Abstract
The quantity, extraction efficiency, and molecular composition of non-volatile oligomeric species in SOA generated by the reaction of α-pinene with ozone were studied. Two different methods of determining the total particulate mass in the reaction chamber were compared and found to be in good agreement when changes in the partitioning of semi-volatile compounds to the particle phase during measurement were properly handled. Almost all of the non-volatile organic carbon formed by the reaction was collected and recovered by extraction with organic solvents; recoveries with water extraction were somewhat lower. The identities of compounds extracted by the various solvents were determined using electrospray ionization Fourier transform ion cyclotron resonance (ESI-FTICR) mass spectrometry. Over 80% of the peaks weighted by mass and intensity were the same in the spectra of samples obtained from different extraction solvents. Standard addition plots were used to determine the amounts of two commercially available monomer compounds in the SOA extracts. When the response factors for those compounds were applied to other monomers detected in the mass spectra, the weight percent of monomers was estimated to be slightly less than 50%, with the remaining mass (over 50%) assigned to oligomers. The oligomer content is sufficiently large that it should be taken into account when modeling the formation and properties of laboratory SOA.
INTRODUCTION
Fine particles in ambient air influence human health and global climate. A substantial portion of this matter is organic in character, much of which has been produced by secondary processes in the atmosphere (CitationTurpin, Saxena, and Andrews 2000). Secondary organic aerosol (SOA) is formed when volatile organic compounds (VOC) react with an oxidizing agent such as OH, O3, and NO3 resulting in semi or non-volatile compounds (SVOC, NVOC) that migrate to the particle-phase (CitationKroll and Seinfeld 2008; CitationPankow 2003). The largest class of SOA precursors is biogenic (CitationHallquist et al. 2009), of which α-pinene is of great importance due to a high level of emission and the relatively low volatilities of its oxidation products. Ozone reacts with α-pinene by attacking the double bond forming an energized Criegee intermediate that can follow two possible pathways: quenching to form a stabilized Criegee intermediate or rearrangement to form a hydroperoxide. The stabilized intermediates from either pathway can form end products through degradation, isomerization, and reaction with other atmospheric species such as water (CitationJohnson and Marston 2008; CitationKroll and Seinfeld 2008). These compounds form the bulk of the products of this reaction and have been characterized by gas chromatography mass spectrometry (GCMS) (CitationYu et al. 1999). However, while they have much lower vapor pressures than α-pinene, even the lowest volatility products (e.g., pinic acid) would exist primarily in the gas-phase under typical ambient conditions. The nonvolatile component of α-pinene SOA is not as well characterized, but to be non-volatile it must be polar (highly oxidized) and/or possess a high molecular weight (oligomerization) (CitationHallquist et al. 2009; CitationPankow 2003).
Because of its molecular complexity, the most common way to study NVOC is to collect particles on a filter, extract organic matter off the filter with a solvent, and characterize the molecular components with a method such as liquid chromatography mass spectrometry (LCMS) (CitationClaeys et al. 2009; CitationHallquist et al. 2009; CitationSamburova et al. 2005), tandem mass spectrometry (MSMS) (CitationTolocka et al. 2004), and/or high accuracy-resolution mass analysis with a time-of-flight, orbitrap, or Fourier transform ion cyclotron resonance instrument (CitationBateman et al. 2008; CitationHeaton et al. 2009; CitationKalberer, Sax, and Samburova 2006; CitationReinhardt et al. 2007; CitationSleighter and Hatcher 2007; CitationWalser et al. 2008; CitationWozniak et al. 2008; CitationShilling et al. 2009). When applied to laboratory-generated SOA produced by oxidation of a biogenic precursor such as α-pinene, more than a thousand unique molecular formulas are typically resolved (CitationBateman et al. 2008; CitationHeaton et al. 2009; CitationReinhardt et al. 2007; CitationTolocka et al. 2004; CitationWalser et al. 2008). The great majority of these compounds have high molecular weights that require formation of dimers and/or higher order oligomers from the initial oxidation products (monomers). Oligomers have been detected in numerous studies (CitationBateman et al. 2008; CitationHeaton et al. 2009; CitationReinhardt et al. 2007; CitationTolocka et al. 2004; CitationWalser et al. 2008), but it is still not clear how large a fraction they represent of the collected SOA. While answering this question may seem straight forward, many factors must be considered, especially with regard to the particle collection and extraction steps. What happens to SVOC during particle collection? Is all of the collected SOA dissolved by the extraction solvent? Is oligomer formation influenced by the extraction solvent, either by selective dissolution of a subset of the compounds present or by inducing oligomer formation in solution?
In the work presented here, SOA is formed by reaction of α-pinene with ozone in a 500 L bag reactor. Direct measures of aerosol mass are compared to the particulate mass that was collected on a filter and extracted into a solvent. Electrospray ionization Fourier transform ion cyclotron resonance (ESI-FTICR) mass spectrometry is used to characterize oligomers isolated from different extraction protocols. Standard addition plots are then used to estimate monomer content of extracted SOA, and by difference the oligomer content.
EXPERIMENTAL
SOA was generated by mixing α-pinene vapor and ozone in a 500 L Tedlar bag. First, the flow from an ozone generator (Prozone, Huntsville, AL, Model PZ5) was directed into the bag (∼1 min flow for 150–200 ppbv ozone). The bag was then filled with clean, dry air to a volume of 500 L (36.4 L/min for 13.75 min), and the ozone concentration was measured with an ozone analyzer (Thermo Scientific, Waltham, MA, Model 49C). α-Pinene vapor was added, in excess to ozone, to the bag by redirecting the air flow into the bag through an impinger spiked with 20 μL of α-pinene liquid at a flow rate of 3.3 L/min for 5 min. The reaction was allowed to proceed for 45 min following the beginning of the α-pinene addition. A scanning mobility particle sizer (SMPS; TSI, St. Paul, MN, Model 3080/3025A) was used to measure the mass concentration and size distribution of SOA in the bag every 5 min during the reaction. At the end of the reaction time, the bag was evacuated with a vacuum pump (Gast, Benton Harbor, MI, model 1HAB-25-M100X) at a flow rate ∼26 L/min, with the aerosol collected on an inline filter (47 mm Fiberfilm, Pall Life Sciences, Port Washington, NY).
A semi-continuous carbon aerosol analyzer (OCEC; Sunset Laboratory, Tigard, OR, Model 3) was used in some experiments as an alternative method to determine the aerosol mass concentration. In these experiments, SOA was prepared as above, with the reaction allowed to proceed for 30 min before taking any measurements. After this time period, aerosol flow from the bag passed through a charcoal denuder to a three-way valve which could direct the flow to either the SMPS or OCEC instrument. Aerosol mass concentration measurements were made sequentially with SMPS and OCEC by switching the valve back and forth. For some OCEC measurements, a flow-tube reactor (FTR), described elsewhere (CitationHeaton et al. 2007; CitationHeaton et al. 2009; CitationTolocka et al. 2006), was placed after the three-way valve to simulate the sample stream dilution in the SMPS (0.3 LPM sample flow and 1.0 LPM sheath flow). Five L/min of clean, dry air was introduced to the FTR as a sheath (dilution) flow where it mixed with 1.5 L/min of aerosol flow from the bag.
To compare the effects of extraction solvent, the filter used to collect particles was divided in to either halves or quarters and the sections were then extracted with one of five solvents or solvent combinations: methanol, acetonitrile, water, and 50:50 water:acetonitrile or methanol (0.46 mL solvent per cm2 of filter, or 2 mL per quarter filter) by sonicating the filter in solvent for 30 min. A Savant speed concentrator (Thermo Scientific, Waltham, MA, Model SC110A) was used to take each sample to near dryness (<0.1 mL) and the sample was reconstituted with acetonitrile to a final volume of 0.5 mL. The acetonitrile solution was then centrifuged to remove any solid material. Over the course of these experiments, 24 filter samples were collected, divided in half and extracted in one of the following combinations: acetonitrile/methanol (8 filters), acetonitrile/water (4 filters), methanol/water (4 filters), 50% acetonitrile/50% methanol (4 filters), acetonitrile/50% acetonitrile (2 filters), and methanol/50% methanol (2 filters). Two additional filters were divided into quarters with one extracted by acetonitrile/water/methanol/water (a different solvent for each quarter), and the other extracted by acetonitrile/50% acetonitrile/methanol/50% methanol. A total of 14 acetonitrile, 14 water, 15 methanol, and 7 each 50% acetonitrile and 50% methanol extracts were prepared. Several of the filters were later re-extracted with either acetonitrile or methanol.
The nonvolatile mass in the filter extracts was measured with an evaporative light scattering detector (ELSD, Shimazdu, Columbia, MD, Model LT-II). In the ELSD, effluent from a liquid chromatograph is atomized into an air flow, the airborne droplets pass through a heated tube to evaporate solvent and other volatile compounds, and the fine particle residue is detected by light scattering. The advantage of an ELSD is that it provides a response based solely on the nonvolatile mass of a sample independent of molecular composition. The sample injection loop and pumps from a liquid chromatograph (Shimazdu, Columbia, MD, Model LC-20AD) were used to deliver a 20 μL aliquot of sample to the ELSD at a flow rate of 1 mL/min with methanol as the mobile phase. In some experiments, 30 μL/min of the mobile phase flow was split to an ESI-QTOF mass spectrometer (Micromass, Cary, NC, Model: Ultima).
The extracts from filters divided into quarters were also analyzed with a 7T electrospray ionization Fourier transform ion cyclotron resonance (ESI-FTICR) mass spectrometer (Bruker, Billerica, MA, Model: Apex-Qe) for accurate and precise mass measurements. The extracts were analyzed in both positive and negative mode, with an aliquot of the sample spiked with a 4 μM solution of ammonium hydroxide (spike volume 25% of the aliquot volume) prior to analysis in the negative ion mode. Only peaks with a signal to noise ratio greater than 5 and a relative intensity greater than 1% were characterized. Peak assignments were restricted to CHO compounds, as (M+H)+ or (M+Na)+ ions in positive mode, and (M-H)− ions in negative mode, with an O:C ratio equal to or less than 1.00, an H:C ratio greater than 0.4 and less than 2N+2 hydrogen atoms per carbon atom (N = number of carbon atoms), and an error of less than 5 ppm ((assigned mass – measured mass)/measured mass).
RESULTS AND DISCUSSION
The goal of this work was to estimate the oligomer concentration in α-pinene SOA that was prepared in a bag reactor, collected on a filter, and extracted into an aliquot of solvent. To determine how relevant the measurement was to SOA within the bag reactor, several questions had to be answered: Does the particulate matter collected on the filter include SVOC? Is all of the particulate matter on the filter extracted into solution? Does the extraction solution modify the oligomer content? The first question was addressed by determining the total suspended particle mass (TSP) within the reactor in two ways: scanning mobility particle sizer (SMPS), and organic carbon elemental carbon analyzer (OCEC). The second question was addressed by comparing the TSPs determined with SMPS and OCEC to the particle mass collected on a filter, extracted with a solvent and analyzed with an evaporative light scattering detector (ESLD). The third question was addressed by high accuracy and resolution mass analysis with ESI-FTICR. The monomer content of the filter extract was estimated by standard addition in ESI-QToF analysis, with the oligomer content inferred by difference.
TSP Determination
Both methods used to determine TSP required assumptions about the particles being measured. The SMPS values assumed a spherical shape and a density of 1.2 g/cm3 based on the work of CitationZelenyuk et al. (2008). The particle size distributions obtained by SMPS showed hardly any particle mass near the upper mobility diameter limit of the instrument (1 μm), suggesting that the entire distribution was contained within the SMPS size range. Therefore, the integrated mass concentration obtained by SMPS was assumed to be equal to TSP. The OCEC values assumed a ratio of the mass of organic matter (OM) to organic carbon (OC) of 1.5 based on the molecular formula assignments from several FTICR measurements described below. This value of the OM/OC ratio is equivalent to the ratio measured by CitationShilling et al. (Shilling et al. 2009) for α-pinene SOA at a similar aerosol mass loading. TSP was determined by dividing OM by the volume of air sampled into the instrument.
When the two methods were used to simultaneously analyze the same SOA sample, OCEC was found consistently to measure a higher TSP than SMPS unless specific sampling conditions were employed. The difference between the two measurements was apparently related to how each method handled semivolatile organic carbon (SVOC). Partitioning of SVOC between the gas and particle phases depends on TSP, which was several hundred μg/m3 in these experiments (). With respect to SVOC, OCEC is subject to a positive artifact in the particulate mass measurement owing to adsorption of gas phase species onto the filter medium and a negative artifact owing to evaporation of semivolatile compounds from the filter prior to analysis. SMPS is subject to a negative artifact owing to dilution of the sampled aerosol with sheath gas—according to partitioning theory, SVOC concentrations are shifted toward the gas phase upon dilution.
TABLE 1 OCEC and SMPS measurements taken from the same SOA sample
summarizes OCEC and SMPS TSP measurements for 8 measurements over 3 experiments and 2 sampling conditions. When aerosol was drawn directly into both instruments, the ratio of OCEC TSP to SMPS TSP was 1.15 ± 0.02 (n = 3). When a denuder was inserted between the aerosol reactor and the inlets to the two instruments and the aerosol flow to the OCEC instrument was diluted by a factor of 3.3 to mimic the effect of sheath flow dilution in the SMPS, the aerosol yield expressed in μg SOA measured/μmol O3 reacted decreased for both methods. The ratio of OCEC TSP to SMPS TSP also decreased to 0.81 ± 0.24 (n = 5). These changes are consistent with loss of SVOC to the gas phase, leaving primarily nonvolatile organic carbon (NVOC). Because of ease of use, TSP was measured by SMPS in the experiments described below. suggests that, if anything, SMPS analysis may have overestimated NVOC relative to OCEC by as much as 20%. The aerosol yield measured with SMPS was 61 ± 5 μg SOA/μmol O3 (n = 33); note that the yields listed in represent only a subset of the 33 total measurements.
Extraction Efficiency
Quantitative characterization of SOA extracted off a filter requires a high extraction efficiency. Extraction efficiencies were investigated with an ELSD to measure the nonvolatile mass in the extracts. The ELSD mass was then compared to the SMPS “mass” (SMPS mass concentration times the volume of air sampled through the filter). A concern with this approach is the response of ELSD to semivolatile compounds. If particulate phase material evaporates when heated in the ELSD, then the measured mass will be lower than the true mass. To explore this possibility, the ELSD response was studied for several test compounds. shows a plot of ELSD recovery (measured mass divided by injected mass) vs. vapor pressure for these compounds. The ELSD signal was found to decrease substantially, almost a step function, when the vapor pressure exceeded about 1 × 10−4 torr. The potential impact this might have on the detection of known monomer products of α-pinene ozonolysis is illustrated in , where the fraction of monomer mass in the particle phase is plotted vs. monomer vapor pressure assuming a total aerosol mass loading of 440 μg/m3. Monomers having a vapor pressure above 1 × 10−4 torr would be expected to partition slightly to the particle phase at this mass loading, but would not be detected by ELSD. Therefore, ELSD may not detect some SVOC in particulate matter.
FIG. 1 (A) ELSD recovery (measured mass/injected mass) of several test compounds (aspartame, diacetyl benzene, and terephthalic, pinic, pinonic, suberic, and succinic acids) vs. vapor pressure. (B) Fraction in the particle phase vs. vapor pressure for several known monomers of α-pinene SOA at an aerosol mass concentration of 440 μg/m3. In both figures, dashed lines denote 1.5 × 10−4 torr. Vapor pressures were calculated with EPI Suite (EPA, www.epa.gov/oppt/exposure/pubs/episuite.htm).
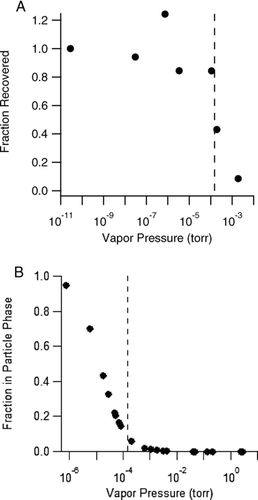
Extraction efficiencies for several solvents and solvent combinations are given in . The recoveries for extraction by either acetonitrile or methanol were on the order of 80 ± 20%. If the above conclusion is true that SMPS is more likely to detect residual SVOC than OCEC with denuder and dilution or ELSD, then the true recoveries for these solvents are likely to be closer 100%. The conclusion of a near 100% extraction efficiency was supported by two additional experiments. First, when a filter previously extracted with acetonitrile or methanol was re-extracted with the solvent indicated in , no additional nonvolatile mass was detected, suggesting that all SOA was removed by the first extraction. Second, when a blank filter was spiked with a known amount of SOA (from a previous extract whose nonvolatile mass was determined by ELSD), the amount of SOA recovered by subsequent extraction was 104 ± 6% (n = 4), showing that no SOA was lost by the experimental procedure.
TABLE 2 Mass recoveries a from filters extracted and reextracted by different solvents
Similar experiments were performed by extraction with a 50/50 mixture of either acetonitrile or methanol with water. Although the 50/50 solvents appeared to give slightly lower recoveries than the pure organic solvents, the uncertainties were large relative to the difference. Furthermore, when filters were re-extracted with pure organic solvent after original extraction with a 50/50 mixture, no additional nonvolatile mass was detected (). This result is consistent with near 100% extraction efficiency of the original 50/50 solvent mixture. The only solvent that may have given a lower extraction efficiency is pure water. When filters originally extracted with water were re-extracted with pure organic solvent, some residual nonvolatile mass was detected. However, the standard deviations for the water extraction experiments were large, making definitive conclusions difficult. Water did not wet the Teflon coated filters very well, and this may have been the source of the uncertainty.
Molecular Composition of Extracted SOA
Accurate mass measurements of molecular components in the SOA extracts were performed with electrospray ionization Fourier transform ion cyclotron resonance (ESI-FTICR) mass spectrometry to determine the influence of extraction solvent on SOA composition. Unique molecular formulas were assigned to more than 1000 peaks for each extract obtained in this study, spanning the molecular mass range of monomers, dimers, and higher order oligomers. The distribution of assigned peaks, for example as expressed in a van Krevelen plot of oxygen to carbon (O:C) ratio vs. hydrogen to carbon (H:C) ratio, was similar to those reported in the literature for SOA samples from α-pinene ozonolysis (CitationHeaton et al. 2007, Citation2009; CitationReinhardt et al. 2007; CitationWalser et al. 2008), and therefore will not be discussed in detail here. provides a succinct summary of the variation of these products by comparing the assigned peaks from different measurements of the same SOA sample.
TABLE 3 Matching molecular formulas a for two extractions of the same sample
In experiment A of , two separate measurements of the same filter extract (using acetonitrile) are compared: 76% of the peaks assigned in measurement #1 were also identified in measurement #2, while 74% of the peaks assigned in measurement #2 were also identified in measurement #1. The peaks that matched generally had high signal intensities, while those that did not generally had low signal intensities. To illustrate this point, the mass weighted intensities that matched between the two measurements are also compared in . The mass-weighted intensity fraction (MIF) of an individual peak (i) is given by
In experiments B–D of , assigned peaks from an extract with one solvent are compared to those of an extract with another solvent obtained from of a different portion of the same filter. In all cases, the mass-weighted intensities of the matching assigned peaks are within the range associated with run-to-run variability (experiment A). These results show that the SOA extracted by each solvent is quite similar, especially among the high intensity peaks in each spectrum. The fraction of assigned peaks in one extraction that matched assigned peaks in the second extraction was a bit less than expected for run-to-run variability, indicating that the solvent does exert some influence on the observed product distribution in the extract, and this difference is expressed mainly in the low intensity peaks. For methanol versus water and acetonitrile, ester formation may contribute to this difference (CitationBateman et al. 2008). For water versus the organic solvents, differing solubilities for different compounds most likely plays a larger role.
The compositional differences among different extracts are summarized in . This table gives mass and intensity weighted averages (MIAs) of O:C ratio, H:C ratio, and double bond equivalents (DBE) calculated by the following equation:
TABLE 4 Average double bond equivalents (DBE), O:C and H:C ratios of matching peaks a
Oligomer Content of SOA
The results of the previous sections suggest that close to 100% of the NVOC was collected and extracted by the procedure used and that the extraction solvent had at most a minor influence on the detected oligomer composition. Therefore, quantifying oligomers in the extract would be expected to give a reasonable approximation of oligomer content in the NVOC portion of SOA in the bag reactor.
Initially, quantification of the oligomer mass in SOA was attempted using size exclusion chromatography (SEC) in combination with ELSD detection by a procedure that was reported elsewhere (CitationSamburova et al. 2005). Chromatograms of the extracts typically showed three resolved peaks. Calibration with polyethylene glycol molecular weight standards suggested that the elution time of the first peak corresponded to higher order oligomers, the second peak to dimers and the third peak to monomers. Unfortunately, additional experiments revealed complexities. First, when a small portion of the effluent from the chromatograph was split to an electrospray ionization mass spectrometer, the oligomer distributions of all three peaks were the same. Second, when pinic and pinonic acid standards were analyzed by the same procedure, their elution times were much shorter than expected and multiple peaks were observed. These results suggested that factors in addition to molecular weight (polarity, variations in hydrodynamic volume with composition, solution phase aggregation, etc.) influenced the separation. Subsequent attempts to separate the chemical constituents by polarity or to derivatize carboxylic groups prior to SEC also were unsuccessful. For these reasons, a different strategy was adopted where monomer concentrations were determined by standard addition and the oligomer content estimated by difference.
shows the standard addition plots (monomer peak intensity vs. μg monomer spiked/total μg SOA) for pinic (2a) and pinonic (2b) acid for three different extracts using negative ion detection by electrospray ionization (ESI). Although the extraction solvent was different for each experiment, all samples were reconstituted in the same solvent (acetonitrile) prior to ESI analysis. The absolute value of the x-intercept for each plot gives the monomer concentration in the corresponding extract (CitationSkoog et al. 2007). For the SOA studied here, pinic acid concentrations were on the order of 15% by mass and pinonic acid concentrations were on the order of 7% by mass. The higher concentrations of pinic acid were not surprising given its lower vapor pressure, which favors partitioning to the particle phase. Negative ion mode was used because of the propensity of carboxylic acids give intense (M–H)− ions by ESI. While the negative ion intensities increased with increasing analyte concentrations, the positive ion intensities of the corresponding cations [(M+Na)++ (M+H)++ (M–H+2Na)+] did not change at all. This observation suggests that most of the monomer signal observed in the positive ion spectrum arose from fragmentation of oligomers in the ESI source rather than direct ionization of monomers.
FIG. 2 Standard addition plots for pinic (a) and pinonic (b) acids based on the signal intensities of (M-H)− in the negative ion ESI mass spectra of samples that were reconstituted in acetonitrile. Triangles indicate the averaged data for water extracted SOA, diamonds for acetonitrile, and squares for a 50/50 mixture of the two. The absolute value of the x-axis intercept is equal to the concentration of analyte in the original sample before the addition of standard. Weight percent is calculated from the mass of analyte added divided by the mass of SOA in the extract as determined by ELSD.
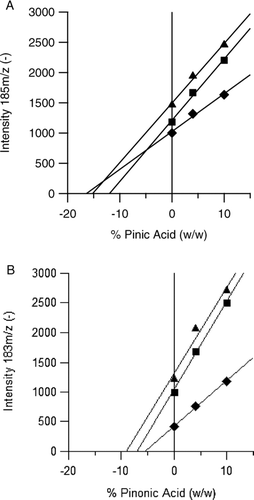
The slopes of the lines in gave the response factors (change in signal intensity divided by change in concentration) for analyte detection in the various sample matrices. It is well known for ESI that the analyte response factors can vary substantially with sample matrix. The plots in show that the response factors for analyte detection in extracts from pure water and 50/50 acetonitrile-water were the same (lines are parallel) while the response factors for pure acetonitrile extracts were lower. This behavior suggests that the SOA sample matrix for the acetonitrile extract was different from the other two solvents, most likely arising from additional impurities that were extracted off the filter by acetonitrile. (The ESI spectrum of the acetonitrile extract of a clean filter shows much more background than extracts with the other two solvents.) For each standard addition, the response for 50/50 acetonitrile-water extracts is between the response for 100% water and 100% acetonitrile extracts.
Although ESI response factors for different compounds in positive ion mode can vary by 6 orders of magnitude (CitationOss et al. 2010), the response factors for pinic and pinonic acids in negative ion mode differed by less than a factor of 1.5. The small difference is not surprising given the presence of similar ionizing groups (carboxylic acids), similar molecular size and polarity. Indeed, one might expect that all monomers encompassed within the collected SOA would possess similar characteristics because substantial partitioning to the particle phase requires a low vapor pressure. For this reason, it is reasonable to assume that all monomers within the SOA extracts studied here contained at least one carboxylic acid and that the response factors were similar to pinic and pinonic acids. With this assumption, it is possible to estimate the total monomer content an SOA extract from the total intensity of ions in the monomer region of the negative ion spectrum (defined here as ions smaller than 232 m/z) in combination with the measured response factors of pinic and pinonic acids. The results are shown in for several extracts. Monomers appear to make up around half or less of the extracted mass, with the remaining mass assigned to oligomers.
TABLE 5 Weight percentage of monomers in SOA filter extracts a
CONCLUSIONS
Nonvolatile SOA derived from α-pinene ozonolysis is quantitatively extracted from a collection filter with an organic solvent. Most (>80%) of this material is water soluble. Of the material extracted with either aqueous or organic solvents, monomers appear to constitute half or less of the total SOA mass, with oligomers accounting for the other half. This estimate agrees well conclusions of CitationGao et al. (2004), who used a different method to quantify monomers. In the present study, use of the standard addition method allows for the determination of monomer concentration while rigorously adjusting for any matrix effects, and use of ELSD allows for the measurement of total SOA mass without having to assume a particle density or shape. The oligomer content of samples prepared in both studies is sufficiently large that it should be taken into account when modeling the formation and properties of laboratory SOA.
Acknowledgments
The authors would like to thank John Dykins, Joseph Klems, and Douglas Ridge for their contributions and suggestions to this work. This research was supported by the National Science Foundation under grant number CHE-0808972.
Related Research Data
REFERENCES
- Bateman , A. P. , Walser , M. L. , Desyaterik , Y. , Laskin , J. , Laskin , A. and Nizkorodov , S. A. 2008 . The Effect of Solvent on the Analysis of Secondary Organic Aerosol Using Electrospray Ionization Mass Spectrometry . Environ. Sci. Technol. , 42 ( 19 ) : 7341 – 7346 .
- Claeys , M. , Iinuma , Y. , Szmigielski , R. , Surratt , J. D. , Blockhuys , F. , Van Alsenoy , C. , Böge , O. , Sierau , B. , Gömez-González , Y. , Vermeylen , R. , Van der Veken , P. , Shahgholi , M. , Chan , A. W. H. , Herrmann , H. , Seinfeld , J. H. and Maenhaut , W. 2009 . Terpenylic Acid and Related Compounds from the Oxidation of α-Pinene: Implications for New Particle Formation and Growth above Forests . Environ. Sci. Technol. , 43 ( 18 ) : 6976 – 6982 .
- Gao , S. , Keywood , M. , Ng , N. L. , Surratt , J. , Varutbangkul , V. , Bahreini , R. , Flagan , R. C. and Seinfeld , J. H. 2004 . Low-Molecular-Weight and Oligomeric Components in Secondary Organic Aerosol from the Ozonolysis of Cycloalkenes and α-Pinene . J. Phys. Chem. A , 108 ( 46 ) : 10147 – 10164 .
- Hallquist , M. , Wenger , J. C. , Baltensperger , U. , Rudich , Y. , Simpson , D. , Claeys , M. , Dommen , J. , Donahue , N. M. , George , C. , Goldstein , A. H. , Hamilton , J. F. , Herrmann , H. , Hoffmann , T. , Iinuma , Y. , Jang , M. , Jenkin , M. E. , Jimenez , J. L. , Kiendler-Scharr , A. , Maenhaut , W. , McFiggans , G. , Mentel , Th. F. , Monod , A. , Prevot , A. S. H. , Seinfeld , J. H. , Surratt , J. D. , Szmigielski , R. and Wildt , J. 2009 . The Formation, Properties and Impact of Secondary Organic Aerosol: Current and Emerging Issues . Atmos. Chem. Phys. , 9 : 5155 – 5236 .
- Heaton , K. J. , Dreyfus , M. A. , Wang , S. and Johnston , M. V. 2007 . Oligomers in the Early Stage of Biogenic Secondary Organic Aerosol Formation and Growth . Environ. Sci. Technol. , 41 ( 17 ) : 6129 – 6136 .
- Heaton , K. J. , Sleighter , R. L. , Hatcher , P. G. , Hall , W. A. IV and Johnston , M. V. 2009 . Composition Domains in Monoterpene Secondary Organic Aerosol . Environ. Sci. Technol. , 43 ( 20 ) : 7797 – 7802 .
- Johnson , D. and Marston , G. 2008 . The Gas-Phase Ozonolysis of Unsaturated Volatile Organic Compounds in the Troposphere . Chemical Society Reviews , 37 ( 4 ) : 699 – 716 .
- Kalberer , M. , Sax , M. and Samburova , V. 2006 . Molecular Size Evolution of Oligomers in Organic Aerosols Collected in Urban Atmospheres and Generated in a Smog Chamber . Environ. Sci. Technol. , 40 ( 19 ) : 5917 – 5922 .
- Kroll , J. H. and Seinfeld , J. H. 2008 . Chemistry of Secondary Organic Aerosol: Formation and Evolution of Low-Volatility Organics in the Atmosphere . Atmos. Environ. , 42 : 3593 – 3624 .
- Oss , M. , Kruve , A. , Herodes , K. and Leito , I. 2010 . Electrospray Ionization Efficiency Scale of Organic Compounds . Analytical Chemistry , 82 ( 7 ) : 2865 – 2872 .
- Pankow , J. F. 2003 . Gas/Particle Partitioning of Neutral and Ionizing Compounds to Single and Multi-Phase Aerosol Particles. 1. Unified Modeling Framework . Atmos. Environment , 37 : 3323 – 3333 .
- Reinhardt , A. , Emmenegger , C. , Gerrits , B. , Panse , C. , Dommen , J. , Baltensperger , U. , Zenobi , R. and Kalberer , M. 2007 . Ultrahigh Mass Resolution and Accurate Mass Measurements as a Tool to Characterize Oligomers in Secondary Organic Aerosols . Anal. Chem. , 79 ( 11 ) : 4074 – 4082 .
- Samburova , V. , Szidat , S. , Hueglin , C. , Fisseha , R. , Baltensperger , U. , Zenobi , R. and Kalberer , M. 2005 . Seasonal Variation of High-Molecular-Weight Compounds in the Water-Soluble Fraction of Organic Urban Aerosols . J. Geophys. Res. , 110 : D23210
- Shilling , J. E. , Chen , Q. , King , S. M. , Rosenoern , T. , Kroll , J. H. , Worsnop , D. R. , DeCarlo , P. F. , Aiken , A. C. , Sueper , D. , Jimenez , J. L. and Martin , S. T. 2009 . Loading-Dependent Elemental Composition of Alpha–Pinene Soa Particles . Atmos. Chem. Phys. , 9 : 771 – 782 .
- Skoog , D. A. , Holler , F. J. and Crouch , S. R. 2007 . Principles of Instrumental Analysis , 6 ed. , Belmont, CA : Thomson Higher Education .
- Sleighter , R. L. and Hatcher , P. G. 2007 . The Application of Electrospray Ionization Coupled to Ultrahigh Resolution Mass Spectrometry for the Molecular Characterization of Natural Organic Matter . J. Mass Spectrometry , 42 ( 5 ) : 559 – 574 .
- Tolocka , M. P. , Heaton , K. J. , Dreyfus , M. A. , Wang , S. , Zordan , C. A. , Saul , T. D. and Johnston , M. V. 2006 . Chemistry of Particle Inception and Growth during α-Pinene Ozonolysis . Environ. Sci. Technol. , 40 ( 6 ) : 1843 – 1848 .
- Tolocka , M. P. , Jang , M. , Ginter , J. M. , Cox , F. J. , Kamens , R. M. and Johnston , M. V. 2004 . Formation of Oligomers in Secondary Organic Aerosol . Environ. Sci. Technol. , 38 ( 5 ) : 1428 – 1434 .
- Turpin , B. J. , Saxena , P. and Andrews , E. 2000 . Measuring and Simulating Particulate Organics in the Atmosphere: Problems and Prospects . Atmos. Environ. , 34 ( 18 ) : 2983 – 3013 .
- Walser , M. L. , Desyaterik , Y. , Laskin , J. , Laskin , A. and Nizkorodov , S. A. 2008 . High-resolution Mass Spectrometric Analysis of Secondary Organic Aerosol Produced by Ozonation of Limonene . Phys. Chem. Chem. Phys. , 10 ( 7 ) : 1009 – 1022 .
- Wozniak , A. S. , Bauer , J. E. , Sleighter , R. L. , Dickhut , R. M. and Hatcher , P. G. 2008 . Technical Note: Molecular Characterization of Aerosol-Derived Water Soluble Organic Carbon Using Ultrahigh Resolution Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry . Atmos. Chem. Phys. , 8 : 5099 – 5111 .
- Yu , J. , Cocker , D. R. , Griffin , R. J. , Flagan , R. C. and Seinfeld , J. H. 1999 . Gas-phase Ozone Oxidation of Monoterpenes: Gaseous and Particulate Products . J. Atmos. Chemistry , 34 : 207 – 258 .
- Zelenyuk , A. , Yang , J. , Song , C. , Zaveri , R. A. and Imre , D. 2008 . A New Real-Time Method for Determining Particles’ Sphericity and Density: Application to Secondary Organic Aerosol Formed by Ozonolysis of α-Pinene . Environ. Sci. Technol. , 42 ( 21 ) : 8033 – 8038 .