Abstract
The organic fraction is a major constituent of fine atmospheric particulate matter, though its chemical composition is complex and not well understood. This complexity presents an extreme analytical challenge and is well suited to analysis by comprehensive two-dimensional gas chromatography (GC × GC). This has recently been coupled to a thermal desorption aerosol gas (TAG) chromatograph instrument to provide improved in-situ, hourly measurement of speciated organic compounds in atmospheric aerosols (2D-TAG). The original 2D-TAG instrument utilized a flame ionization detector, dual-stage modulator, and a second-dimension column that proved to be thermally unstable under optimal analysis conditions, which limited compound identification, instrument robustness, and time resolution. In this paper, we address these shortcomings by demonstrating the successful integration of a time-of-flight mass spectrometer (TOFMS), development of a simplified hybrid thermo-pneumatic modulator, and incorporation of a more thermally stable secondary column. These improvements resulted in an instrument capable of providing detailed speciated information of organic tracer compounds in atmospheric aerosols in near real time.
INTRODUCTION
Atmospheric aerosols impact visibility, directly and indirectly affect the global radiation budget, and are detrimental to human health (Charlson et al. Citation1992; Schwartz et al. Citation1996; Ramanathan et al. Citation2001). The organic fraction is a major constituent of airborne particles, comprising 20–90% of the mass of PM2.5, particles with aerodynamic diameters smaller than 2.5 μm (Kanakidou et al. Citation2005; Zhang et al. Citation2007). The chemical composition of organic aerosols is complex and not well understood. Identifying the chemical composition of atmospheric particulate matter is critical for tracing sources, elucidating formation and transformation processes, and assessing its effects on global climate.
Until recently, identification and quantification of specific organic compounds in aerosols involved integrated sample collection by filtration or impaction followed by off-line extraction and analysis by gas or liquid chromatography (Schauer et al. Citation1996; Gao et al. Citation2006; Surratt et al. Citation2008; Sheesley et al. Citation2010; Offenberg et al. Citation2011). Our approach has been to combine collection by particle impaction with analysis by thermal desorption and gas chromatography/mass spectrometry (GC/MS) (Williams et al. Citation2006). Our instrument, the thermal desorption aerosol gas (TAG) chromatograph, provides hourly quantitative speciation of the organic fraction of atmospheric aerosols by collecting and thermally desorbing ambient aerosol samples onto a GC column for immediate analysis. The sample collection is done concurrently with the analysis of the prior sample to provide hourly measurements. The TAG instrument has, to date, provided weeks of hourly information on hundreds of individual organic compounds in ambient aerosols in both urban and remote regions (Williams et al. Citation2007, Citation2010; Worton et al. Citation2011).
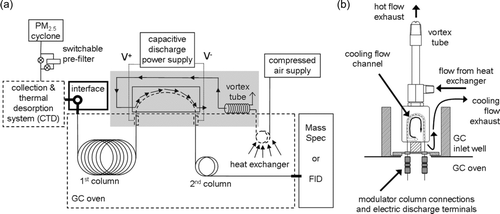
FIG. 1 (a) Components of the 2D-TAG system (originally described in Goldstein et al. Citation2008), including the thermal modulation system illustrating the use of a vortex cooler to provide forced air cooling for the single-stage pressure pulse modulator. Bold elements remain heated and dashed elements are thermally cycled during collection–desorption–analysis cycling. A heat exchanger inside the oven creates a differentially heated air stream that passively tracks the oven temperature. Components inside the shaded gray box are shown in more detail in (b).
However, even with the high resolution of GC, there are still peak co-elutions, interfering matrix constituents, and a large band of material that elutes as part of an unresolved complex mixture (UCM), which complicates identification and quantification of individual compounds. Even spectral deconvolution software has been shown to fail when co-eluting peaks have common mass fragments or when a “constant” background is present (Schnelle-Kreis et al. Citation2005). Comprehensive two-dimensional GC (GC × GC), first introduced by Liu and Phillips (Citation1991), utilizes two chromatographic columns connected in series by a modulator to produce higher peak capacity and greater selectivity than single-column GC, allowing it to overcome some of the above limitations.
The higher chromatographic resolution and peak capacity of GC × GC results from employing two columns coated with stationary phases as different from one another as possible (so called “orthogonal phases”) (Liu et al. Citation1995). Most commonly, GC × GC is designed to provide a slow volatility separation (primary column) followed by a fast polarity separation (secondary column). The modulator collects analytes eluting from the primary column and injects them into the second column at regular intervals (e.g., every 5 s). The secondary column yields individual short chromatograms for each modulated sample eluting from the primary column. The end result is a technique capable of separating and quantifying a significantly larger number of organic compounds than single-column GC. GC × GC has the further advantage that the primary column bleed is not retained on the secondary column and appears as a narrow band well separated from small peaks of more polar organic compounds.
Since its inception, GC × GC has been applied to the analysis of a variety of demanding and complex environmental samples, such as petrochemicals (Gaines et al. Citation1999), soils and sediments (Frysinger et al. Citation2003; Ventura et al. Citation2008), food (Adahchour et al. Citation2003) and atmospheric volatile organic compounds (Lewis et al. Citation2000; Xu et al. Citation2003). The complexity of ambient aerosol provides many analytical challenges that are well suited to analysis by GC × GC. In particular, GC × GC overcomes the common co-elution problem of peaks with similar volatility but different functionalities, e.g., with a polar second column, alkanes are clearly separated from aromatics, ketones and acids due to differences in their secondary retention times.
Previous work using thermal desorption coupled to GC × GC for offline analysis of ambient air filter samples has shown that this method can separate several thousand individual compounds present in ambient aerosols (Kallio et al. Citation2003; Welthagen et al. Citation2003; Hamilton et al. Citation2004; Schnelle-Kreis et al. Citation2005; Ochiai et al. Citation2007; Vogt et al. Citation2007; Ho et al. Citation2008). Welthagen et al. (Citation2003) and Vogt et al. (Citation2007) also suggested approaches to separate observed species into compound classes based on GC × GC retention times and organic compound fragmentation patterns that potentially provide a useful data classification for environmental and epidemiological studies. For more details of the types of compounds observed and the methods employed, the reader is directed to a recent review of the application of GC × GC to studying atmospheric organics by Hamilton (Citation2010).
In this paper, we report improvements to our original 2D-TAG instrument (Goldstein et al. Citation2008), most notably the development of a robust single-stage hybrid thermo-pneumatic modulator interface and the integration of a time-of-flight mass spectrometer (TOFMS). These improvements led to an in-situ instrument with hourly time resolution capable of identifying and quantifying a larger number of individual organic compounds in ambient aerosols.
INSTRUMENT IMPROVEMENTS
System Description and Methodology
The 2D-TAG system is shown schematically in . In brief, atmospheric particles are humidified and deposited into the collection and thermal desorption cell (CTD) by inertial impaction, followed by thermal desorption onto a GC column with subsequent detection by MS or flame ionization detection (FID). An Agilent 6890 GC was used to house both columns and constant column flow methods were used with helium at normal rates of 1.5 mL/min. The trapping capillary was mounted in a custom-built housing located outside the GC oven in a free injection port well (). The custom-built housing was machined out of glass-filled polytetrafluoroethylene (PTFE) with a narrow 3-mm2 channel, where the trapping capillary was located and through which the cooling air was directed across the length of the trapping capillary. Major advances reported here include: (1) optimization of the columns set through the use of a more thermally stable secondary column, (2) a novel modulator design enabling trapping and desorbing of analytes in a single stage with no loss of resolution compared with a dual-stage modulator, and (3) incorporation of a TOFMS to detect and identify a broader array of analytes. In this paper, we have chosen to go into more detail than might be necessary for analytical chemists because this paper is targeting an aerosol chemistry audience that is probably less familiar with the methods involved.
System improvements were evaluated through liquid standards deposited directly by syringe injection into the CTD through a customized port. These were allowed to dry under a constant flow of helium and were thermally desorbed onto the column using the same procedures as for ambient aerosol analyses (Kreisberg et al. Citation2009). Injections were either done manually (Kreisberg et al. Citation2009) or automatically using a custom-built injection system, described in detail by Isaacman et al. (Citation2011a). Five test standards, typical of the volatility range observable by thermal desorption GC for organic species in ambient aerosols, were used: a three n-alkane mix (hexadecane, eicosane, and tetracosane; Sigma-Aldrich), diesel fuel from a local gas station, an n-alkane (C8–C40) windowing standard (Accustandard Inc.), and two custom made 76- and 110-component mixtures of n-alkanes (Accustandard Inc.), polycyclic aromatic hydrocarbons (PAH) (Accustandard Inc.), oxygenated PAHs (Sigma-Aldrich, Alfa Aesar), carboxylic acids (Supelco), hopanes, steranes (NIST), and ketones (Supelco) prepared in our laboratory. Various configurations of the modulator and column sets were evaluated with principal focus on the semi-volatile range of compounds found in ambient aerosols. The primary column was the same in all cases.
Chromatographic Column Selection
There are several important factors in GC × GC that need to be considered when selecting the primary and secondary columns: namely column diameters and stationary phases. Narrower-bore second-dimension columns are routinely used for GC × GC as they are expected to give the highest chromatographic efficiency. However, because carrier gas velocities are strongly dependent on the internal diameter of the column, the greater the difference between the diameter of the primary and that of the secondary column, the larger the difference between their flow velocities. This usually results in a non-optimal flow velocity in the secondary column even for low velocities in the primary column. Typically, this effect can be tolerated due to the broad minimum of the Van Deemter curves for helium with these narrow-bore columns (McNair and Miller Citation2009). Nevertheless, as a result of zonal compression in the modulator, major analyte bands in narrow-bore columns can be large enough to exceed the capacity in the secondary column, which results in column overloading that affects peak shapes and overall chromatographic resolution. Harynuk et al. (Citation2005; Harynuk and Gorecki Citation2007) showed that column overloading was greatly reduced when using larger-bore secondary columns (≥ 0.25 mm inner diameter [ID]) relative to narrower ones (≤ 0.18 mm ID), while chromatographic performance was comparable. Based on these considerations, the primary and secondary columns were selected with matching inner diameters of 0.25 mm. This is important for minimizing column overloading during analysis of organics in atmospheric aerosols due to the very large range and variability in the abundances of observable compounds.
A critical consideration in this work was a high thermal tolerance for the stationary phases of both columns due to the high oven temperatures needed for elution of the semi- and non-volatile organic compounds present in atmospheric aerosols. Goldstein et al. (Citation2008) used a Varian Factor Four VF-5ms (30 m × 0.25 mm, 1 μm) as the primary column and an SGE Solgel Wax (1.5 m × 0.25 mm, 0.25 μm) as the secondary column. The Solgel Wax column exhibited minimal bleed at a maximum temperature of 280°C. This thermal limit led to longer analysis times (greater than four hours to elute tetracontane, C40H82) and significant band broadening in both dimensions as a result of an extended isothermal period at the end of each run (Panić et al. Citation2011). The isothermal period at 280°C led to an undesirable upward drift in the secondary retention times of the n-alkanes larger than C30. The shift in retention time for non-polar compounds compresses the secondary-dimension space, causing many analytes eluting during this isothermal period to have secondary retention times longer than the modulation period (a phenomenon commonly called “wrap-around”), which complicates chromatogram analysis and is thus undesirable. Two thermally stable intermediate polarity phases (RTX-200, Restek Corporation and BPX-50, SGE Analytical Science) were evaluated for their suitability as replacements for the Solgel Wax column.
Requirements for the Ideal Modulator for In-Situ Measurements
The key to any GC × GC system is the modulator located between the two chromatographic columns (Adahchour et al. Citation2006; Gorecki et al. Citation2006). The modulator traps material eluting from the first column and then rapidly injects it onto the second column, providing additional separation for co-eluting peaks after they exit the first column. The ideal modulator is one that completely traps compounds eluting from the first column until the modulator is triggered without significantly impeding carrier gas flow. Once triggered, the ideal modulator would instantly release the trapped compounds onto the second column and then just as instantly would return to the state of trapping. The efficiency of trapping and the quickness of the release determine the sharpness of the introduction of compounds onto the second column and the quickness of return to efficient trapping determines the amount of analyte breakthrough during the trigger step; ideally there should be no breakthrough.
Peak widths observed at the detector depend largely on the modulator performance along with diffusional broadening in the second column. As a result, the main criterion for evaluating modulator performance was the full width at half maximum height (FWHM) of the detected peaks. The modulator performance can be directly measured by replacing the second column with non-retaining guard column (Libardoni et al. Citation2005). However, weakly retained compounds, such as n-alkanes, exhibit minimal diffusional broadening in the second column and therefore offer a good approximation to peak widths exiting the second column.
The width of the peaks directly affects the peak capacity of that dimension, i.e., the narrower the peaks, the higher the capacity. Peak capacity of the second dimension in GC × GC is essentially the number of peaks that, when placed side by side, with a given minimum resolution add up to the modulation period. The ideal FWHM for maximum capacity of the second dimension depends on the data acquisition rate. The narrower the peaks, the higher the required acquisition rate of the detector in order to adequately define the peak shapes. Ideally, at least 10 data points should be acquired across a peak to properly define it (Marriott and Kinghorn 1998). A further constraint for a semi-continuous field instrument is the tradeoff between the sensitivity of the TOFMS and the data acquisition rate. Faster data acquisition rates result in lower sensitivity as fewer spectra are averaged. In laboratory analysis, this is easily compensated for by increasing the analyte loading–a solution that could only be replicated in an in-situ field instrument with longer collection times, and thus lower temporal resolution.
The ideal peak capacity is therefore defined by the realistic acquisition rate of the TOFMS. At 50 Hz–a fast but achievable TOFMS acquisition rate–a peak must have a base width of 200 ms to be properly defined. This corresponds to an FWHM of approximately 120 ms (FWHM = base width × 2.354/4) (McNair and Miller Citation2009). For a 5-s modulation period and complete baseline separation (distance between peak maxima/base width = 1.5), the ideal peak capacity of the second dimension is 16. This peak capacity is usually adequate to resolve the various potential functional group classes observable by thermal desorption GC for organic species in ambient aerosols using the column set employed in this work.
Single-Stage Modulation
Liu and Phillips (Citation1991) originally demonstrated that dual-stage modulation was necessary in order to avoid analyte breakthrough. Earlier, resistive heater-based interfaces, including dual-stage resistively heated metal capillaries, were shown to be inferior to cryogenic modulators as they yielded much broader peaks (FWHM of 350 ms vs. less than 200 ms) and were considered non-optimal for GC × GC separations (Lee et al. Citation2000; Marriott et al. Citation2000, Citation2003). The most commonly used modulators in atmospheric chemistry have been cryogenic and valve-based modulators (Hamilton Citation2010). However, because robustness, simplicity, and low operating costs are attractive features, recent work has focused on the development of improved versions of this type of modulator (Libardoni et al. Citation2005, Citation2006, Citation2010; Panić et al. Citation2011). Successful single-stage modulation has been reported using recirculated cold air (−30°C) (Libardoni et al. Citation2005) and liquid ethylene glycol (−30°C) (Libardoni et al. Citation2006) to rapidly cool the trapping capillary between heat pulses, producing very narrow peaks (FWHM < 40 ms) for analytes with boiling points of up to the equivalent of n-eicosane (C20H42; bp = 344°C).
The modulator employed in our 2D-TAG system has been specifically tailored for field analysis of semi- and non-volatile organics in atmospheric aerosols, where mechanical robustness and minimal consumables are an important design constraint. The modulator employed here, a simplified version of the modulator described by Panić et al. (Citation2011), is a thermal modulator most closely related to the early work of Liu and Phillips (Citation1991). The modulator consists of a short (15 cm) segment of flattened, 0.53 mm OD × 0.28 mm ID, deactivated Silcosteel™ tubing continuously cooled by forced air convection and heated by 100-ms-duration capacitive electric discharge pulse. This heating temporarily overcomes the cooling of the air stream, eliminating the need to turn off the cooling air flow during desorption. The trapping capillary is located outside the GC oven and connects to the two columns inside the GC oven. The electrical connections are made at the unions between the modulator and the columns inside the GC oven to ensure no cold spots exist across the entire length of the modulator capillary. The capacitive discharge is controlled by a custom external power supply synchronized to the detector acquisition clock, thus enabling proper deconvolution of the data into two chromatographic dimensions. The modulator requires no consumables and does not incorporate any valves or flow splits, making it a robust and simple interface ideal for in-situ GC × GC field measurements of trace-level semi-volatile and non-volatile compounds.
Integration of a TOFMS
A fast scanning detector is essential for GC × GC as a result of the much narrower peak widths (e.g., 0.1 s) compared with 1D chromatography (e.g., 10 s). Therefore, data acquisition rates faster than 50 Hz are required in order to adequately capture the peak shapes. In an earlier version of this instrument, Goldstein et al. (Citation2008) used an FID operating at 50 Hz. Quadrupole mass spectrometers, similar to that utilized in the original TAG instrument, typically have scanning rates of between 2000 and 10,000 atomic mass units per second (amu/s). This results in data acquisition rates of between 4 and 20 Hz when scanning a typical mass range of 30–500 amu, which are suboptimal for quantification purposes. TOFMS offers high data acquisition rates with no spectral bias, such as is seen with quadrupole mass filters, because the TOFMS acquires the entire mass spectrum for each group of extracted ions every few microseconds as opposed to scanning through the mass range once or twice a second. Two different TOFMS instruments were employed in this work depending upon their availability. One was from Scientific Analysis Instruments (Kronus model, Manchester, UK) and the other was from Tofwerk (HTOF model, Thun, Switzerland). These compact detectors both feature an ∼1-m-long reflectron flight path with orthogonal ion extraction with a 70V electron impact ionization source and similar technical specifications. The Kronus uses a high-speed dynode electron multiplier coupled to a high-speed A/D data acquisition system capable of 100 Hz sampling rates. The HTOF uses a high-speed dual multi-channel plate detector system capable of 500 Hz sampling rates and high mass resolution. In this research, the instruments were normally operated at 50 Hz.
RESULTS: SYSTEM EVALUATION
Chromatographic Column Selection
In this work, the primary column (VF-5ms) used by Goldstein et al. (Citation2008) was replaced with a Rxi-5ms (Restek Corporation) of identical dimensions. The Rxi-5ms and VF-5ms phases are identical but the Rxi column has improved inertness and exhibits lower bleed at high temperatures. The phase thickness was also reduced from 1 μm to 0.5 μm in order to further minimize the amount of column bleed. The need for high thermal tolerance stationary phases places significant limitations on the choice of higher polarity columns, which tend to be less thermally stable than other stationary phase types. Two intermediate polarity columns with high thermal stability were evaluated: BPX-50 (SGE Analytical Science) and RTX-200 (Restek Corporation). Both eliminated the upward drift in secondary retention times for late eluting compounds by allowing the GC oven to be ramped to 330 °C, thus minimizing the isothermal period. However, the BPX-50 phase produced undesirable wrap-around for many aromatic compounds, e.g., PAHs, most likely as a result of significant interactions between the analytes and the phenyl groups in the stationary phase. Therefore, the RTX-200 was selected as the secondary column for all subsequent work. With this column set, the chromatographic run times were reduced to two hours for a C12–C40 n-alkane equivalent volatility window.
Modulator Improvements
The critical processes underlying the successful implementation of single-stage modulation with our modulator were the flattening (Panić et al. Citation2011) and rapid cooling of the trapping capillary. Flattening of the capillary minimizes the internal volume without significantly increasing the pneumatic resistance of the trap. This reduces the amount of carrier gas expansion inside the trap during thermal desorption and results in narrower injection pulses into the secondary column, leading to reduced band broadening observed at the detector. Rapid cooling reduces the transition time between desorption and trapping phases, thus minimizing analyte breakthrough between injections onto the secondary column.
Rapid cooling of the capillary was achieved through forced air convection in a narrow channel. The cooling rate was strongly dependent on the velocity and temperature of the forced air flow across the capillary. The air velocity inside the cooling channel was ∼50 m/s when operated with a typical 30 Lpm flow rate. A Ranque–Hilsch “vortex” tube-based cooler (Exair Corporation) was used to condition the input flow to the cooling channel and was directed attached to the modulator assembly. When inputting compressed air, the vortex tube generates two separate air streams, one hotter and one colder relative to the input temperature, as a result of the very rapid vortex that is formed within the tube (Hilsch Citation1947). The hotter air stream was vented as exhaust into the room and the cooler air stream was used to cool the trapping capillary. The temperature differential between the hot and the cold flow depended on both the pressure of the compressed air supply and the fraction of the total flow exiting the cold end. A compressed air flow of 30 Lpm at 25 psig, with three quarters of the flow exiting the cold side and flowing across the trapping capillary generating a temperature differential of 20°C between input and output.
The cold and hot temperatures of the trapping capillary are important factors controlling the trapping and injection of analytes between the primary and the secondary column. Trapping temperatures only need to be sufficiently low to trap compounds eluting at a given time. Since the boiling points of compounds eluting from the primary column steadily increase during the course of a GC run, the trapping temperature could also increase without affecting the trapping efficiency. At the same time, higher trapping temperatures significantly improve desorption efficiencies of the lower-volatility compounds from the modulator and thus the sharpness of their injection onto the secondary column. Similarly, desorption temperatures only need to be sufficient to revolatilize the components that are currently trapped on the capillary. As the GC run progresses and the oven temperatures increases, the trapped components become less volatile and higher desorption temperatures are desirable to minimize band broadening during injection onto the second column. Libardoni et al. (Citation2006) ramped the discharge voltage of the heating pulse to increase the desorption rate of higher molecular weight analytes and maintain sharp injections onto the second column. Therefore, ideally, the trapping and desorption temperatures of the most efficient modulator should track changes in the GC oven temperature.
The heating rate of our capillary was instantaneous on the time scale of peak elution (i.e., milliseconds versus seconds) and the maximum temperature of the trapping capillary was proportional to the applied voltage squared. The voltage required to increase the trapping capillary temperature by ΔT = 280°C from room temperature (∼25°C) was estimated by spot welding a 50-μm thermocouple to the trapping capillary (Panić et al. Citation2011). It is not practical to measure the desorption temperature of the trapping capillary in place because the shallow thermal boundary layer created by the strong forced air cooling interferes with the surface temperature measurement. However, by assuming constant heat capacity, a valid assumption since variations are <10% for this temperature range, it is possible to estimate the desorption temperature of the trapping capillary as a function of the forced air temperature that cools the capillary by adding this ΔT.
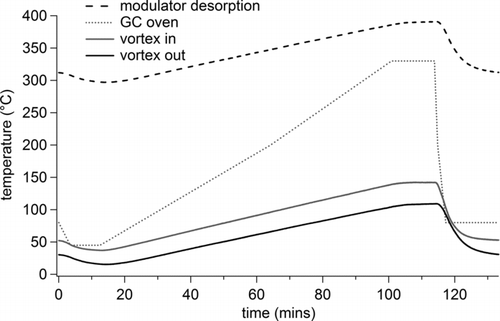
In this work, the input air to the vortex tube was heated through a 1.2 m long × 9.5 mm OD coiled aluminum tubing heat exchanger within the GC oven (), which allows the cooling flow of the modulator and the desorption temperature of the trapping capillary to track the ramping oven temperature (). The combined vortex cooler and heat exchanger provides a very reliable, simple, inexpensive, and passively controlled air stream to cool the modulator after each heating cycle without any moving parts.
FIG. 2 Variation in the temperature of the cooling flow across the trapping capillary (vortex out), the temperature of the supply air from the heat exchanger (vortex in), the desorption temperature of the trapping capillary (modulator desorption) and the GC oven temperature during the course of a standard GC run for an air supply of 25 psi and with 75% of the flow exiting the cold side of the vortex tube.
The trapping capillaries were flattened over most of their length except at the tubular ends (Panić et al. Citation2011). In order to optimize the performance of the single-stage modulator, five capillaries with flattened sections of different lengths were evaluated using injections of the three n-alkane standard solution and diesel fuel to determine the required length of the restriction in the trapping capillary for optimal performance. The main criterion for good performance was minimum FWHM. In addition, peak shapes were carefully examined to ensure no analyte breakthrough occurred. FWHM of 150, 210 and 210 ms for hexadecane, eicosane and tetracosane, respectively, were obtained for the original 15-cm-long modulator with a 14.2-cm restriction. Shortening of the restricted zone invariably led to broader second-dimension peaks and obvious signs of analyte breakthrough. As a result, the original modulator geometry was used in all further analyses as it proved to be optimal for single-stage modulation.
FIG. 3 A GC × GC chromatogram of a 76-component standard (center) along with 1D cross-sections of 10 selected compounds that span both the volatility and the polarity windows of the 2D-TAG instrument. This chromatogram shows the extracted ions of m/z 57, 60, 178, 180, 202, 218, 219, 228, 252, 276, 278, and 300. Peak widths in milliseconds at half maximum height (FWHM) are given for the major slices. Each cross-section is 15 s wide.
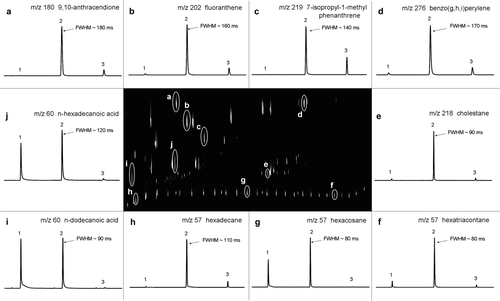
TABLE 1 Precision of the primary (t R 1) and secondary (t R 2) retention times, full peak width at half maximum height (FWHM) for 10 compounds spanning 2D-TAG's volatility and polarity windows. Mean and standard deviations based on four repeat injections, over a seven-day period, of a 76-component standard mixture of alkanes, polycyclic aromatic hydrocarbons, oxygenated polycyclic aromatic hydrocarbons, carboxylic acids, hopanes, steranes, and ketones. Per compound injection level given in nanograms (ng) on-column
The trapping capillaries used by Goldstein et al. (Citation2008) were coated with various thicknesses of polydimethylsiloxane (PDMS) stationary phase (3-μm phase coating, 1-μm phase coating, and deactivated tubing with a minimal coating of stationary phase necessary for surface passivation) on the inside. The purpose of the stationary phase was to enhance the retention of the more volatile components, thus extending the volatility range of trappable species. The volatility range for the 3 μm, 1 μm and deactivated tubing were determined using an n-alkane windowing standard and a traditional dual-stage modulator with separate hot and cold blowers. The efficient volatility ranges for these various phase thickness were found to be C6–C30, C7–C40 and C16–C40, respectively (Panić et al. Citation2011). In this work, we achieved successful modulation of n-tetradecane (C14H30) using the deactivated trapping capillary. We attribute this increased modulation range to the use of higher velocity cooling air, the result of the restrictive channel of the custom-built modulator housing, relative to the more dispersed nature of the cooling air supply used by Panić et al. (Citation2011). The performance of the coated and deactivated trapping capillaries, defined as a function of peak widths and shapes, were nearly equivalent for compounds in the semi-volatile and non-volatile range. As a result, we exclusively used the deactivated capillary in this paper as it had the advantage of increased longevity as a result of a lack of a degradable stationary phase and reduced tendency to introduce peak broadening of the least volatile compounds. It has proved to be extremely robust and exhibited no degradation in performance through more than 300,000 heat cycles, or several weeks of continuous operation.
With this modulator, it is possible to efficiently modulate compounds with boiling points in the semi-volatile (e.g., tetradecane, bp = 254°C) to non-volatile ranges (e.g., tetracontane, bp = 516°C). shows the reproducibility, from repeat injections of the 76-component standard, of the primary (t R 1) and secondary (t R 2) retention times and the FWHM for 10 selected compounds that span the volatility and polarity windows of the chromatographic system. Retention time reproducibility was excellent and FWHM ranged from 80 ms to 160 ms. FWHM were most consistent for the least retained and least functionalized analytes, i.e., n-alkanes (2%), and slightly higher for the more retained PAHs (4%) and more functionalized analytes, e.g., carboxylic acids (6%). shows a GC × GC chromatogram of the 76-component standard mixture together with the corresponding 1D cross-sections (raw chromatograms) for the 10 selected compounds listed in both the tables, demonstrating good peak shapes and the absence of breakthrough between the modulated slices.
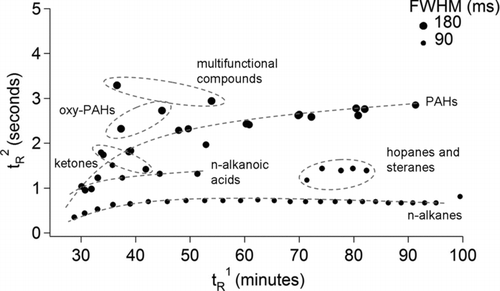
shows the FWHM as a function of t R 1 and t R 2 for the full range of n-alkanes and PAHs, and a selection of oxygenated PAHs, ketones, n-alkanoic acids, hopanes, and steranes from four repeated injections of the 76-component standard. The peak widths were strongly correlated with their retention in the second dimension, e.g., PAH peaks were wider than n-alkanes. FWHM were much narrower (<190 ms) than the 350 ms reported for stainless steel capillaries by Lee et al. (Citation2000). Furthermore, for species retained only slightly in the second dimension (e.g., n-alkanes) FWHM were similar to the 100 ms reported for at least one type of cryogenic (Marriott et al. Citation2000, Citation2003) and fluidic modulator (Bueno and Seeley Citation2004).
TABLE 2 Detection and quantification limits and full peak width at half maximum height (FWHM) for 10 compounds spanning 2D-TAG's volatility and polarity windows. Matches to the NIST08 mass spectral library are shown as the forward match (FM) and reverse match (RM). Limits of quantification (LOQ) were determined from five-point calibration curves of a 110-component standard mixture of alkanes, polycyclic aromatic hydrocarbons, oxygenated polycyclic aromatic hydrocarbons, carboxylic acids, hopanes, steranes, and ketones. Calibration curves showed a power law dependence of the form (y = axb + c), similar to previous quantification work of the TAG system (Kreisberg et al. Citation2009)
FIG. 4 Peak widths in milliseconds at half maximum heights (FWHM) as a function of the primary (t R 1) and secondary (t R 2) retention times for the full range of C14–C40 n-alkanes and 3- to 7-ring polycyclic aromatic hydrocarbons (PAHs) and for a selection of C12–C18 n-alkanoic acids, hopanes, steranes, ketones, oxygenated PAHs (oxy-PAHs), and two multifunctional compounds. Each data point represents an individual compound and the differences in the marker sizes illustrate the FWHM, with larger marker sizes reflecting broader peaks.
TOFMS Linearity and Dynamic Range
An MS used in GC × GC applications must be characterized by its linearity of response through a sufficient dynamic range to accommodate the significant increase in peak amplitude caused by zonal compression in the modulator. The actual dynamic range was determined from the maximum signal-to-noise ratio observed for very large single-compound mass loadings with near or at detector saturation. The signal-to-noise ratio was taken as the peak height divided by the root mean square of the baseline of the ion of interest just before or after the eluting peak. If the chosen peak had an unusually flat peak top, this was taken as evidence of detector saturation. At saturation, the signal-to-noise ratio represents the maximum usable dynamic range. This analysis was performed on multiple peaks from several different chromatograms that showed evidence of detector saturation using both TOFMS instruments. The average measured signal-to-noise ratios were 1.5 × 105 and 1.9 × 105 for the Kronus and the HTOF, respectively, which equated to dynamic ranges of approximately five decades for both detectors. The linearity of the detectors was evaluated through an injection of a five n-alkane standard spanning nearly four orders of magnitude in concentration (0.02, 0.1, 1.0, 10, and 100 ng for nonadecane (C19H40), octadecane (C18H38), eicosane (C20H42), docosane (C22H44), and tetracosane (C24H46), respectively). The observed linear regressions yielded slopes of 1.01 ± 0.07 and 1.05 ± 0.08 for the Kronus and HTOF, respectively, following a log transformation to provide uniform weighting across this dynamic range, in good agreement with the desired value of unity.
Mass Spectral Library Matches of Authentic Standards
The data acquisition and analysis software for both the Kronus (custom-written software; SAI) and HTOF (GC Image) link directly with the widely used NIST mass spectrometer database search routine developed by Stein (Citation2008). shows two metrics (forward match, FM; reverse match, RM) outputted from the NIST08 database for spectra from data acquired with the Kronus TOFMS. The reverse match (RM) parameter excludes possible contaminants present in the unknown spectra at the cost of an increased chance of returning a false positive. If the FM and RM are very close in value, as shown for all species in , this indicates that the obtained spectra are very “clean,” i.e., free of background interference or spectral fragments that are not associated with this compound, which increases confidence in the assignment of an unknown compound. Matches were good for all species with the exception of the n-alkanes, which was not unexpected, given the similarity in their mass spectral fragmentation patterns.
FIG. 5 (a) Calibration response curve of n-tetracosane in the external standard (ES) to perdeuterated n-tetracosane in the internal (IS) from multiple (17 in total) co-injections at seven different concentration levels. (b) Time series of detector response for n-tetracosane (C24H50) in ambient air (AMB) sampled in Berkeley, CA, and detector response to 10 ng of perdeuterated n-tetracosane (C24D50) injected with each ambient run as part of a 21-component internal standard mixture. (c) Ratio of n-tetracosane in ambient air (AMB) to perdeuterated n-tetracosane (IS) injected in each run. (d) Calibrated time series of n-tetracosane (ng m−3) in ambient air determined by applying the calibrated detector response shown in (a) to the observed ratio of ambient n-tetracosane to perdeuterated n-tetracosane.
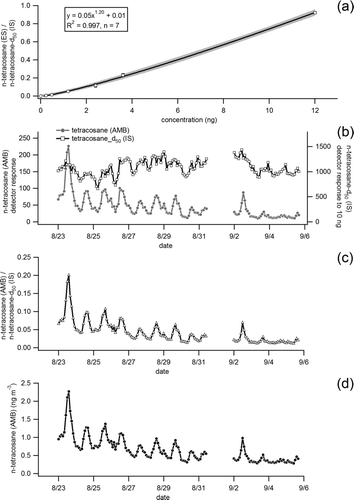
TOFMS Quantification and Stability
shows the quantification limits for the same 10 selected compounds shown in and . Quantification limits, defined as being a signal-to-noise ratio of 10:1, were determined from best-fit power law curves fitted to data from five different concentration levels, spanning the ranges shown in the table, of the 110-component standard. These were in the 0.01–10 ng range and were generally similar to those reported for 1D-TAG (Williams et al. Citation2006; Kreisberg et al. Citation2009), suggesting that gains in the signal-to-noise ratio from zonal compression in the modulator were approximately cancelled out by the loss in sensitivity caused by faster mass spectrometer acquisition rates for the narrower peaks. The exceptions were later eluting compounds, e.g., n-hexatriacontane (C36H74), where quantification limits were approximately an order of magnitude better. This was most likely the result of the peaks being separated from high background noise in this region of the chromatogram, primarily caused by column bleed from the first column.
For calibration purposes, during in-situ sampling, internal and external standards were both applied using a custom-built auto injection system, which has been described and shown to be stable in a recent publication (Isaacman et al. Citation2011a). In brief, pressurized reservoirs, selected using a multi-port selector valve, deliver liquid standards to fill a sample loop of known volume that is injected into the CTD cell of TAG via a six-port valve. The reservoirs are kept cold (∼5°C) in a custom-built heat exchanger consisting of a machined aluminum block and a thermoelectric cooler (Custom Thermoelectric). A 21-component deuterated “internal” standard was routinely injecting on top of every ambient sample to monitor both detector drift and the transfer efficiency of compounds through the analytical system. This is equivalent to the standardized method used for offline filter analyses where filters are spiked with deuterated liquid standards prior to solvent extraction (Schauer et al. Citation1996).
Multi-point calibration response curves of known standards relative to the “internal” standard were generated by co-injection of a 110-component “external” standard with the “internal” standard. Each component of the “external” standard was ratioed to a component in the internal standard based on elution proximity in the chromatogram as well as functional group similarity producing a calibration response curve of ES j /IS k (component j in the “external” standard/component k in the “internal” standard) versus concentration (nanograms). Observed analytes in the atmosphere (AMB j ) were ratioed to specific components in the internal standard in the same fashion in each run (AMB j /IS k ) and this ratio was used along with the appropriate response curve (ES j /IS k ) to assign calibrated values. An example of this calibration methodology as applied to n-tetracosane (C24H50) measured in ambient air collected in Berkeley, CA, is shown in .
The stability of the overall system during typical ambient sampling operation including the TOFMS response is illustrated by the normalized detector response for 10 ng of n-hexadecane-d34 using the HTOF over a two-week period of ambient sampling in Berkeley (). This component of the internal standard was chosen because it is not affected by changes in transfer efficiency resulting from variations in aerosol loading. Overall, the instrument stability was very good with a standard deviation of <7% over two weeks of continuous operation with no observable decline in instrument response.
FIG. 6 Plot of normalized detector response for 10 ng of perdeuterated n-hexadecane (C16D34) injected on top of ambient air for a two-week period in Berkeley, CA, illustrating overall system stability. The solid line represents the average response over the measurement period and the dashed lines the standard deviation.
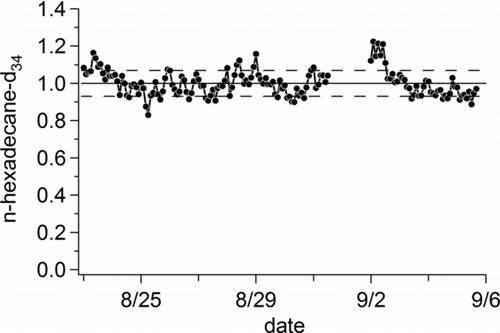
FIG. 7 (a) A GC × GC chromatogram of oxidized diesel exhaust acquired at the Carnegie-Mellon University smog chamber. The enlarged region (inset) shows heavier compounds thought to be primary organic aerosol. Prominent classes of observed compounds are labeled. (b) A GC × GC chromatogram of ambient air from Pasadena, CA, acquired during the California at the Nexus between Climate and Air Quality (CalNex) atmospheric experiment. Commonly observed classes of observed compounds are labeled.
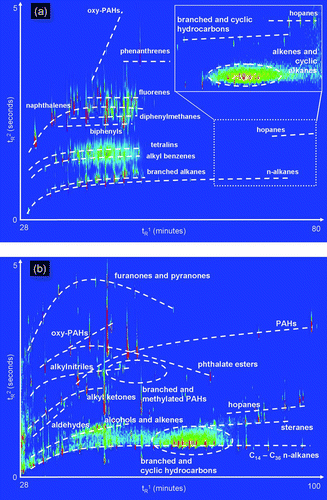
Data Examples: Ambient and Smog Chamber Studies
Thus far, the 2D-TAG instrument has been successfully deployed twice; first at the Carnegie Mellon University's smog chamber in May and June 2009 (shown next and in Isaacman et al. (Citation2011b)) and then as part of the California at the Nexus between Climate and Air Quality (CalNex) experiment, Pasadena, CA, during May and June 2010. Here, we present example chromatograms from both of these deployments. In both cases, the x-axis represents the primary retention time (t R 1) and the y-axis the secondary retention time (t R 2). In these chromatograms, 10 “families” or classes of compounds are clearly separated in the second dimension, suggesting an exhibited peak capacity of at least 10 and indicating that the estimated ideal peak capacity of ∼16 was sufficient in these cases.
Chamber Study: Diesel Exhaust Photo-oxidation
shows a GC × GC chromatogram acquired by TAG of diesel exhaust exposed to 40 min of ultraviolet light, showing ion extraction of m/z 92, 99, 115, 128, 154, 166, 168, 178, 180, 182, 191, 192, 194, 196, 198, 200, and 206. Sample collection occurred for 5 min at a sample flow rate of 9 Lpm for a total sample size of 0.05 m3. Prominent peaks labeled in include: oxygenated PAHs; phenanthrene and mono- and di-methylated isomers; fluorene and mono- and di-methylated isomers; naphthalene and mono-, di-, tri-, tetra-, and penta-methylated isomers, as well as branched isomers of equal carbon number; diphenylmethane and mono- and di-methylated isomers, as well as branched isomers of equal carbon number; biphenyl and mono-, di-, and tri-methylated isomers, as well as branched isomers of equal carbon number; mono-, di-, and tri-methyltetralin; alkylated benzenes with alkyl chains between five and 11 carbons; a complex mixture of branched alkanes; normal alkanes spanning a range of tetradecane to hexatriacontane; and hopanes. While many of these compounds are known to be partitioned in the gas phase, the enlarged area shows heavier compounds known to be primary organic aerosol (POA), with ions at m/z 81, 97, 113, and 191 extracted. It is clear in this enlargement that there are significant particle phase compounds, including various as-yet-unidentified branched and cyclic hydrocarbons, and a mixture of mostly alkenes and cyclic alkanes.
Ambient Study: CalNex
shows a GC × GC chromatogram acquired by TAG of ambient air in Pasadena during the CalNex experiment. This chromatogram shows the extracted ions of m/z 57, 58, 60, 85, 97, 149, 178, 180, 191, 196, 202, 206, 208, 217, 218, 234, and 262. Sample collection occurred for 85 min, from 14:34 to 15:59 on June 14, 2010, with a flow rate of 9 Lpm for a total sample size of 0.77 m3. The commonly observed compound classes highlighted in were: furanones and pyranones; oxygenated PAHs; mono- and di-phthalate esters; PAHs, largely phenanthrene, anthracene, pyrene, and fluoranthene, and dueterated PAHs used as internal standards; alkylnitriles, most notably tetra-, hexa-, and octa-decanonitrile; methylated and other branched PAHs, including 7-isopropyl-1-methylphenanthrene (retene), a biomass burning marker; straight-chain and branched alkyl ketones; straight-chain aldehydes; a mixture of alcohols and alkenes; hopanes; stearanes; various as-yet-unidentified branched and cyclic hydrocarbons; and alkanes, including normal alkanes spanning a range of tetradecane to hexatriacontane, and dueterated alkanes used as an internal standard. Additional well-resolved peaks that are often observed but not highlighted in this figure include phthalic acid, fatty acids, methylated fatty acid esters, alkyl benzenes, chlorobenzenes, phthalimide, and some amines.
SUMMARY
Important improvements to our 2D-TAG instrument have enhanced the capability to separate and identify organics in atmospheric aerosols with in-situ measurements. The two most notable improvements were: (1) development of a single-stage modulator interface, and (2) integration of a TOFMS detector. The modulator employed in the 2D-TAG system is a hybrid thermo-pneumatic modulator. This modulator was shown to be capable of efficiently modulating compounds that span the semi-volatile to non-volatile range (bp = 250–520°C) of organic compounds present in atmospheric aerosols. The modulator traps and desorbs analytes in a single stage, requires no consumables, and has proved to be extremely robust and reliable. In general, optimized peak widths were less than 200 ms at half maximum height with good reproducibility (<10%). Peak widths were dependent on the elution time from the secondary column, with less retained compounds, e.g., n-alkanes, being narrower. Retention time reproducibility in both dimensions was very good. Detection limits for early eluting compounds were similar to 1D- TAG and improved by an order of magnitude for later eluting compounds (>C30), mainly as a result of the reduction in signal interference caused by column bleed. The integration of the TOFMS provides significant improvements for unknown compound identification relative to FID. Mass spectral library matches were very good with extremely strong agreement between the forward (FM) and reverse matches (RM), indicating the “clean” nature of the mass spectra. Overall, the instrument stability, including the response of the TOFMS, was very good with a standard deviation of <7% over two weeks of continuous operation. This instrument can provide more detailed information on the molecular composition of organic aerosols at a higher time resolution than previously studied by offline filter analyses. This time resolution provides a more direct approach to study the sources and aging of atmospheric aerosol on timescales that are comparable with other core measurements typically used in atmospheric chemistry field campaigns, e.g., volatile organic compounds, ozone, and carbon monoxide.
ACKNOWLEDGMENTS
This work was supported by the US Department of Energy STTR program, DEFG02–05ER-86235, and the National Science Foundation Atmospheric Chemistry Program grants 0922562 and 0931934. Restek Corporation is gratefully acknowledged for providing Silcosteel® tubing free of charge. T.G. acknowledges the financial support of the Natural Sciences and Engineering Research Council of Canada. The authors thank Neil Donahue, Allen Robinson, Christopher Hennigan, Albert Presto, and Manish Ranjan from the Center for Particle Studies at Carnegie Mellon University, Pittsburgh, PA, for their time and use of their smog chamber to obtain the diesel exhaust chamber data.
Related Research Data
REFERENCES
- Adahchour , M. , Beens , J. , Vreuls , R. J. J. and Brinkman , U. A. T. 2006 . Recent Developments in Comprehensive Two-Dimensional Gas Chromatography (GC × GC) II. Modulation and Detection . TracTrend. Anal. Chem., , 25 : 540 – 553 .
- Adahchour , M. , van Stee , L. L. P. , Beens , J. , Vreuls , R. J. J. , Batenburg , M. A. and Brinkman , U. A. T. 2003 . Comprehensive Two-Dimensional Gas Chromatography with Time of Flight Mass Spectrometric Detection for the Trace Analysis of Flavour Compounds in Food . J. Chromatogr. A , 1019 : 157 – 172 .
- Bueno , P. A. and Seeley , J. V. 2004 . Flow-Switching Device for Comprehensive Two-Dimensional Gas Chromatography . J. Chromatogr. A , 1027 : 3 – 10 .
- Charlson , R. J. , Schwartz , S. E. , Hales , J. M. , Cess , R. D. , Coakley , J. A. , Hansen , J. E. and Hofmann , D. J. 1992 . Climate Forcing by Anthropogenic Aerosols . Science , 255 : 423 – 430 .
- Frysinger , G. S. , Gaines , R. B. , Xu , L. and Reddy , C. M. 2003 . Resolving the Unresolved Complex Mixture in Petroleum-Contaminated Sediments . Environ. Sci. Technol. , 37 : 1653 – 1662 .
- Gaines , R. B. , Frysinger , G. S. , Hendrick-Smith , M. S. and Stuart , J. D. 1999 . Oil Spill Source Identification by Comprehensive Two-Dimensional Gas Chromatography . Environ. Sci. Technol. , 33 : 2106 – 2112 .
- Gao , S. , Surratt , J. D. , Knipping , E. M. , Edgerton , E. S. , Shahgholi , M. and Seinfeld , J. H. 2006 . Characterization of Polar Organic Components in Fine Aerosols in the Southeastern United States: Identity, Origin, and Evolution . J. Geophys. Res. , 111
- Goldstein , A. H. , Worton , D. R. , Williams , B. J. , Hering , S. V. , Kreisberg , N. M. , Panić , O and Górecki , T . 2008 . Thermal Desorption Comprehensive Two-Dimensional Gas Chromatography for In Situ Measurements of Organic Aerosols . J. Chromatogr. A , 1186 : 340 – 347 .
- Gorecki , T. , Panić , O. and Oldridge , N. 2006 . Recent Advances in Comprehensive Two-Dimensional Gas Chromatography (GC × GC) . J. Liq. Chromatogr. R. T. , 29 : 1077 – 1104 .
- Hamilton , J. F. 2010 . Using Comprehensive Two-Dimensional Gas Chromatography to Study the Atmosphere . J. Chromatogr. Sci. , 48 : 274 – 282 .
- Hamilton , J. F. , Webb , P. J. , Lewis , A. C. , Hopkins , J. R. , Smith , S. and Davy , P. 2004 . Partially Oxidised Organic Components in Urban Aerosol Using GC × GC-TOF/MS . Atmos. Chem. Phys. , 4 : 1279 – 1290 .
- Harynuk , J. , and Gorecki , T. 2007 . Experimental Variables in GC × GC: A complex interplay . Am. Lab. , 39 : 36 – 39 .
- Harynuk , J. , Gorecki , T. , and de Zeeuw , J. 2005 . Overloading of the Second-Dimension Column in Comprehensive Two-Dimensional Gas Chromatography . J. Chromatogr. A , 1071 : 21 – 27 .
- Hilsch , R. 1947 . The Use of the Expansion of Gases in a Centrifugal Field as Cooling Process . Rev. Sci. Instrum. , 18 : 108 – 113 .
- Ho , S. S. H. , Yu , J. Z. , Chow , J. C. , Zielinska , B. , Watson , J. G. , Sit , E. H. L., and Schauer , J. J. 2008 . Evaluation of an In-Injection Port Thermal Desorption-Gas Chromatography/Mass Spectrometry Method for Analysis of Non-Polar Organic Compounds in Ambient Aerosol Samples . J. Chromatogr. A , 1200 : 217 – 227 .
- Isaacman , G. , Kreisberg , N. M. , Worton , D. R. , Hering , S. V., and Goldstein , A. H. 2011a . A Versatile and Reproducible automatic Injection System for Liquid Standard Introduction: Application to In-Situ Calibration . Atmos. Meas. Tech. , 4 : 1937 – 1942 .
- Isaacman , G. , Worton , D. R. , Kreisberg , N. M. , Hennigan , C. J. , Teng , A. P. , Hering , S. V. , Robinson , A. L. , Donahue , N. M., and Goldstein , A. H. 2011b . Understanding Evolution of Product Composition and Volatility Distribution through In-Situ GC x GC Analysis: A Case Study of Longifolene Ozonolysis . Atmos. Chem. Phys. , 11 : 5335 – 5346 .
- Kallio , M. , Hyotylainen , T. , Lehtonen , M. , Jussila , M. , Hartonen , K. , Shimmo , M. , and Riekkola , M. L. 2003 . Comprehensive Two-Dimensional Gas Chromatography in the Analysis of Urban Aerosols . J. Chromatogr. A , 1019 : 251 – 260 .
- Kanakidou , M. , Seinfeld , J. H. , Pandis , S. N. , Barnes , I. , Dentener , F. J. , Facchini , M. C. , Van Dingenen , R. , Ervens , B. , Nenes , A. , Nielsen , C. J. , Swietlicki , E. , Putaud , J. P. , Balkanski , Y. , Fuzzi , S. , Horth , J. , Moortgat , G. K. , Winterhalter , R. , Myhre , C. E. L. , Tsigaridis , K. , Vignati , E. , Stephanou , E. G., and Wilson , J. 2005 . Organic Aerosol and Global Climate Modelling: A Review . Atmos. Chem. Phys. , 5 : 1053 – 1123 .
- Kreisberg , N. M. , Hering , S. V. , Williams , B. J. , Worton , D. R., and Goldstein , A. H. 2009 . Quantification of Hourly Speciated Organic Compounds in Atmospheric Aerosols, Measured by an In-Situ Thermal Desorption Aerosol Gas Chromatograph (TAG) . Aerosol Sci. Technol. , 43 : 38 – 52 .
- Lee , A. L. , Lewis , A. C. , Bartle , K. D. , McQuaid , J. B. and Marriott , P. J. 2000 . A Comparison of Modulating Interface Technologies in Comprehensive Two-Dimensional Gas Chromatography (GC × GC) . J. Microcolumn. Sep. , 12 : 187 – 193 .
- Lewis , A. C. , Carslaw , N. , Marriott , P. J. , Kinghorn , R. M. , Morrison , P. , Lee , A. L. and Bartle , K. D . 2000 . A Larger Pool of Ozone-Forming Carbon Compounds in Urban Atmospheres . Nature , 405 : 778 – 781 . and Pilling, M. J.
- Libardoni , M. , Fix , C. , Waite , J. H. and Sacks , R. 2010 . Design and Performance Evaluation of a Two-Stage Resistively-Heated Thermal Modulator for GC × GC . Anal. Method. , 2 : 936 – 943 .
- Libardoni , M. , Hasselbrink , E. , Waite , J. H. and Sacks , R. 2006 . At-Column Heating and a Resistively Heated, Liquid-Cooled Thermal Modulator for a Low-Resource Bench-Top GC × GC . J. Sep. Sci. , 29 : 1001 – 1008 .
- Libardoni , M. , Waite , J. H. and Sacks , R. 2005 . Electrically Heated, Air-Cooled Thermal Modulator and At-Column Heating for Comprehensive Two-Dimensional Gas Chromatography . Anal. Chem. , 77 : 2786 – 2794 .
- Liu , Z. , Patterson , D. G. Jr and Lee , M. L. 1995 . Geometric Approach to Factor Analysis for the Estimation of Orthogonality and Practical Peak Capacity in Comprehensive Two-Dimensional Separations . Anal. Chem. , 67 : 3840 – 3845 .
- Liu , Z. Y. and Phillips , J. B. 1991 . Comprehensive Two Dimensional Gas Chromatography Using an on Column Thermal Modulator Interface . J. Chromatogr. Sci. , 29 : 227 – 231 .
- Marriott , P. J. , Dunn , M. , Shellie , R. and Morrison , P. 2003 . Targeted Multidimensional Gas Chromatography Using Microswitching and Cryogenic Modulation . Anal. Chem. , 75 : 5532 – 5538 .
- Marriott , P. J. and Kinghorn , R. M. 1998 . Modulation and Manipulation of Gas Chromatographic Bands by Using Novel Thermal Means . Analytical Sciences , 14 : 651 – 659 .
- Marriott , P. J. , Ong , R. C. Y. , Kinghorn , R. M. and Morrison , P. D. 2000 . Time-Resolved Cryogenic Modulation for Targeted Multidimensional Capillary Gas Chromatography Analysis . J. Chromatogr. A , 892 : 15 – 28 .
- McNair , H. M. and Miller , J. M. 2009 . “ Basic Concepts and Terms ” . In Basic Gas Chromatography , 29 – 52 . Hoboken, , NJ : John Wiley & Sons .
- Ochiai , N. , Ieda , T. , Sasamoto , K. , Fushimi , A. , Hasegawa , S. , Tanabe , K. and Kobayashi , S . 2007 . Comprehensive Two-Dimensional Gas Chromatography Coupled to High-Resolution Time-of-Flight Mass Spectrometry and Simultaneous Nitrogen Phosphorous and Mass Spectrometric Detection for Characterization of Nanoparticles in Roadside Atmosphere . J. Chromatogr. A , 1150 : 13 – 20 .
- Offenberg , J. H. , Lewandowski , M. , Jaoui , M. and Kleindienst , T. E. 2011 . Contributions of Biogenic and Anthropogenic Hydrocarbons to Secondary Organic Aerosol during 2006 in Research Triangle Park, NC . Aerosol Air Qual. Res. , 11 : 99 – 108 .
- Panić , O. , Górecki , T. , McNeish , C. , Goldstein , A. H. , Williams , B. J. , Worton , D. R. , Hering , S. V. and Kreisberg , N. M. 2011 . Development of a New Consumable-Free Thermal Modulator for Comprehensive Two-Dimensional Gas Chromatography (GC × GC) . J. Chromatogr. A , 1218 : 3070 – 3079 .
- Ramanathan , V. , Crutzen , P. J. , Kiehl , J. T. and Rosenfeld , D. 2001 . Atmosphere - Aerosols, Climate, and the Hydrological Cycle . Science , 294 : 2119 – 2124 .
- Schauer , J. J. , Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. and Cass , G. R. 1996 . Source Apportionment of Airborne Particulate Matter Using Organic Compounds as Tracers . Atmos. Environ. , 30 : 3837 – 3855 .
- Schnelle-Kreis , J. , Welthagen , W. , Sklorz , M. and Zimmermann , R. 2005 . Application of Direct Thermal Desorption Gas Chromatography and Comprehensive Two-Dimensional Gas Chromatography Coupled to Time of Flight Mass Spectrometry for Analysis of Organic Compounds in Ambient Aerosol Particles . J. Sep. Sci. , 28 : 1648 – 1657 .
- Schwartz , J. , Dockery , D. W. and Neas , L. M. 1996 . Is Daily Mortality Associated Specifically with Fine Particles? . J. Air Waste Manage., , 46 : 927 – 939 .
- Sheesley , R. J. , Deminter , J. T. , Meiritz , M. , Snyder , D. C. and Schauer , J. J. 2010 . Temporal Trends in Motor Vehicle and Secondary Organic Tracers Using in Situ Methylation Thermal Desorption GCMS . Environ. Sci. Technol. , 44 : 9398 – 9404 .
- Stein , S. E. 2008 . National Institute and Standards and Technology (NIST) Mass Spectral Search Program, Version 2.0f . NIST, Gaithersburg, MD
- Surratt , J. D. , Gomez-Gonzalez , Y. , Chan , A. W. H. , Vermeylen , R. , Shahgholi , M. and Kleindienst , T. E. 2008 . Organosulfate Formation in Biogenic Secondary Organic Aerosol . J. Phys. Chem. A , 112 : 8345 – 8378 . Edney, E. O., Offenberg, J. H., Lewandowski, M., Jaoui, M., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.
- Ventura , G. T. , Kenig , F. , Reddy , C. M. , Frysinger , G. S. , Nelson , R. K. and Van Mooy , B. , . 2008 . Analysis of Unresolved Complex Mixtures of Hydrocarbons Extracted from Late Archean Sediments by Comprehensive Two-Dimensional Gas Chromatography (GC × GC) . Org. Geochem. , 39 : 846 – 867 . and Gaines, R. B.
- Vogt , L. , Groger , T. and Zimmermann , R. 2007 . Automated Compound Classification for Ambient Aerosol Sample Separations using Comprehensive Two-Dimensional Gas Chromatography-Time-of-Flight Mass Spectrometry . J. Chromatogr. A , 1150 : 2 – 12 .
- Welthagen , W. , Schnelle-Kreis , J. and Zimmermann , R. 2003 . Search Criteria and Rules for Comprehensive Two-Dimensional Gas Chromatography-Time-of-Flight Mass Spectrometry Analysis of Airborne Particulate Matter . J. Chromatogr. A , 1019 : 233 – 249 .
- Williams , B. J. , Goldstein , A. H. , Kreisberg , N. M. and Hering , S. V. 2006 . An In Situ Instrument for Speciated Organic Composition of Atmospheric Aerosols: Thermal Desorption Aerosol GC/MS-FID (TAG) . Aerosol Sci. Technol. , 40 : 627 – 638 .
- Williams , B. J. , Goldstein , A. H. , Kreisberg , N. M. , Hering , S. V. , Worsnop , D. R. and Ulbrich , I. M. 2010 . Major Components of Atmospheric Organic Aerosol in Southern California as Determined by Hourly Measurements of Source Marker Compounds . Atmos. Chem. Phys. Discuss. , 10 : 6567 – 6639 . Docherty, K. S., and Jimenez, J. L.
- Williams , B. J. , Goldstein , A. H. , Millet , D. B. , Holzinger , R. , Kreisberg , N. M. and Hering , S. V. 2007 . Chemical Speciation of Organic Aerosol during the International Consortium for Atmospheric Research on Transport and Transformation 2004: Results from In Situ Measurements . J. Geophys. Res-Atmos. , 112 White, A. B., Worsnop, D. R., Allan, J. D., and Jimenez, J. L. (, D10526.
- Worton , D. R. , Goldstein , A. H. , Farmer , D. K. , Docherty , K. S. , Jimenez , J. L. and Gilman , J. B. 2011 . Origins and Composition of Fine Atmospheric Carbonaceous Aerosol in the Sierra Nevada Mountains, California . Atmos. Chem. Phys. , 11 : 10219 – 10241 . Kuster, W. C., de Gouw, J., Williams, B. J., Kreisberg, N. M., Hering, S. V., Bench, G., McKay, M., Kristensen, K., Glasius, M., Surratt, J. D., and Seinfeld, J. H.
- Xu , X. , van Stee , L. L. P. , Williams , J. , Beens , J. , Adahchour , M. and Vreuls , R. J. J. 2003 . Comprehensive Two-Dimensional Gas Chromatography (GC × GC) Measurements of Volatile Organic Compounds in the Atmosphere . Atmos. Chem. Phys. , 3 : 665 – 682 . Brinkman, U. A. Th., and Lelieveld, J.
- Zhang , Q. , Jimenez , J. L. , Canagaratna , M. R. , Allan , J. D. , Coe , H. and Ulbrich , I. 2007 . Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenically-Influenced Northern Hemisphere midlatitudes . Geophys. Res. Lett. , 34 Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R. (, L13801.