Environ. , 39 : 6015 – 6026 .
- Shukla , S. P. and Sharma , M. 2008 . Source Appointment of Atmospheric PM10 in Kanpur, India . Environ. Eng. Sci. , 25 : 849 – 862 .
- Singh , R. P. , Sagnik , D. , Tripathi , S. N. and Tare , V. 2004 . Variability of Aerosol Parameters Over Kanpur, Northern India. . J. Geophys. Res. , 109 : D23206 doi: 10.1029/2004JD004966
- Strader , R. , Lurmann , F. and Pandis , S. N. 1999 . Evaluation of Secondary Organic Aerosol Formation in Winter. . Atmos. Environ. , 33 : 4849 – 4863 .
- Turpin , B. J. , Cary , R. A. and Huntzicker , J. J. 1990 . An InSitu, Time–Resolved Analyzed for Aerosol Organic and Elemental Carbon . Aerosol Sci. Technol. , 12 : 161 – 171 .
- Turpin , B. J. and Huntzicker , J. J. 1995 . Identification of Secondary Organic Aerosol Episodes and Quantitation of Primary and Secondary Organic Aerosol Concentrations During SCAQS . Atmos. Environ. , 29 : 3527 – 3544 .
- Turpin , B. J. , Huntzicker , J. J. , Larsod , S. M. and Cass , G. R. 1991 . Los Angeles Summer Midday Particulate Carbon: Primary and Secondary Aerosol . Environ. Sci. Technol. , 25 : 1788 – 1793 .
- Turpin , B. J. and Lim , H.-J. 2001 . Species Contributions to PM2.5 Mass Concentrations: Revisiting Common Assumptions for Estimating Organic Mass . Aerosol Sci. Technol. , 35 : 602 – 610 .
- US Environmental Protection Agency (US EPA) . 1998 . “ Quality Assurance Guidance Document 2.12. Monitoring PM2.5 in ambient air using designated reference or class I equivalent methods. US Environmental Protection Agency (US EPA) (1999) ” . Compendium of methods for the determination of inorganic compounds in ambient air, Compendium method IO-4.2: Determination of reactive acidic and basic gases and strong acidity of atmospheric fine particles (<2.5 μm). EPA/625/R–96/010a.
- US Environmental Protection Agency (US EPA) . 1999 . Compendium of Methods for the Determination of Inorganic Compounds in Ambient Air, Compendium Method IO-3.1: Selection, Preparation and Extraction of Filter Material. EPA/625/R–96/010a.
- Vallius , M. J. , Ruuskanen , J. , Mirme , A. and Pekkanen , J. 2000 . Concentrations and Estimated Soot Content of PM1, PM2.5, and PM10 in a Subarctic Urban Atmosphere. . Environ. Sci. Technol. , 34 : 1919 – 1925 .
- Yuan , Z. B. , Yu , J. Z. , Lau , A. K. H. , Louie , P. K. K. and Fung , J. C. H. 2005 . Application of Positive Matrix Factorization in Estimating Aerosol Secondary Organic Carbon in Hong Kong and Insights into the Formation Mechanisms. . Atmos. Chem. Phys. , Discussions, 5 : 5299 – 5324 .
- Zhang , J. , Wu , Y. , Liu , C. L. , Shen , Z. B. and Zhang , Y. 2002 . Major Components of Aerosols in North China: Desert Region and the Yellow Sea in the Spring and Summer of 1995 and 1996. . J. Atmos. Sci. , 59 : 1515 – 1532 .