Abstract
Particle mass spectrometers of two types—a time-of-flight aerosol mass spectrometer (AMS) of Aerodyne Research Inc. and a laser desorption/ionization single particle aerosol mass spectrometer (LISPA-MS) developed at Nagoya University—were deployed to characterize aerosol particles in the Tokyo metropolitan area during the summer of 2008. Based on the ensemble measurements by AMS, equivalent mass concentration of organic aerosol, traced by mass-to-charge ratio (m/z) 44, showed a closer correlation with particulate nitrate and gas-phase odd oxygen, [O 3 +NO 2 ], whereas equivalent mass concentration of organic aerosol, traced by m/z 57, did not. On a particle-by-particle basis, the relative signal peak area of various target species in the LISPA-MS spectra, which was calculated as the ion-signal fraction of the species relative to the total signal peak area summed over all the ion peaks in each spectrum, was used as a measure of the relative amount of the species. A rough qualitative agreement was obtained between the temporal variation observed in the LISPA-MS RCOO– signal and that in the AMS m/z 44, but not the AMS m/z 57, in which the LISPA-MS RCOO– signal was defined as the sum of the relative signal peak areas of 17 different negative-ion mass peaks used as markers of oxygenated organics. Analysis of the LISPA-MS spectra also showed that approximately 95% of the oxygen-containing organic particles contained nitrate, which is expected to be responsible in part for the correlation between AMS m/z 44 and AMS nitrate.
1. INTRODUCTION
The metropolis of Tokyo is well known as extremely crowded metropolitan areas in terms of not only population density but also traffic density, in which photochemical smog is a phenomenon observed during hot sunny days in summer. This kind of smog is generated by photochemical reactions involving highly concentrated pollutants of organic compounds and NO x , and secondary aerosol particles consisting of atmospheric oxidants are also included (CitationSeinfeld and Pandis 2006). Our current understanding of the formation processes and the chemical and physical properties of ambient aerosol remains insufficient (e.g., CitationKanakidou et al. 2005; CitationJimenez et al. 2009). Additional research on the characteristics of ambient aerosol is essential for accurate evaluation of the impact of urban aerosols on visibility, climate, and human health and is a prerequisite to implementing more effective regulations.
In the summer of 2008, we conducted a comprehensive observation campaign in the Tokyo metropolitan area to measure aerosol and gas-phase compounds with the goal of characterizing the chemical and optical properties of urban particulate matters related to photochemical smog episodes. We used an originally developed cavity ring-down spectrometer as well as commercially available instruments of a skyradiometer, a nephelometer, and a particle soot absorption photometer for investigating optical properties of urban aerosol (CitationNakayama et al. 2010). We also used an originally developed laser-based single-photon ionization time-of-flight mass spectrometer (CitationTonokura et al. 2010) as well as a commercially available proton-transfer reaction mass spectrometer, and O3 and NO x analyzers for monitoring gas-phase molecules act as aerosol precursors. State-of-the-art instruments for on-line analysis of aerosol were also employed to explore chemical properties of urban aerosol. In this article, we describe results of the chemical analysis of ambient aerosol using on-line particle mass spectrometers of two different types—an Aerodyne high-resolution-Time-of-Flight Aerosol Mass Spectrometer (ToF-AMS) (CitationJimenez et al. 2003; CitationDeCarlo et al. 2006; CitationZhang et al. 2007 and references therein) and an originally developed laser-desorption/ionization single-particle aerosol mass spectrometer (LISPA-MS) (CitationMatsumoto et al. 2006; CitationNarukawa et al. 2007a, b, 2008).
The chemical complexity and labile nature of ambient aerosol strongly favors real-time instrumental analysis techniques that characterize relevant physical and chemical properties without having to collect, store, and transport samples. The field of online aerosol mass spectrometry has emerged to provide a real-time method for the chemical analysis of aerosol particles. Such techniques overcome the problems associated with bulk collection and offline analysis by analyzing compositions in situ. The basic principle of aerosol mass spectrometry is to introduce airborne particles into the instrument, vaporize and ionize the material and then analyze the produced ions using mass spectrometry. Although some different designs for aerosol mass spectrometers have been introduced over the years, as given in review articles (CitationCanagaratna et al. 2007; CitationMurphy 2007), widely used instruments can be categorized roughly into two types based on their technical aspects. The first major type involves the use of lasers to both vaporize and ionize individual particles sampled into the mass spectrometer source region, which delivers quantitative size and qualitative composition information on individual particles. The other major type involves thermal desorption of individual or collected particles, followed by various ionization techniques such as electron impact and vacuum-ultraviolet single-photon ionization methods, which provides quantitative data related to both the size and composition of the entire aerosol ensemble, but provides only limited data about specific particles and which does not examine refractory components. Therefore, combined deployment of instruments of both types can be expected to provide much deeper insights into the nature of aerosol properties than by the use of either instrument alone. Actually, since the pioneering work of CitationMiddlebrook et al. (2003), a variety of work in which particle mass spectrometers of both types were used together has been increasingly available (CitationCubison et al. 2008; CitationDrewnick et al. 2008; CitationDall’Osto et al. 2009a, b; CitationMurphy et al. 2006; CitationSalcedo et al. 2010). In our study, on-line particle mass spectrometers of two different types were thus deployed together to characterize time- and size-resolved chemical properties of urban aerosol.
Urban aerosol in Tokyo has recently been studied by Kondo and co-workers (CitationTakegawa et al. 2005, Citation2006, Citation2007; CitationMiyakawa et al. 2007; CitationMorino et al. 2006), in which an early developed model of the Aerodyne instrument with quadrupole mass spectrometry detection (Q-AMS) was deployed. Typical chemical characteristics of Tokyo urban aerosol they found are that (1) the organic composition is predominant during all seasons, (2) the sulfate composition is high in summer and low in winter, and (3) the nitrate composition is low in summer and high in winter. In the present study, we investigated the chemical properties of aerosol particles by using two different types of particle mass spectrometers in combination, on a particle-by-particle basis using LISPA-MS and on an ensemble basis using ToF-AMS (hereinafter called AMS).
2. EXPERIMENTAL
2.1. Sampling Site
Intensive measurements of aerosol and trace gases were made in an urban site in Tokyo during 28 July–29 August, 2008. The two particle mass spectrometers were deployed together during 11–15 August in a room at the 3rd floor of a laboratory building in the Hongo campus of the University of Tokyo (35.71°N, 139.76°E), located within the urban center of metropolitan Tokyo. Ambient aerosol was sampled through a copper tube 4.4 mm in inner diameter and approximately. 3 m in length, with the inlet facing downward and set 1 m away from the building wall. Several major industrial areas are situated in the suburbs of Tokyo city close to our observation site including the Keihin industrial area, about 20 km south, and the Keiyo industrial area, about 15–30 km east-to-southeast from the observation site, both of which are located on the coast of Tokyo Bay. The weather during the campaign was characterized by typical Asian monsoon, which provided warm and humid southerly winds (temperature 26–34°C, relative humidity 49–82%). No precipitation was observed at the measurement site during 11–15 August. All dates and times in the text without notation are based on Japan Standard Time (JST; UTC + 09:00).
2.2. Instruments
A primary difference in technique between the two mass spectrometers is their volatilization/ionization method: LISPA-MS uses laser desorption/ionization technique to volatilize and ionize a particle simultaneously using a single laser shot, whereas AMS vaporizes particles by impaction onto a heated surface, with subsequent electron-impact ionization of the vaporized component. A mass spectrum from AMS has the information of an ensemble of particles sampled, whereas that from LISPA-MS gives information on an individual particle.
Details of the AMS instrument and its mechanism have been described in CitationDrewnick et al. (2005) and DeCarlo et al. (2006). For that reason, only a brief description is given here. Aerosol particles are introduced into the instrument through an aerodynamic lens assembly at a nominal flow rate of 1.46 ± 0.1 cm3s–1, where the particles are focused into a narrow beam. The particle beam travels across the vacuum chamber, and most of the gas component is pumped away. The AMS instrument can observe non-refractory particles of PM1.0, and particles in the size range 50–600 nm are focused with an efficiency of almost 100% (CitationLiu et al. 2007). Smaller and larger particles were also collected with lower efficiency. At the end of the chamber, the non-refractory components in the aerosol particles are flash-vaporized on a resistively heated surface (912 K during this observation) and the evolved vapor molecules are then ionized under a 70-eV electron impact (EI) ionization source. The resulting positive ions are transported into an orthogonal extraction ToF-MS for mass analysis. The AMS instrument was operated in “V-mode” in this work. Aerodynamic particle size data is obtained by chopping the particle beam and collecting mass spectra as a function of particle flight time between the chopper and the vaporizer. The instrument provides 10-min averages of non-refractory aerosol components as well as species-resolved size distributions. The data are analyzed using the IGOR-based AMS analysis software package. Collection efficiency (CE) of 0.5 is adopted for the analysis. The relative ionization efficiency values for ammonium, nitrate, sulfate, and organics used for this study are 4.0, 1.1, 1.2, and 1.4, respectively. The organic mass concentration is obtained by subtracting gaseous species and inorganic aerosol species from the total. Because of an instrumental problem, the AMS could only be operated with a person in attendance (mostly daytime).
The LISPA-MS instrument was developed in Nagoya University, which enables real-time measurements of chemical composition of individual particles. Instrumental design of the LISPA-MS is similar to those of some other related instruments of single particle mass spectrometry (SPMS) and the general capabilities of SPMS for field studies have been reviewed previously (CitationSuess and Prather 1999; CitationNoble and Prather 2000; CitationSullivan and Prather 2005; CitationMurphy 2007). Details of our home-made LISPA-MS instrument have been described elsewhere (CitationMatsumoto et al. 2006, CitationNarukawa et al. 2007a, b, 2008). For that reason, we only give a brief description of the LISPA-MS instrument. Aerosol particles are introduced into an aerodynamic lens and are focused into a narrow particle beam. After exiting the focusing lens, the particle beam is transmitted into the ion-source region, where a continuous-wave laser beam of a frequency-doubled Nd:YAG laser at 532 nm is set to intersect with the particle beam to detect particles reaching the ion-source region. The intensity of the light scattered from individual particles at 532 nm is used to estimate the particle size. The size distributions of detectable particles with the LISPA-MS instrument range from 250 nm to 1.3 μm in diameter, as estimated by laboratory experiments using standard polystyrene latex particles (CitationNarukawa et al. 2008). The instrument mainly analyzed submicron particles during the observation campaign. The scattered light of an intensity above a controlled threshold then acts to trigger a pulsed Excimer laser (λ= 248 nm, Lambda Physik, OPTex) that evaporates and ionizes the detected particle. The laser power density was ∼1 × 107 W cm–2. The resulting ions are measured using a time-of-flight mass spectrometer with a microchannel plate detector. In our experiments, the voltage polarity of the instrument was automatically changed periodically with a time interval of 1 minute to detect either the positive- or negative-ions. Over the measurement period from 11–15 August, the LISPA-MS analyzed 7459 negative and 6898 positive single-particle mass spectra.
3. RESULTS AND DISCUSSION
3.1. General Trend in AMS Observation
Figures and 1b show the temporal variation in the individual and summed-mass concentrations of organics, nitrate, sulfate, ammonium, and chloride along with their weight-based fraction observed by AMS during 11–15 August. Organic aerosol comprised 24–85% (average 53%) of the non-refractory submicron particle mass and was the dominant constituent. Sulfate (10–54%, avg. 32%) followed by ammonium (3–20%, avg. 13%) and nitrate (1–4%, avg. 2%) accounted for the remainder of major constituents. The chloride fraction was very low, accounting for <0.5%, throughout the observation. The total mass concentration varied in the range 6.4–20 μg/m3 for 10-min average, giving an average of 10.6 μg/m3 over the entire observation period. CitationZhang et al. (2007) summarized the observations using an Aerodyne AMS (mainly Q-AMS) at many sites around the world. Results show that the mass-based constituent fraction we have measured in Tokyo resembles some of the summer data measured in urban areas such as New York, Houston, Manchester, and Tokyo. Specifically, these similarities are that: (1) organic aerosol comprises more than 50% of the total aerosol mass concentration; and (2) the order of major components follows organics > sulfate > ammonium > nitrate > chloride.
FIG. 1 Temporal variation in (a) weight-based fraction of non-refractory components in aerosol particles and (b) mass concentration summed over organic aerosol (green), nitrate (blue), sulfate (red), ammonium (orange), and chloride (pink), as observed by AMS. AMS measurements were conducted mostly in daytime. Temporal variation in the three-hour integrated relative peak areas of (c) m/z 97 (HSO– 4) and (d) m/z 18 (NH+ 4), as measured by LISPA-MS. For discussion purposes, the observation period was divided into two periods named Period I (14:40, 11 August to 12:00, 13 August) and Period II (10:50, 14 August to 20:30, 15 August) as indicated (see the text). The numbers of particles counted both in positive and negative ion measurement modes are plotted for each three-hour bin in panel (e).
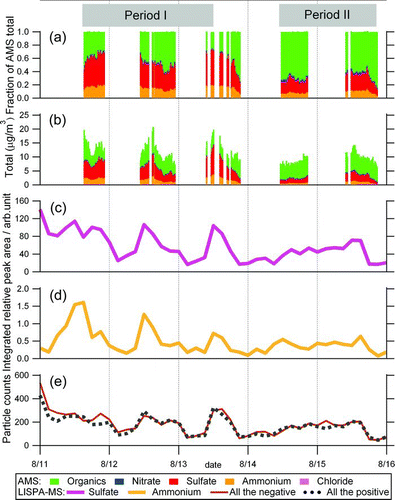
The Q-AMS study in Tokyo in the summer of 2003 (site at 35°39′N, 139°40′E) gave results of: organic aerosol 47% (average of the entire observation period), sulfate 29%, ammonium 14%, nitrate 8%, and chloride 2% (CitationMiyakawa et al. 2007). The variation in total mass concentration in 2003 varied from 8.3 to 18.3 μg/m3 (CitationTakegawa et al. 2006), which is in a similar range to our 2008 data (6.4–20 μg/m3). It is noteworthy that CitationTakegawa et al (2006) attached a temperature–humidity controller at the AMS inlet although we did not, thus the measurement conditions were different. The absolute accuracy of the data reported here is limited by uncertainty in AMS CE factor. Most field studies use a CE value around 0.4–0.5 (CitationAllan et al. 2004; CitationDrewnick et al. 2004; CitationTakegawa et al. 2005; CitationWeimer et al. 2006) and AMS measurements with CE = 0.5 compared favorably with other collocated data in several different environments (Alfarra et al. 2004; Takegawa et al. 2005; Zhang et al. 2005a; Salcedo et al. 2007). In the present study, we assumed a constant value of CE = 0.5 over the entire observation period. However, recent studies have shown that CE values may be influenced by factors such as particle phase, composition, and water content (CitationAllan et al. 2004; CitationMatthew et al. 2008; CitationZhang et al. 2005a). Temperature and relative humidity of the outside air in the daytime when the AMS instrument was working in our study ranged 28–34°C and 48–77%, respectively, whereas the laboratory building was air-conditioned at 28°C. Thus we cannot completely rule out the possibility that under some conditions the mass concentrations reported here might be somewhat overestimated.
The total mass concentration of non-refractory submicron aerosol particles was calculated as the sum of sulfate, nitrate, ammonium, chloride, and organic aerosol, as shown in and b. The total mass concentration showed a diurnal variation during 11–13 August. However, on 14 August, such a variation was unclear, and on 15 August, the variation was weak with a smaller maximum than seen during 11–13 August. The average total mass concentration of Period I (14:40, 11 August–12:00, 13 August) and Period II (10:50, 14 August–20:30, 15 August) were, respectively, 12.4 and 9.0 μg/m3. We calculate that this decrease was mainly caused by a decrease in the amounts of sulfate and ammonium. The sulfate and ammonium mass loadings averaged over period I were 4.8 and 1.8 μg/m3, respectively, whereas those averaged over period II were 1.9 and 0.8 μg/m3, respectively. Consequently, from period I to period II, the sum of sulfate and ammonium decreased by 3.9 μg/m3, which is close to the reduction in total aerosol over this period (3.4 μg/m3). In contrast, the organics mass concentration showed a minimal increase from 5.4 to 6.0 μg/m3 from period I to period II, and its relative fraction of the total mass concentration increased from 59% to 67%. The mass concentrations of nitrate and chloride remained almost constant throughout the observation period.
The AMS instrument measures mass size distributions of non-refractory components. Our data analysis showed that the size distributions of sulfate and ammonium were similar, with a mode at 650 nm in Period I; and the mode shifted to 500 nm in Period II. To discuss the changes in size distribution of sulfate and ammonium between Period I and II, as well as changes in the sulfate and ammonium mass loadings averaged over each period, we investigated near-surface wind fields in and around the Tokyo area over this time. The dataset of wind fields was referred from the European Centre for Medium-Range Weather Forecasts (ECMWF) Interim Re-Analysis data. The near-surface wind over Tokyo region was relatively gentle from 11 August until the morning of 13 August, but shifted suddenly to fairly strong southwesterly wind in the afternoon of 13 August and remained strong on 14 August. Such a change in the near-surface wind field might be related to the changes in the sulfate and ammonium mass concentrations and their mass size distributions over this time.
3.2. General Trend in LISPA-MS Observation
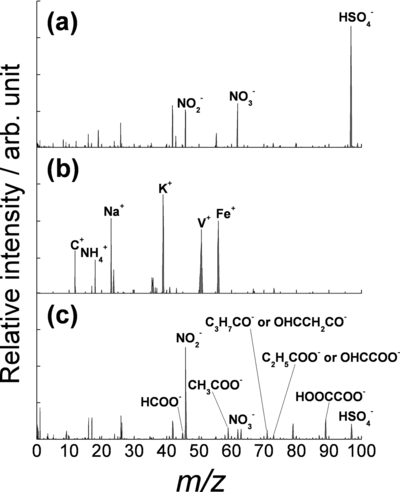
For the LISPA-MS observations, a total of 7459 negative and 6898 positive spectra were obtained over the entire observation period. shows a representative negative-ion mass spectrum of an individual particle containing nitrite (m/z 46), nitrate (m/z 62), and sulfate (m/z 97). We use nitrite and nitrate signals as a marker of particulate nitrate in the analysis of LISPA-MS spectra. The number of spectra giving nitrate and nitrite signals was 5361, whereas those giving a sulfate signal was 5994, which accounts for 72% and 80% of the total negative mass spectra, respectively, where the minimum detectable peak area was determined as the signal giving peak height which was seven times the baseline noise level. Fraction of negative ion mass spectra containing both nitrate and sulfate was 56% in Period I and 59 % in Period II. In a previous laboratory experiment using standard compounds, we detected peaks at m/z46 and 62 from particles of (NH4)NO3 and NaNO3, and a peak at m/z 97 from (NH4)2SO4 (CitationNarukawa 2007a). Also in many other studies using SPMS, m/z 46 and 62 and m/z 97 have been identified as markers of nitrate and sulfate, respectively (CitationLee et al. 2002, Citation2003; CitationMatsumoto et al. 2006; CitationMiddlebrook et al. 2003; CitationMurphy et al. 2006; CitationSullivan and Prather 2005, Citation2007; CitationPratt and Prather 2010; CitationReinard and Johnston 2008). shows a positive-ion mass spectrum of an individual particle containing ammonium ion, with a peak at m/z 18 (NH+ 4), as well as a variety of other species at m/z 12 (C+), 23 (Na+), 39 (K+), 51 (V+), and 56 (Fe+). The number of the LISPA-MS spectra containing ammonium was 1099, accounting for 16% of the total positive mass spectra. Analysis of the alkali metals and other species detected in the positive-ion mass spectra was beyond the scope of this study. shows a representative negative-ion mass spectrum of an individual particle giving ion signals originating from organics, with peaks at m/z 45 (HCOO– and/or C2H5O–), 59 (CH3COO– and/or C3H7O–), 71 (OHCCH2CO– and/or C3H7CO–), 73 (OHCCOO– and/or C2H5COO–), 89 (HOOCCOO–), as well as nitrate (NO– 2 and NO– 3) and sulfate (HSO– 4). Although organic molecules undergo fragmentation during ablation and/or ionization, some typical ion signals originating from organic molecules can be detected in the negative spectra, as seen in our laboratory measurements (CitationNarukawa et al. 2007b, Citation2008). The spectrum in is a direct evidence of the coexistence of organics, nitrate, and sulfate in an individual particle observed in the Tokyo metropolitan area. The capability of SPMS to investigate the aerosol mixing state has been widely demonstrated (CitationSuess and Prather 1999; CitationNoble and Prather 2000; CitationLee et al. 2002, Citation2003; CitationMiddlebrook et al. 2003; CitationSullivan and Prather 2005, Citation2007; CitationMurphy, 2007; CitationPratt and Prather 2010), whereas generally the results arising from the ensemble analysis of the AMS instrument alone cannot conclusively prove that the nitrate, sulfate, and organics are internally mixed. Formation of organic nitrate aerosol during the atmospheric oxidation of volatile organic compounds has recently received increasing attention from both laboratory and air sampling studies (CitationBruns et al. 2010; CitationGarnes and Allen 2002; CitationJordan et al. 2008; CitationSato et al. 2010; CitationWang et al. 2009). We, however, did not observe any ions characteristic of aerosol-phase organic nitrogen species in our LISPA-MS spectra. Nevertheless this does not exclude the possibility that some organic nitrogen species might be present in urban aerosol in Tokyo, since they may have been fragmented during the ablation/ionization under our experimental conditions. The photon energy at 248 nm (482 kJ mol–1) is well above the dissociation energies of the weakest RO-NO2bond in alkyl nitrates, RONO2 (170–200 kJ mol–1) (CitationAtkinson et al. 2006).
FIG. 2 Typical negative- and positive-ion mass spectra of individual particles observed by LISPA-MS, with assignments of the corresponding ions: (a) sulfate (m/z 97) and nitrate (m/z 46 and 62), (b) ammonium (m/z 18) and C+, Na+, K+, V+, and Fe+, and (c) organics as well as sulfate and nitrate.
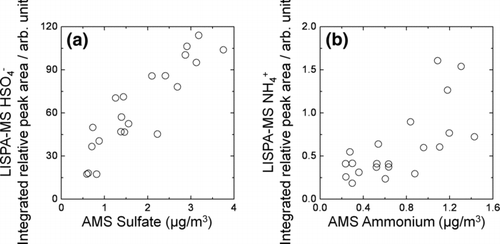
To further investigate the chemical properties of individual particles, we use relative peak area of a selected ion signal in each spectrum as a measure of the relative amount of chemical component. The relative peak area of a target species in an individual particle was calculated as the ion-signal fraction of the species relative to the total signal peak area summed over all ion peaks in each spectrum. The relative peak area is more reproducible than absolute peak area, owing to spectrum-to-spectrum fluctuations in the interaction of the sampled individual particle with the ionization laser beam, as reported in many studies (CitationLee et al. 2002, Citation2003; CitationMurphy et al. 2006; CitationSullivan et al. 2007; CitationSullivan and Prather 2007; CitationReinard and Johnston 2008). Figures and 1d show the temporal variation in the 3-h integrated relative peak areas of the m/z 97, and m/z 18 signals. The total numbers of the LISPA-MS positive and negative ion mass spectra obtained for each 3-h bin are depicted in . The three-hour integrated relative peak areas of sulfate and ammonium in Figures and 1d showed similar diurnal profiles, peaking in the daytime during period I, however, in period II this trend was unclear. These trends are analogous to the temporal variations in the AMS observed sulfate and ammonium mass concentrations presented in . Correlations of sulfate and ammonium between LISPA-MS and AMS are depicted in (R= 0.90 and 0.69 for sulfate and ammonium, respectively), suggesting that the three-hour integrated relative peak areas of LISPA-MS m/z 97 and 18 trace the relative amounts of sulfate and ammonium in aerosol particles.
FIG. 3 Correlation plots for LISPA-MS relative peak area and AMS mass loading: (a) sulfate and (b) ammonium. Note that LISPA-MS peak areas were integrated over each 3-h bin, whereas AMS mass concentrations (originally logged as 10-min interval data) were averaged over each three-hour bin for comparison.
3.3. Characteristics of Individual Aerosol Components
3.3.1. Ammonium
In examining the ion balance of aerosol particles observed by AMS (CitationDrewnick et al. 2005; CitationTakami et al. 2005; CitationTakegawa et al. 2006; CitationZhang et al. 2005a, b), we assumed that ammonium would arise from neutral compounds formed from sulfate, nitrate, and chloride, i.e., (NH4)2SO4, NH4NO3, and NH4Cl, respectively. Specifically, we calculated the sum of 2×[SO2– 4], [NO– 3], and [Cl–] and compared it with the observed [NH+ 4]. The slope was almost unity (0.998) with a correlation factor of R= 0.99. We found a plot of the molar concentration of NH+ 4 versus SO2– 4 showed tight correlations with a molar ratio of 2.0:1 and 2.1:1 for Periods I and II, respectively. Consequently, we concluded that most of the ammonium signal observed during the campaign arose from ammonium sulfate. This can explain similar behaviors of the temporal variations observed for the mass concentrations of sulfate and ammonium ().
3.3.2. Sulfate
In the AMS data, sulfate was the most abundant inorganic component and was found in the form of ammonium sulfate as discussed above. Sulfate exhibited a diurnal variation during 12–13 August, which contributed mainly to the variation in total aerosol mass concentration on both days, whereas such variation was not apparent in Period II. In the LISPA-MS data, a similar temporal variation is evident in the three-hour integrated relative peak area of HSO– 4, as shown in Figures and 3a, which suggests that both techniques provide consistent information on the temporal variation in sulfate loading, although the fundamental measurement principle of each technique differs and AMS measures the amounts of chemical species more quantitatively.
Anthropogenic sulfate aerosol is mainly produced from oxidation of SO2. SO2 reacts under tropospheric conditions via both gas- and aqueous-phase processes and is also removed via dry and wet deposition (CitationSeinfeld and Pandis 2006). With respect to gas-phase reaction, reaction with the OH radical is dominant. However, the lifetime of SO2 based on the OH reaction, at typical levels of OH, is about one week (CitationSeinfeld and Pandis 2006). Localized in-situ gas-phase formation thus cannot fully account for temporal variations in sulfate loading observed in this work. In-situ oxidation of SO2 by aqueous phase reactions would have been slow during the campaign, since neither rainfall nor fog was observed. We explored whether hydroxymethanesulfonate (HMS) was identified by a deprotonated molecular ion signal at m/z= 111 (CH2(OH)SO– 3) in the LISPS-MS negative ion spectra, because in previous SPMS studies (CitationDall’Osto et al. 2009a; CitationLee et al. 2002, Citation2003; CitationWhiteaker and Prather 2003) HMS was examined as a marker of aqueous-phase chemistry. HMS is formed by SO2 reacting with formaldehyde in aqueous solutions such as clouds or fogs (CitationBoyce and Hoffmann 1984). The formation and stability of HMS has been shown to be highly dependent on the pH of the particle or droplet, as well as the concentrations of formaldehyde, bisulfite (or sulfite) ions, and oxidants present (CitationBoyce and Hoffmann 1984; CitationSeinfeld and Pandis 2006). We found that the proportion of LISPA-MS spectra giving a sulfate signal in the total number of negative ion spectra was 81.0% in Period I and 81.3% in Period II, of which only a few percent of particles gave the m/z 111 mass peak. Relatively low detection frequencies of HMS throughout the observation period are likely to indicate that in-situ aqueous processes are unimportant in controlling the sulfate loading in this work, although for more detailed spectral analysis detection sensitivity of LISPA-MS to HMS needs to be characterized with laboratory experiments. The interpretation of temporal variations in sulfate loading should be related to photochemical processing of gas-phase SO2 in these polluted air masses and the regional scale production and transport of sulfates, though a definitive analysis would require accounting for the detailed meteorology.
3.3.3. Organic Aerosol
In contrast to sulfate, the AMS data suggested that the mass concentration of organics did not change distinctly between the two periods, as shown in . Meanwhile, a significant correlation was found between the organics and nitrate (R= 0.59) observed by AMS, as shown in . To provide an in-depth interpretation of the complex organic spectra, we examine the equivalent mass concentrations of organic aerosol represented by AMS signals at m/z 44 and 57. Previous AMS studies have indicated that AMS m/z 44 signal is dominated by COO+, which typically exhibits a much greater oxygen content mainly because of SOA formation, whereas AMS m/z 57 signals in urban area are typically dominated by aliphatic organic compounds (mostly C4H+ 9) (CitationAiken et al. 2008; CitationMohr et al. 2009; CitationZhang et al. 2005a, and references therein). As shown in and c, AMS m/z 44 had a much closer correlation with nitrate (R= 0.87), whereas AMS m/z 57 did not (R= −0.16). Furthermore, as shown in –, AMS m/z 44 was well correlated with gas-phase potential ozone [= O3+
NO2] (R= 0.86), whereas AMS m/z 57 was not (R= −0.33). The results of these correlation analyses indicate that the correlations between organics and nitrate/potential ozone are not due to primary organic aerosol, but to a rapid growth of SOA in urban air that overwhelms the direct emissions of primary organic aerosol within a few hours of the relevant photochemical processes. Such a close correlation between oxygenated organics and potential ozone has been observed previously in several urban areas as well as in Tokyo (CitationAiken et al. 2009; CitationKondo et al. 2008; CitationHerndon et al. 2008; CitationZhang et al. 2005a; CitationWood et al. 2010). A very recent work by CitationWood et al. (2010) investigated literature data reporting a wide range of correlation in the relationship between oxygenated organics and potential ozone. Our correlation value is comparable to previously reported values of 0.86 (CitationKondo et al. 2008) and 0.89 (11:00–14:00 LT in Matsui et al. 2009) in summer in Tokyo, where it should be noted that oxygenated organic aerosol (OOA) component used in their correlation analyses was derived from the recorded AMS spectra using a deconvolution algorithm and thus the OOA component was not just proportional to m/z 44.
FIG. 4 Correlation plots for the mass concentrations observed by AMS and the potential ozone of [O3] + [NO2]: (a) Nitrate versus organics, (b) nitrate versus m/z 44, (c) nitrate versus m/z 57, (d) potential ozone versus organics, (e) potential ozone versus m/z 44, and (f) the potential ozone versus m/z 57. Those plots include all data over Periods I and II.
![FIG. 4 Correlation plots for the mass concentrations observed by AMS and the potential ozone of [O3] + [NO2]: (a) Nitrate versus organics, (b) nitrate versus m/z 44, (c) nitrate versus m/z 57, (d) potential ozone versus organics, (e) potential ozone versus m/z 44, and (f) the potential ozone versus m/z 57. Those plots include all data over Periods I and II.](/cms/asset/8d526443-a733-456c-a2cb-76c1cfdecb51/uast_a_533720_o_f0004g.gif)
The AMS m/z 44/total organic aerosol ratio can be a measure for the oxidation level of organic aerosol, namely, the larger the proportion of m/z 44 to the total organic aerosol, the larger the oxidation level of the aerosol (CitationDrewnick et al. 2007; CitationSchneider et al. 2004; CitationTakegawa et al. 2005). For example, CitationTakegawa et al. (2005) reported that the ratio observed in the Tokyo urban area in summer 2003 was in the range of 2–13% for all one-hour averages. In the present study, the ratio averaged over the entire observation period was 20%, and no obvious change was observed from Period I (21%) to Period II (18%). Our ratio is higher than those in previous study of CitationTakegawa et al. (2005), which might be attributable to the fact that our AMS observations were conducted mainly during afternoons whereas their data included the results of morning and midnight observations.
From the viewpoint of single particle analysis, the LISPA-MS instrument sometimes gave signals of organic fragments, together with a nitrate signal in identical particles, as shown in , which is a direct evidence that organics and nitrate coexist in the particle in Tokyo. We estimated the amount of aerosol that contained both organics and nitrate on a particle-by-particle basis. In the LISPA-MS spectra, we were able to identify 23 different mass peaks attributable to organic fragments, as presented in , of which some had also been identified in our laboratory experiment (CitationNarukawa et al. 2007b). CitationNarukawa et al. (2007b) showed that organics containing carboxyl group in aerosol particles provide intense signals of deprotonated ions, [M-H]–, with a few weak fragment ions in the negative ion mass spectra, so that their molecular weights, namely, parent molecules can be inferred through observations of deprotonated ions. For example, dicarboxylic acids and oxoacids are thus expected to contribute to the distinct peaks observed at m/z 89 (oxalic acid), 101 (4-oxo-butanoic acid), 103 (maloic acid), 115 (5-oxo-pentanoic acid), 117 (succinic acid), 129 (6-oxo-pentanoic acid), 131 (glutaric acid), and 145 (adipic acid) in the present study. The m/z 45 signal of HCOO–/C2H5O– and the m/z 99 signal of HOCC3H6CO–/C5H11CO– were intentionally excluded because they are sometimes confused with the strong m/z 46 signal of NO– 2 and the m/z 99 signal of H34SO– 4. Consequently, any of 21 mass peaks was used as a marker of organics. This analysis revealed that organics were detected in 744
particles in Period I and in 685 particles during Period II, respectively, of which 707 and 652 particles contained nitrate. This result infers that a considerable proportion of oxygen-containing organics were likely to be mixed with nitrate in individual particles observed in Tokyo during the campaign, which is expected to be the reason, in part, for the correlation between m/z 44 and nitrate on an ensemble basis, as shown in . Some laboratory and field studies have particularly addressed the uptake of organic nitrates, HNO3, NO3, and N2O5, and/or the heterogeneous reaction of NO2 on the surface of hydrophilic organic aerosol particles (CitationBroske et al. 2003; CitationMorino et al. 2006; CitationStemmler et al. 2006; CitationThornton et al. 2003; CitationYabushita et al. 2009).
TABLE 1 Mass peaks (m/z) explored as candidate markers of organics in negative-ion mass spectra measured by LISPA-MS and their possible assignment of corresponding ions
The possible criticism to the choice of 21 mass peaks as makers of oxygen-containing organic fragments concerns alternative assignment of inorganic fragments at the same ion peaks. For example, the m/z= 43, 59, and 103 ion peaks can be AlO–, AlO– 2, and AlSiO– 3, respectively. Potential interferences of those aluminosilicate ions with organic peaks were ruled out by examining other aluminosilicate ions at m/z= 76 (SiO– 3) and 119 (AlSiO– 4) within the same spectrum, as reported in previous studies (CitationLee et al 2002, Citation2003). Mass peaks at m/z= 129, 131, 133 can be MgCl– 3 and m/z= 145, 147, 149 can be CaCl– 3. However, careful analysis of the LISPA-MS spectra revealed that mass peaks at m/z= 129 and 131 were not observed simultaneously, and similarly mass peaks at m/z= 145 and 147 were not. Therefore, it was unlikely that MgCl– 3 and CaCl– 3 contributed significantly at those mass peaks. The m/z= 159 ion peak can arise from ammonium nitrate dimer. Laboratory measurements of aerosol particles generated from standard ammonium nitrate solution did not provide the m/z= 159 peak with a remarkable intensity (CitationNarukawa et al. 2007a). It is unlikely that ammonium nitrate dimer contributed significantly at the mass peak under our experimental conditions in the present study.
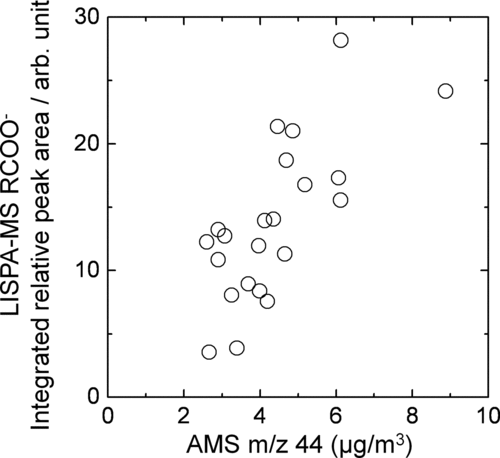
shows a comparison of the temporal variations in mass concentrations of total organics, and equivalent mass concentrations of organics represented by m/z 44 and 57 (Figures and c), observed by AMS, against the variation in the relative peak area of the RCOO– signal observed by LISPA-MS (), along with potential ozone (). The RCOO– signal, exhibiting the oxygenated organics observed by LISPA-MS, was defined as the sum of the peak areas of 17 negative-ion fragments (m/z 59, 73, 75, 87, 89 101, 103, 115, 117, 129, 131, 133, 143, 145, 147, 159, 161). A rough qualitative agreement can be observed between the temporal variations in the LISPA-MS RCOO– and the AMS m/z 44 signals rather than the AMS m/z 59 signals, although the AMS data were available mainly during afternoons. shows a correlation plot between LISPA-MS RCOO– and AMS m/z 44, indicating a linear correlation with R= 0.72. The LISPA-MS RCOO– signal showed daytime maxima, with the exception of the 14 August data, and such a diurnal behavior was in phase with the potential ozone, suggesting that the RCOO– signal provides a signature of the relative amount of oxidized organics in the particulate phase. Recently CitationTakegawa et al. (2007) reported that about 14% of the AMS m/z 44 signal on average originated from dicarboxylic and ω-oxocarboxylic acids in the summer of 2003 in Tokyo. CitationKawamura and Yasui (2005) reported that total aerosol concentrations of diacids, ketoacids and dicarbonyls in Tokyo showed a concentration maximum in daytime with a dynamic change in their molecular composition, based on the aerosol collections using a high-volume air sampler followed by gas-chromatography (GC) and GC/MS analysis. It seems likely that such observations are related to the diel variations observed in the RCOO– signal and the significant correlation between LISPA-MS RCOO– and AMS m/z 44. We suggest that detailed analysis of the LISPA-MS RCOO– data with appropriate laboratory calibration experiments would be useful for investigating temporal changes in molecular composition and mixing state of organics containing carboxyl group in aerosol particles in various locations. For example, a recent field study using SPMS detected that oxalic and malonic acids were internally mixed with mineral dust and aged sea salt particles in Asian outflow (CitationSullivan and Prather 2007).
FIG. 5 Temporal variations in total organics, m/z 44, and m/z 57, as measured by AMS (10-min interval), and the three-hour integrated signals of RCOO–, as measured by LISPA-MS, along with gaseous O3 and NO2 concentrations. The definition of LISPA-MS RCOO– is described in the text. Measurements of gas-phase ozone (Thermo Electron, 49C) and NO x (Ecophysics, CLD88yp) were performed at the same sampling site.
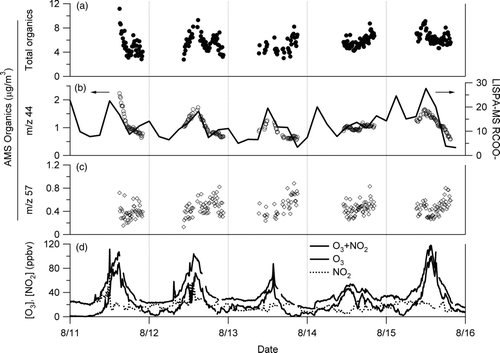
FIG. 6 Same as , for a correlation between LISPA-MS RCOO– and AMS m/z 44.
In some other SPMS studies, m/z+ 43 (CH3CO+ and/or C3H+ 7) in the positive mass spectra has been identified as a marker for oxidized organics both in the field and laboratory measurements (CitationDall’Osto et al. 2009b; CitationHuang et al. 2007; CitationLee et al. 2002; CitationMoffet et al. 2008; CitationMurphy et al. 2006; CitationNarukawa et al. 2008; CitationQin and Prather 2006). In the aircraft measurements of tropospheric aerosol, CitationMurphy et al. (2006) showed that the relative carbonaceous signal for SPMS correlated with both AMS organics and PILS (Particle Into Liquid Sampler) water-soluble organic carbon. We analyzed a relationship in the temporal variations between the LISPA-MS m/z+ 43 relative peak area and AMS m/z 44, and found a moderate linear correlation (R= 0.39). A relatively weaker correlation compared to the RCOO–/AMS m/z 44 plot might be explained by a possible overlap, at the same mass peak, of CH3CO+ and C3H+ 7 that can arise from aliphatic hydrocarbons. On the other hand, Dall’Osto et al. (2009a) reported that, during a fog event in London, high mass organic carbon signals in SPMS did not show any obvious correlation with AMS organic mass loading. Such high mass organic carbon signals were assumed to be humic-like substances that were considered refractory organic molecules. In general, a direct comparison between the data of SPMS and AMS is not straightforward because of major differences in the volatization/ionization method and detectable particle size biases. Nevertheless, this work present a new marker of LISPA-MS RCOO– for tracing relative amount of oxygenated organics as being broadly consistent with AMS m/z 44.
Finally, we will discuss briefly that the RCOO– marker showed second maxima on the midnight of 11–12 August, as shown in . As stated in section 2.1. this campaign was conducted from 28 July–29 August in which the LISPA-MS instrument worked properly, and nighttime peaks were sometimes observed in the LISPA-MS spectra. It would be intriguing to identify whether nighttime ozonolysis and NO3 reactions were responsible for such nighttime peaks. However, it is difficult to provide conclusive evidence for the assignment of RCOO– signal related to nighttime oxidation processes. Not only a careful investigation of the single particle mass spectra obtained in the field measurements but also laboratory analysis of aerosol particles generated from ozonolysis, NO3 reactions and OH reactions of volatile organic compounds will be essential to discuss the issue. Such laboratory studies using SPMS are now increasingly available (CitationGross et al. 2006; CitationNarukawa et al. 2007b, Citation2008, CitationSmith et al. 2002; CitationVaden et al. 2010; CitationYu et al. 2008; CitationZhang et al. 2010), but much still remains to be done.
4. CONCLUSIONS
Combining different types of on-line analysis instruments served a large body of self-consistent data for chemical characteristics of aerosol particles in the Tokyo metropolitan area during the summer of 2008. Temporal variations of the three-hour integrated relative peak areas of m/z 97 (HSO– 4), m/z
18 (NH+ 4) obtained from the single particle analysis using LISPA-MS were found to be analogous to sulfate and ammonium mass concentrations estimated from the ensemble measurements by AMS, whereas stoichiometric analysis of the AMS data could conclude that ammonium was in a form neutralized with sulfate for most of the measurement period. The AMS m/z 44 signals had a much closer correlation with the AMS nitrate signals, whereas the AMS m/z 57 signals did not. Also the AMS m/z 44 signals were well correlated with gas-phase potential ozone, whereas the AMS m/z 57 signals were not. The correlation analysis of the temporal variations in the LISPA-MS RCOO– signal defined as the sum of relative signal peak areas of the selected 17 negative-ion mass peaks and the AMS m/z 44 signals showed a significant linear correlation. Such a correlation will be reasonable considering that di-, mono-, and oxoacids consist mainly of both AMS m/z 44 signals (CitationZhang et al. 2005a) and LISPA-MS RCOO– signals. Our study suggests that the LISPA-MS RCOO– signal could be a measure of relative abundance of some oxygenated organics in ambient aerosol. Detailed analysis of the LISPA-MS RCOO– data with appropriate laboratory calibration experiments would be useful for investigating temporal changes in molecular composition of organics containing carboxyl group in aerosol particles in various locations. On a particle-by-particle basis, approximately 95% of the organic-containing particles contained nitrate, which was expected to be responsible, in part, for the correlation between m/z 44 and nitrate observed by AMS. Thus, combinatorial use of the data on a particle-by-particle basis and an ensemble basis can provide deeper insights into the analysis of chemical properties of ambient aerosol. Note that a very recent study by CitationCross et al. (2009) has achieved single particle characterization using a light scattering module coupled to AMS.
Acknowledgments
The authors would like to thank Eriko Nishimoto and Yusuke Yamane (Kyoto Univ.) for helpful discussion regarding the meteorological data. Financial support from the Asahi Glass Foundation, Program for Improvement of Research Environment for Young Researchers from Special Coordination Funds for Promoting Science and Technology (SCF) commissioned by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, Grants-in-Aid from the MEXT for Scientific Research on Innovative Areas Impacts of Aerosols in East Asia on Plants and Human Health, and Japan Society for the Promotion of Science, are acknowledged. The authors are grateful to anonymous referees for fruitful comments.
REFERENCES
- Alfarra , M. R. , Coe , H. , Allan , J. D. , Bower , K. N. , Boudries , H. , Canagaratna , M. R. , Jimenez , J. L. , Jayne , J. T. , Garforth , A. A. , Li , S. M. and Worsnop , D. R. 2004 . Characterization of Urban and Rural Organic Particulate in the Lower Fraser Valley Using Two Aerodyne Aerosol Mass Spectrometers . Atmos. Environ. , 38 : 5745 – 5758 .
- Allan , J. D. , Bower , K. N. , Coe , H. , Boudries , H. , Jayne , J. T. , Canagaratna , M. R. , Millet , D. B. , Goldstein , A. H. , Quinn , P. K. , Weber , R. J. and Worsnop , D. R. 2004 . Submicron Aerosol Composition at Trinidad Head, California, during ITCT 2K2: Its Relationship with Gas Phase Volatile Organic Carbon and Assessment of Instrument Performance . J. Geophys. Res.—Atmos. , 109 ( D23 ) doi:10.1029/2003JD004208
- Aiken , A. C. , Decarlo , P. F. , Kroll , J. H. , Worsnop , D. R. , Huffman , J. A. , Docherty , K. S. , Ulbrich , I. M. , Mohr , C. , Kimmel , J. R. , Sueper , D. , Sun , Y. , Zhang , Q. , Trimborn , A. , Northway , M. , Ziemann , P. J. , Canagaratna , M. R. , Onasch , T. B. , Alfarra , M. R. , Prevot , A. S.H. , Dommen , J. , Duplissy , J. , Metzger , A. , Baltensperger , U. and Jimenez , J. L. 2008 . O/C and OM/OC Ratios of Primary, Secondary, and Ambient Organic Aerosols with High-Resolution Time-of-Flight Aerosol Mass Spectrometry . Environ. Sci. Technol. , 42 : 4478 – 4485 .
- Aiken , A. C. , Salcedo , D. , Cubison , M. J. , Huffman , J. A. , DeCarlo , P. F. , Ulbrich , I. M. , Docherty , K. S. , Sueper , D. , Kimmel , J. R. , Worsnop , D. R. , Trimborn , A. , Northway , M. , Stone , E. A. , Schauer , J. J. , Volkamer , R. M. , Fortner , E. , de Foy , B. , Wang , J. , Laskin , A. , Shutthanandan , V. , Zheng , J. , Zhang , R. , Gaffney , J. , Marley , N. A. , Paredes-Miranda , G. , Arnott , W. P. , Molina , L. T. , Sosa , G. and Jimenez , J. L. 2009 . Mexico City Aerosol Analysis during MILAGRO Using High Resolution Aerosol Mass Spectrometry at the Urban Supersite (T0). Part 1: Fine Particle Composition and Organic Source Apportionment . Atmos. Chem. Phys. , 9 : 6633 – 6653 .
- Atkinson , R. , Baulch , D. L. , Cox , R. A. , Crowley , J. N. , Hampson , R. F. , Hynesm , R. G. , Jenkins , M. E. , Rossi , M. J. and Troe , J. 2006 . Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry. Volume II: Gas Phase Reactions of Organic Species . Atmos. Chem. Phys. , 6 : 3625 – 4055 .
- Boyce , S. D. and Hoffmann , M. R. 1984 . Kinetics and Mechanism of the Formation of Hydroxymethanesulfonic Acid at Low pH . J. Phys. Chem. , 88 : 4740 – 4746 .
- Broske , R. , Kleffmann , J. and Wiesen , P. 2003 . Heterogeneous Conversion of NO2 on Secondary Organic Aerosol Surfaces: A Possible Source of Nitrous Acid (HONO) in the Atmosphere . Atmos. Chem. Phys. , 3 : 469 – 474 .
- Bruns , E. A. , Perraud , V. , Zelenyuk , A. , Ezell , M. J. , Johnson , S. N. , Yu , Y. , Imre , D. , Finlayson-Pitts , B. J. and Alexander , M. L. 2010 . Comparison of FTIR and Particle Mass Spectrometry for the Measurement of Particulate Organi Nitrates . Envion. Sci. Technol. , 44 : 1056 – 1061 .
- Canagaratna , M. R. , Jayne , J. T. , Jimenez , J. L. , Allan , J. D. , Alfarra , M. R. , Zhang , Q. , Onasch , T. B. , Drewnick , F. , Coe , H. , Middlebrook , A. , Delia , A. , Williams , L. R. , Trimborn , A. M. , Northway , M. J. , DeCarlo , P. F. , Kolb , C. E. , Davidvits , P. and Worsnop , D. R. 2007 . Chemical and Microphysical Characterization of Ambient Aerosols with the Aerodyne Aerosol Mass Spectrometer . Mass Spectrom. Rev. , 26 : 185 – 222 .
- Cross , E. S. , Onasch , T. B. , Canagaratna , M. , Jayne , J. T. , Kimmel , J. , Yu , X. -Y. , Alexander , M. L. , Worsnop , D. R. and Davidovits , P. 2009 . Single Particle Characterization Using a Light Scattering Module Coupled to a Time-of-Flight Aerosol Mass Spectrometer . Atmos. Chem. Phys. , 9 : 7769 – 7793 .
- Cubison , M. J. , Ervens , B. , Feingold , G. , Docherty , K. S. , Ulbrich , I. M. , Shields , L. , Prather , K. , Hering , S. and Jimenez , J. L. 2008 . The Influence of Chemical Composition and Mixing State of Los Angeles Urban Aerosol on CCN Number and Cloud Properties . Atmos. Chem. Phys. , 8 : 5649 – 5667 .
- Dall’Osto , M. , Harrison , R. M. , Coe , H. and Williams , P. I. 2009a . Real-Time Secondary Aerosol Formation during a Fog Event in London . Atmos. Chem. Phys. , 9 : 2459 – 2469 .
- Dall’Osto , M. , Harrison , R. M. , Coe , H. , Williams , P. I. and Allan , J. D. 2009b . Real-Time Chemical Characterization of Local and Regional Nitrate Aerosols . Atmos. Chem. Phys. , 9 : 3709 – 3720 .
- DeCarlo , P. F. , Kimmel , J. R. , Trimborn , A. , Northway , M. J. , Jayne , J. T. , Aiken , A. C. , Gonin , M. , Fuhrer , K. , Horvath , T. , Docherty , K. S. , Worsnop , D. R. and Jimenez , J. L. 2006 . Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer . Anal. Chem. , 78 : 8281 – 8289 .
- Drewnick , F. , Dall’Osto , M. and Harrison , R. 2008 . Characterization of Aerosol Particles from Grass Mowing by Joint Deployment of ToF-AMS and ATOFMS Instruments . Atmos. Environ. , 42 : 3006 – 3017 .
- Drewnick , F. , Jayne , J. T. , Canagaratna , M. , Worsnop , D. R. and Demerjian , K. L. 2004 . Measurement of Ambient Aerosol Composition during the PMTACS-N . Part 2: Chemically Speciated Mass Distributions. Aerosol Sci. Technol. , 38 : 104 – 117 . 200, Using an Aerosol Mass Spectrometer
- Drewnick , F. , Hings , S. S. , DeCarlo , P. , Jayne , J. T. , Gonin , M. , Fuhrer , K. , Weimer , S. , Jimenez , J. L. , Demerjian , K. L. , Borrmann , S. and Worsnop , D. R. 2005 . A New Time-of-Flight Aerosol Mass Spectrometer (TOF-AMS)—Instrument Description and First Field Deployment . Aerosol Sci. Technol. , 39 : 637 – 658 .
- Drewnick , F. , Schneider , J. , Hings , S. S. , Hock , N. , Noone , K. , Targino , A. , Weimer , S. and Borrmann , S. 2007 . Measurement of Ambient, Interstitial, and Residual Aerosol Particles on a Mountaintop Site in Central Sweden using an Aerosol Mass Spectrometer and a CVI . J. Atmos. Chem. , 56 : 1 – 20 .
- Garnes , L. A. and Allen , D. T. 2002 . Size Distributions of Organonitrates in Ambient Aerosol Collected in Houston, Texas . Aerosol Sci. Technol. , 36 : 983 – 992 .
- Gross , D. S. , Gälli , M. E. , Kalberer , M. , Prevot , A. S.H. , Dommen , J. , Alfarra , M. R. , Duplissy , J. , Gaeggeler , K. , Gascho , A. , Metzger , A. and Baltensperger , U. 2006 . Real-Time Measurement of Oligomeric Species in Secondary Organic Aerosol with the Aerosol Time-of-Flight Mass Spectrometer . Anal. Chem. , 78 : 2130 – 2137 .
- Herndon , S. C. , Onasch , T. B. , Wood , E. C. , Kroll , J. H. , Canagaratna , M. R. , Jayne , J. T. , Zavala , M. A. , Knighton , W. B. , Mazzoleni , C. , Dubey , M. K. , Ulbrich , I. M. , Jimenez , J. L. , Seila , R. , Gouw , J. A.D. , Foy , B. D. , Fast , J. , Molina , L. T. , Kolb , C. E. and Worsnop , D. R. 2008 . Correlation of Secondary Organic Aerosol with Odd Oxygen in Mexico City . Geophys. Res. Lett. , 35 ( L15804 ) doi:10.1029/2008GL034058
- Huang , M. , Zhang , W. , Hao , L. , Wang , Z. , Zhao , W. , Gu , X. , Guo , X. , Liu , X. , Long , B. and Fang , L. 2007 . Laser Desorption/Ionization Mass Spectrometric Study of Secondary Organic Aerosol Formed from the Photooxidation of Aromatics . J. Atmos. Chem. , 58 : 237 – 252 .
- Jimenez , J. L. , Jayne , J. T. , Shi , Q. , Kolb , C. E. , Worsnop , D. R. , Yourshaw , I. , Seinfeld , J. H. , Flagan , R. C. , Zhang , X. F. , Smith , K. A. , Morris , J. W. and Davidovits , P. 2003 . Ambient Aerosol Sampling Using the Aerodyne Aerosol Mass Spectrometer . J. Geophys. Res.—Atmos. , 108 ( D7 ) : 8425 doi:10.1029/2001JD001213
- Jimenez , J. L. 2009 . Evolution of Organic Aerosols in the Atmosphere . Science. , 326 : 1525 – 1529 . (64 authors)
- Jordan , C. E. , Ziemann , P. J. , Griffin , R. J. , Lim , Y. B. , Atkinson , R. and Arey , J. 2008 . Modeling SOA Formation from OH Reactions with C8-C17 n-Alkanes . Atmos. Environ. , 42 : 8015 – 8026 .
- Kawamura , K. and Yasui , O. 2005 . Diurnal Changes in the Distribution of Dicarboxylic Acids, Ketocarboxylic Acids and Dicarbonyls in the Urban Tokyo Atmosphere . Atmos. Environ. , 39 : 1945 – 1960 .
- Kanakidou , M. , Seinfeld , J. H. , Pandis , S. N. , Barnes , I. , Dentener , F. J. , Facchini , M. C. , Dingenen , R. V. , Ervens , B. , Nenes , A. , Nielsen , C. J. , Swietlicki , E. , Putaud , J. P. , Balkanski , Y. , Fuzzi , S. , Horth , J. , Moortgat , G. K. , Winterhalter , R. , Myhre , C. E.L. , Tsigaridis , K. , Vignati , E. , Stephanou , E. G. and Wilson , J. 2005 . Organic Aerosol and Global Climate Modelling: A Review . Atmos. Chem. Phys. , 5 : 1053 – 1123 .
- Kondo , Y. , Morino , Y. , Fukuda , M. , Kanaya , Y. , Miyazaki , Y. , Takegawa , N. , Tanimoto , H. , McKenzie , R. , Johnston , P. , Blake , D. R. , Murayama , T. and Koike , M. 2008 . Formation and Transport of Oxidized Reactive Nitrogen, Ozone, and Secondary Organic Aerosol in Tokyo . J. Geophys. Res. Atomos. , 113 doi:10.1029/2008JD010134
- Lee , S. -H. , Murphy , D. M. , Thomson , D. S. and Middlebrook , A. M. 2002 . Chemical Components of Single Particles Measured with Particle Analysis by Laser Mass Spectrometry (PALMS) during the Atlanta SuperSite Projects: Focus on Organic/Sulfate, Lead, Soot, and Mineral Particles . J. Geophys. Res.—Atmos. , 107 ( D1 ) doi:10.1029/2000JD000011
- Lee , S.-H. , Murphy , D. M. , Thomson , D. S. and Middlebrook , A. M. 2003 . Nitrate and Oxidized Organic Ions in Single Particle Mass Spectra during the 1999 Atlanta Supersite Project . J. Geophys. Res. -Atmos. , 108 ( D7 ) doi:10.1029/2001JD001455
- Liu , P. S.K. , Deng , R. , Smith , K. A. , Williams , L. R. , Jayne , J. T. , Canagaratna , M. R. , Moore , K. , Onasch , T. B. , Worsnop , D. R. and Deshler , T. 2007 . Transmission Efficiency of an Aerodynamic Focusing Lens System: Comparison of Model Calculations and Laboratory Measurements for the Aerodyne Aerosol Mass Spectrometer . Aerosol Sci. Technol. , 41 : 721 – 733 .
- Matsui , H. , Koike , M. , Takegawam , N. , Kondo , Y. , Griffin , R. J. , Miyazaki , Y. , Yokouchi , Y. and Ohara , T. 2009 . Secondary Organic Aerosol Formation in Urban Air: Temporal Variations and Possible Contributions from Unidentified Hydrocarbons . J. Geophys. Res.—Atmos. , 114 doi:10.1029/2008JD010164
- Matsumoto , J. , Takahashi , K. , Matsumi , Y. , Yabushita , A. , Shimizu , A. , Matsui , I. and Sugimoto , N. 2006 . Scavenging of Pollutant Acid Substances by Asian Mineral Dust Particles . Geophys. Res. Lett. , 33 ( L07816 ) doi:10.1029/2006GL025782
- Matthew , B. M. , Middlebrook , A. M. and Onasch , T. B. 2008 . Collection Efficiencies in an Aerodyne Aerosol Mass Spectrometer as a Function of Particle Phase for Laboratory Generated Aerosols . Aerosol Sci. Technol. , 42 : 884 – 898 .
- Middlebrook , A. M. , Murphy , D. M. , Lee , S.-H. , Thomson , D. S. , Prather , K. A. , Wenzel , R. J. , Liu , D.-Y. , Phares , D. J. , Jayne , J. T. , Worsnop , D. R. , Yourshaw , I. , Seinfeld , J. H. and Flaga , R. C. 2003 . A Comparison of Particle Mass Spectrometers during the 1999 Atlanta Supersite Project . J. Geophys. Res.—Atmos. , 108 ( D7 ) : 8424 doi10.1029/2001JD000660
- Miyakawa , T. , Takegawa , N. and Kondo , Y. 2007 . Removal of Sulfur Dioxide and Formation of Sulfate Aerosol in Tokyo . J. Geophys. Res.—Atmos. , 112 ( D13209 ) doi:10.1029/2006JD007896
- Moffet , R. C. , de Foy , B. , Molina , L. T. , Molina , M. J. and Prather , K. A. 2008 . Measurement of Ambient Aerosols in Northern Mexico City by Single Particle Mass Spectrometry . Atmos. Chem. Phys. , 8 : 4499 – 4516 .
- Mohr , C. , Huffman , J. A. , Cubison , M. J. , Aiken , A. C. , Docherty , K. S. , Kimmel , J. R. , Ulbrich , I. M. , Hannigan , M. and Jimenez , J. L. 2009 . Characterization of Primary Organic Aerosol Emissions from Meat Cooking, Trash Burning, and Motor Vehicles with High-Resolution Aerosol Mass Spectrometry and Comparison with Ambient and Chamber Observations . Environ. Sci. Technol. , 43 : 2443 – 2449 .
- Morino , Y. , Kondo , Y. , Takegawa , N. , Miyazaki , Y. , Kita , K. , Komazaki , Y. , Fukuda , M. , Miyakawa , T. , Moteki , N. and Worsnop , D. R. 2006 . Partitioning of HNO3 and Particulate Nitrate over Tokyo: Effect of Vertical Mixing . J. Geophys. Res.—Atmos. , 111 ( D15215 ) doi:10.1029/2005JD006887
- Murphy , D. M. 2007 . The Design of Single Particle Laser Mass Spectrometers . Mass Spectrom. Rev. , 26 : 150 – 165 .
- Murphy , D. M. , Cziczo , D. J. , Froyd , K. D. , Hudson , P. K. , Matthew , B. M. , Middlebrook , A. M. , Peltier , R. E. , Sullivan , A. , Thomson , D. S. and Weber , R. J. 2006 . Single-Particle Mass Spectrometry of Tropospheric Aerosol Particles . J. Geophys. Res.—Atmos. , 111 doi:10.1029/2006JD007340
- Nakayama , T. , Hagino , R. , Matsumi , Y. , Sakamoto , Y. , Kawasaki , M. , Yamazaki , A. , Uchiyama , A. , Kudo , R. , Moteki , N. and Kondo , Y. 2010 . Measurements of Aerosol Optical Properties in Central Tokyo during Summertime Using Cavity Ring-Down Spectroscopy: Comparison with Conventional Techniques . Atmos. Environ. , 44 : 3034 – 3042 .
- Narukawa , M. , Matsumi , Y. , Takahashi , K. and Yabushita , Y. 2007a . Measurements of Ammonium and Sodium Salt Aerosol Particles Using a Laser Ionization Single-Particle Aerosol Mass Spectrometer . Chem Lett. , 36 : 904 – 905 .
- Narukawa , M. , Matsumi , Y. , Matsumoto , J. , Takahashi , K. , Yabushita , A. , Sato , K. and Imamura , T. 2007b . Real-Time Analysis of Secondary Organic Aerosol Particles Formed from Cyclohexene Ozonolysis Using a Laser Ionization Single-Particle Aerosol Mass Spectrometer . Anal. Sci. , 23 : 507 – 512 .
- Narukawa , M. , Matsumi , Y. , Matsumoto , J. , Takahashi , K. , Yabushita , A. , Sato , K. and Imamura , T. 2008 . Single Particle Analysis of Secondary Organics Aerosols Formed from 1,4-Cyclohexadiene Ozonolysis Using a Laser Ionization Single-Particle Aerosol Mass Spectrometer . Bull. Chem. Soc. Jpn. , 81 : 120 – 126 .
- Noble , C. A. and Prather , K. A. 2000 . Real-Time Single Particle Mass Spectrometry: A Historical Review of a Quarter Century of the Chemical Analysis of Aerosols . Mass Spectrom. Rev. , 19 : 248 – 274 .
- Pratt , K. A. and Prather , K. A. 2010 . Aircraft Measurements of Vertical Profiels of Aerosol Mixing States . J. Geophys. Res. , 115 ( D11305 ) doi:10.1029/2009JD013150
- Qin , X. and Prather , K. A. 2006 . Impact of Biomass Emissions on Particle Chemistry during the California Regional Particulate Air Quality Study . Int. J. Mass Spectrom , 258 : 142 – 150 .
- Reinard , M. S. and Johnston , M. V. 2008 . Ion Formation Mechanism in Laser Desorption Ionization of Individual Nanoparticles . J. Am. Soc. Mass Spectrom. , 19 : 389 – 399 .
- Salcedo , D. , Onasch , T. B. , Aiken , A. C. , Williams , L. R. , de Foy , B. , Cubison , M. J. , Worsnop , D. R. , Molina , L. T. and Jimenez , J. L. 2010 . Determination of Particulate Lead Using Aerosol Mass Spectrometry: MILAGRO/MCMA-2006 Observations . Atmos. Chem. Phys. , 10 : 5371 – 5389 .
- Salcedo , D. , Onasch , T. B. , Canagaratna , M. R. , Dzepina , K. , Huffman , J. A. , Jayne , J. T. , Worsnop , D. R. , Kolb , C. E. , Weimer , S. , Drewnick , F. , Allan , J. D. , Delia , A. E. and Jimenez , J. L. 2007 . Technical Note: Use of a Beam Width Probe in an Aerosol Mass Spectrometer to Monitor Particle Collection Efficiency in the Field . Atmos. Chem. Phys. , 7 : 549 – 556 .
- Sato , K. , Takami , A. , Isozaki , T. , Hikida , T. , Shimono , A. and Imamura , T. 2010 . Mass Spectrometric Study of Secondary Organic Aerosol Formed from the Photo-Oxidation of Aromatic Hyrdocarbons . Atmos. Environ. , 44 : 1080 – 1087 .
- Schneider , J. , Borrmann , S. , Wollny , A. G. , Blasner , M. , Mihalopoulos , N. , Oikonomou , K. , Sciare , J. , Teller , A. , Levin , Z. and Worsnop , D. R. 2004 . Online Mass Spectrometric Aerosol Measurements during the MINOS Campaign (Crete, August 2001) . Atmos. Chem. Phys. , 4 : 65 – 80 .
- Seinfeld , J. H. and Pandis , S. N. 2006 . Atmospheric Chemistry and Physics: From Air Pollution to Climate Change , 2nd , 1 – 1203 . New York : John Wiley & Sons .
- Smith , G. D. , Woods , E. III , DeForest , C. L. , Baer , T. and Miller , R. E. 2002 . Reactive Uptake of Ozone by Oleic Acid Aerosol Particles: Application of Single-Particle Mass Spectrometry to Heterogeneous Reaction Kinetics . J. Phys. Chem. , A106 : 8085 – 8095 .
- Stemmler , K. , Ammann , M. , Donders , C. , Kleffmann , J. R. and George , C. 2006 . Photosensitized Reduction of Nitrogen Dioxide on Humic Acid as a Source of Nitrous Acid . Nature , 440 : 95 – 198 .
- Suess , D. T. and Prather , K. A. 1999 . Mass Spectrometry of Aerosols . Chem. Rev. , 99 : 3007 – 3036 .
- Sullivan , R. C. and Prather , K. A. 2005 . Recent Advances in Our Understanding of Atmospheric Chemistry and Climate Made Possible by On-Line Aerosol Analysis Instrumentation . Anal. Chem. , 77 : 3861 – 3885 .
- Sullivan , R. C. and Prather , K. A. 2007 . Investigations of the Diurnal Cycle and Mixing State of Oxalic Acid in Individual Particles in Asian Aerosol Outflow . Environ. Sci. Technol. , 41 : 8062 – 8069 .
- Sullivanm , R. C. , Guazzotti , S. A. , Sodeman , D. A. and Prather , K. A. 2007 . Direct Observations of the Atmospheric Processing of Asian Mineral Dust . Atmos. Chem. Phys. , 7 : 1213 – 1226 .
- Takami , A. , Miyoshi , T. , Shimono , A. and Hatakeyama , S. 2005 . Chemical Composition of Fine Aerosol Measured by AMS at Fukue Island, Japan during APEX Period . Atmos. Environ , 39 : 4913 – 4924 .
- Takegawa , N. , Miyazaki , Y. , Kondo , Y. , Komazaki , Y. , Miyakawa , T. , Jimenez , J. L. , Jayne , J. T. , Worsnop , D. R. , Allan , J. D. and Weber , R. J. 2005 . Characterization of an Aerodyne Aerosol Mass Spectrometer (AMS): Intercomparison with Other Aerosol Instruments . Aerosol Sci. Technol. , 39 : 760 – 770 .
- Takegawa , N. , Miyakawa , T. , Kondo , Y. , Jimenez , J. L. , Zhang , Q. , Worsnop , D. R. and Fukuda , M. 2006 . Seasonal and Diurnal Variations of Submicron Organic Aerosol in Tokyo Observed Using the Aerodyne Aerosol Mass Spectrometer . J. Geophys. Res.—Atmos. , 111 ( D11206 ) doi:10.1029/2005JD006515
- Takegawa , N. , Miyakawa , T. , Kawamura , K. and Kondo , Y. 2007 . Contribution of Selected Dicarboxylic and ω-Oxocarboxylic Acids in Ambient Aerosol to the m/z 44 Signal of an Aerodyne Aerosol Mass Spectrometer . Aerosol Sci. Technol. , 41 : 418 – 437 .
- Thornton , J. A. , Braban , C. F. and Abbatt , J. P. D . 2003 . N2O5 Hydrolysis on Sub-Micron Organic Aerosols: The Effect of Relative Humidity, Particle Phase, and Particle Size . Phys. Chem. Chem. Phys. , 5 : 4593 – 4603 .
- Tonokura , K. , Kanno , N. , Yamamoto , Y. and Yamada , H. 2010 . Development of a Compact Laser-Based Single Photon Ionization Time-of-Flight Mass Spectrometer . Int. J. Mass. Spectrom. , 290 : 9 – 13 .
- Vaden , T. D. , Song , C. , Zaveri , R. A. , Imre , D. and Zelenyuk , A. 2010 . Morphology of Mixed Primary and Secondary Organic Particles and the Adsorption of Spectator Organic Gases during Aerosol Formation . PNAS. , 107 : 6658 – 6663 .
- Wang , X. F. , Zhang , Y. , Chen , H. , Yang , X. and Chen , J. 2009 . Particulate Nitrate Formation in a Highly Polluted Urban Area: A Case Study by Single-Particle Mass Spectrometry in Shanghai . Environ. Sci. Technol. , 43 : 3061 – 3066 .
- Weimer , S. , Drewnick , F. , Hogrefe , O. , Schwab , J. J. , Rhoads , K. , Orsini , D. , Canagaratna , M. , Worsnop , D. R. and Demerjian , K. L. 2006 . Size-Selective Nonrefractory Ambient Aerosol Measurements during the Particulate Matter Technology Assessment and Characterization Study—New York 2004 Winter Intensive in New York City . J. Geophys. Res.—Atmos. , 111 doi:10.1029/2006JD007215
- Whiteaker , J. R. and Prather , K. A. 2003 . Hydroxymethanesulfonate as a Tracer for Fog Processing of Individual Aerosol Particles . Atmos. Environ. , 37 : 1033 – 1043 .
- Wood , E. C. , Canagratna , M. R. , Herndon , S. C. , Kroll , J. H. , Onasch , T. B. , Kolb , C. E. , Worsnop , D. R. , Knighton , W. B. , Selia , R. , Zavala , M. , Molina , L. T. , DeCarlo , P. F. , Jimenez , J. L. , Weinheimer , A. J. , Knapp , D. J. , Jobson , B. T. , Stutz , J. , Kuster , W. C. and Williams , E. J. 2010 . Investigation of the Correlation between Odd Oxygen and Secondary Organic Aerosol in Mexico City and Huston . Atmos. Chem. Phys. Discuss. , 10 : 3547 – 3604 .
- Yabushita , A. , Enami , S. , Sakamoto , Y. , Kawasaki , M. , Hoffmann , M. R. and Colussi , A. J. 2009 . Anion-Catalyzed Dissolution of NO2 on Aqueous Microdroplets . J. Phys. Chem. , A113 : 4844 – 4848 .
- Yu , Y. , Ezell , M. J. , Zelenyuk , A. , Imre , D. , Alexander , L. , Ortega , J. , D’Anna , B. , Harmon , C. W. , Johnson , S. N. and Finlayson-Pitts , B. J. 2008 . Photooxidation of α-Pinene at High Relative Humidity in the Presence of Increasing Concentrations of NO x . Atmos. Environ. , 42 : 5044 – 5060 .
- Zhang , Q. , Alfarra , M. R. , Worsnop , D. R. , Allan , J. D. , Coe , H. , Canagaratna , M. R. and Jimenez , J. L. 2005a . Deconvolution and Quantification of Hydrocarbon-Like and Oxygenated Organic Aerosols Based on Aerosol Mass Spectrometry . Environ. Sci. Technol. , 39 : 4938 – 4952 .
- Zhang , Q. , Canagaratna , M. R. , Jayne , J. T. , Worsnop , D. R. and Jimenez , J. L. 2005b . Time- and Size-Resolved Chemical Composition of Submicron Particles in Pittsburgh: Implications for Aerosol Sources and Processes . J. Geophys. Res.—Atmos. , 110 ( D07S09 ) doi:10.1029/2004JD004649
- Zhang , Q. , Jimenez , J. L. , Canagaratna , M. R. , Allan , J. D. , Coe , H. , Ulbrich , I. , Alfarra , M. R. , Takami , A. , Middlebrook , A. M. , Sun , Y. L. , Dzepina , K. , Dunlea , E. , Docherty , K. , DeCarlo , P. F. , Salcedo , D. , Onasch , T. , Jayne , J. T. , Miyoshi , T. , Shimono , A. , Hatakeyama , S. , Takegawa , N. , Kondo , Y. , Schneider , J. , Drewnick , F. , Borrmann , S. , Weimer , S. , Demerjian , K. , Williams , P. , Bower , K. , Bahreini , R. , Cottrell , L. , Griffin , R. J. , Rautiainen , J. , Sun , J. Y. , Zhang , Y. M. and Worsnop , D. R. 2007 . Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenically Influenced Northern Hemisphere Midlatitudes . Geophys. Res. Lett. , 34 ( L13801 ) doi:10.1029/2007GL0 29979
- Zhang , Y. , Yang , B. , Meng , J. , Gao , S. , Dong , X. and Shu , J. 2010 . Homogeneous and Heterogeneous Reactions of Phenanthrene with Ozone . Atmos. Environ. , 44 : 697 – 702 .