Abstract
We describe a new instrument, chemical ionization (CI) high-resolution time-of-flight mass spectrometer (ToFMS) coupled to a micro-orifice volatilization impactor (MOVI-HRToF-CIMS). The MOVI-HRToF-CIMS instrument is unique in that, within a compact field-deployable package, it provides (1) quantifiable molecular-level information for both gas and particle-phase organic species on timescales ranging from ≤1 s for gases to 10–60 min for particle-phase compounds that can be used to efficiently probe oxidation and secondary organic aerosol (SOA) formation mechanisms, and (2) relative volatility information of the detected compounds simultaneously estimated using the programmed thermal desorption information obtained from the MOVI. We demonstrate the capabilities of a prototype instrument using known test compounds and complex mixtures generated from the oxidation of biogenic and anthropogenic hydrocarbons. We present spectra obtained using both negative and positive ion CI with acetate (CH3C(O)O−) and protonated water clusters (H3O+·(H2O) n ), respectively, as reagent ions. The instrument has high mass resolving power (R = 5000 above m/Q 250 Th) and mass accuracy (±20 ppm) enabling estimation of compound elemental composition. Instrument sensitivity in negative ion mode was tested using formic acid as a representative gas-phase compound, and that for particle-phase compounds was tested using palmitic, azelaic, and tricarballylic acids. With a heated MOVI inlet, an ion count rate of ∼15 Hz is achieved when sampling 1 pptv (= 1 pmol/mol) of formic acid (or other monocarboxylic acids) under typical operating conditions. This sensitivity translates to detection limits less than 1 ng/m3 for carboxylic acids in the particle-phase. We also discuss the remaining challenges with this instrument to broadly characterizing gaseous and particulate oxygenated organic compounds in situ.
Copyright 2012 American Association for Aerosol Research
1. INTRODUCTION
Organic species (organic aerosols [OA] and organic gases [OG]) are ubiquitous in Earth's atmosphere. Through contributions to aerosol particle formation and growth, organic species affect human health, visibility, cloud formation and climate (Novakov and Penner Citation1993; Quinn et al. Citation2005; Mochida et al. Citation2008; Pope et al. Citation2009; Sorooshian et al. Citation2009). Understanding OA formation and loss, and the partitioning of organic species between gas and particle-phases is key to understanding and mitigating its impacts. Current estimates of primary OA (POA, emitted directly into the atmosphere in the particle-phase) and secondary OA (SOA, produced from chemical reactions in the atmosphere) budgets are highly uncertain (Goldstein and Galbally Citation2007; Hallquist et al. Citation2009; Heald et al. Citation2010). Recent studies have improved our understanding of OA and OG (Donahue et al. Citation2006; Robinson et al. Citation2007; Goldstein et al. Citation2009; Jimenez et al. Citation2009; Paulot et al. Citation2009; Ng et al. Citation2011), but critical parameters remain to be adequately quantified for models to accurately simulate regional and global OA distributions (Hallquist et al. Citation2009). Significant uncertainties related to particle sources, formation and growth mechanisms, loss rates, and direct and indirect effects (changes in light scattering and absorption, and cloud albedo) persist, in part, due to a lack of an understanding of the species which make up OG and OA.
Detailed discussions of various offline and online techniques for OA analysis have been published recently (Sullivan and Prather Citation2005; Nash et al. Citation2006; Chow et al. Citation2008; Farmer and Jimenez Citation2010), therefore, only the most relevant research is summarized below. The application of time-of-flight mass spectrometers (ToFMS) for real-time study of OA and atmospheric OG composition has been demonstrated previously (Prather et al. Citation1994; Murphy and Thomson Citation1995; Blake et al. Citation2004; Drewnick et al. Citation2005; Zelenyuk and Imre Citation2005; DeCarlo et al. Citation2006; Bertram et al. Citation2011), but its coupling with chemical ionization (CI) is only recently becoming more widespread.
The main advantages of ToFMS are (1) the ability to acquire an entire mass spectrum, for example, from 15–1500 Th (Th = thomson = u/e), at rates exceeding 1000 Hz (Kimmel et al. Citation2010), leading to a superior duty cycle for broad mass range, without any a priori assumptions on which m/Q to monitor, (2) high mass resolving power, R, with field deployable instruments routinely reaching R > 3000 Th Th−1, and (3) high relative mass accuracy in the range of 20 μTh Th−1. While ToFMS instruments for gas-phase composition analysis of atmospheric samples now exist (Jordan et al. Citation2009; Bertram et al. Citation2011), the more common application of ToFMS instruments to atmospheric composition studies has been analysis of particles. Two main subgroups are single particle laser-ablation instruments (Prather et al. Citation1994; Murphy and Thomson Citation1995; Zelenyuk and Imre Citation2005) and ensemble particle analysis instruments (Drewnick et al. Citation2005; DeCarlo et al. Citation2006; Holzinger et al. Citation2010). Early designs of single particle instruments caused extensive fragmentation of organic compounds due to laser desorption and ionization, but recent improvements reduce fragmentation by using single photon ionization and variable energy desorption lasers (Oktem et al. Citation2004; Vaden et al. Citation2010). The Aerodyne time-of-flight aerosol mass spectrometer (ToF-AMS) is the most widely used ensemble particle analysis instrument providing nonrefractory particle composition (DeCarlo et al. Citation2006). This instrument uses electron impact ionization, which induces extensive fragmentation of organic molecules. As a consequence, identification and quantification of specific molecular species with the Aerodyne AMS is not possible. However, by determining the elemental composition of all fragments the average oxidation state of carbon in the aerosol particles can be inferred (Aiken et al. Citation2008), and groups of species representing different sources of OA can be identified through factor analysis of the ensemble spectra (Zhang et al. Citation2011). Recently, proton-transfer-reaction (PTR) ionization, a softer ionization scheme than EI, was coupled to a high-resolution ToFMS for OA analysis (Holzinger et al. Citation2010). This work reported detection of un-oxidized and oxidized organic compounds, inorganic compounds, and organonitrate and organosulfate compounds at a high altitude site.
The MOVI-HRToF-CIMS instrument is unique in that it provides, within a compact field-deployable package, quantifiable molecular-level information for both gas and particle-phase oxygenated organic species on a semicontinuous timescale across a large range of volatilities. When combined with the fast acquisition, high mass resolving power and mass accuracy of a high-resolution ToFMS, and the soft and selective ionization scheme, this technique can be used to efficiently probe oxidation and SOA formation mechanisms by estimating the elemental composition of the detected compounds. In addition, using the programmed thermal desorption information from the MOVI, relative volatility information of the detected compounds can be simultaneously estimated. The CI source design used here is similar to that described previously by Bertram et al. (Citation2011, and reference therein), and the HRToFMS is largely similar to the atmospheric pressure interface (API-ToF) described by Junninen et al. (Citation2010). Therefore, we focus our description and characterization on the unique aspects of the MOVI and CI combination.
2. INSTRUMENT DESCRIPTION
The instrument contains four main components. First, a MOVI enables analysis of gas-phase compounds while aerosol particles are simultaneously collected with a cut-point diameter (d 50, transition-regime aerodynamic diameter (DeCarlo et al. Citation2004) of 130 nm. A cartridge heater of the MOVI operates at regular intervals to thermally desorb the collected aerosol mass. The analyte molecules from the MOVI enter an ion-molecule reactor (IMR) where the gas-phase compounds (OG or thermally desorbed OA) undergo CI. A collisional dissociation chamber (CDC) after the IMR is used to optionally break apart ion-molecule clusters with tunable declustering strengths. Finally, the resulting parent ions are focused and guided through differentially pumped regions into a high-resolution ToFMS (HTOF, TOFWERK AG, Switzerland) where they are mass analyzed and detected.
The MOVI-CIMS technique using a quadrupole mass spectrometer has been described by Yatavelli and Thornton (Citation2010), therefore, only modifications since that publication are reported here. The specific high-resolution ToFMS employed here is the same used in the Aerodyne ToF-AMS (DeCarlo et al. Citation2006) and the Ionicon proton-transfer-reaction ToFMS (PTR-TOF-MS (Jordan et al. Citation2009). The atmospheric pressure interface (API) was first described for the detection of atmospheric ions with the high-resolution ToFMS (Junninen et al. Citation2010) and more recently, Bertram et al. (Citation2011) used the same API for coupling of a CI IMR to a compact ToFMS (CTOF, TOFWERK) for analysis of gas-phase organic acids. For simplicity, we refer to the instrument described in Bertram et al. (Citation2011) as a CToF-CIMS. The instrument reported in this article represents, to our knowledge, the first application of high-resolution ToFMS to neutral organic compounds in both gas and particle-phases that are selectively ionized by positive and negative CI schemes.
FIG. 1 Schematic of the improved micro-orifice volatilization impactor. Numbers 1–4 correspond to different pressure regions with region 3 at 863 mbar and region 4 at 90 mbar.
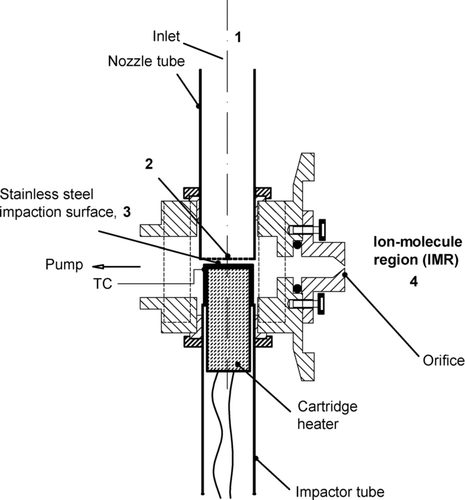
2.1. Micro Orifice Volatilization Impactor
shows a schematic of the MOVI design used in the work described here, slightly revised from the design described previously (Yatavelli and Thornton Citation2010). The impactor characteristics remain largely the same with 100 nozzles, each 150 μm in diameter, and the jet-to-plate distance at 0.6 mm. The design changes described here were aimed at decreasing the condensation of semivolatile and low-volatility compounds in the MOVI prior to CI. Toward this goal, the total surface area of the manifold block was reduced by nearly 50% by eliminating unnecessary dead space. The MOVI manifold now couples directly to the IMR region, so that desorbed molecules have a shorter residence time and a reduced number of wall collisions before ionization. Up to four cartridge heaters programmatically heat the manifold block during the heating cycle to enhance transfer of low-volatility compounds evolving from the collection post.
The manifold block is also coated with an inert material (Inertium, AMCX Corp., State College, PA, USA for Aluminium or SilicoNert 2000, Restek Corp., Bellefonte, PA, USA for stainless steel manifold blocks) to further decrease the adsorption of condensable species inside the MOVI. Similarly, the stainless steel impaction surface also has inert coating (SilicoNert 2000) to minimize adsorption of semivolatile organic compounds. Finally, the critical orifice between the IMR and MOVI is pushed further into the IMR compared to the previous design where the orifice was closer to the impaction surface. This change has proven critical in increasing the sensitivity of the instrument presumably due to better mixing between the reagent ion and sample flows (induced by the relatively high flow velocity at the orifice exit). Apart from these design changes, the manifold block is held at 50°C during aerosol sampling and gas phase analysis to reduce the absorption of gases to the walls during sampling. The impaction surface is kept near ambient temperature by the flow of ambient air through the micro-orifices. Pressure in the MOVI during sampling is slightly below atmospheric pressure, for example, 863 mbar if atmospheric pressure is 1013 mbar.
The particle collection efficiency (CE) of the MOVI can be expressed as the product of several terms:
2.2. Chemical Ionization
The instrument allows analysis with various negative or positive reagent ions. Here, we present data with the acetate anion (CH3C(O)O−), which selectively ionizes acids (Veres et al. Citation2008; Roberts et al. Citation2010, Citation2011; Bertram et al. Citation2011), and protonated water clusters H3O+·(H2O) n . Bertram et al. (Citation2011) described the CH3C(O)O− ion chemistry in detail using the same IMR and CDC designs used here and at similar though not identical pressures; therefore, no discussion of that ion chemistry is presented here.
CI-MS analysis using H3O+ is commonly referred to as PTR-MS, and it has been extensively used to detect volatile organic compounds (VOCs) in the atmosphere (Lindinger et al. Citation1998; de Gouw and Warneke Citation2007). Our implementation does not use H3O+ ions but rather H3O+·(H2O) n cluster ions, due to the relatively high pressures (90 mbar) and low electric field strengths in the ionization region. Hearn and Smith (Citation2004) also observed H3O+·(H2O) n ions in their ionization region maintained at 20 mbar. As a result of the higher proton affinity of H3O+·(H2O) n compared to base H3O+, ionization is more selective and does not ionize species with dipole moment = 0 (Spanel and Smith Citation1995).
In this study, protonated water cluster ions were generated by flowing 2000 sccm of humidified N2 through the 210Po radioactive ion source. The ion source and the IMR are maintained at 50 V relative to ground. Humidified N2 flow was generated by bubbling 2300 sccm N2 through 18 MΩ water maintained at room temperature (no temperature control). A critical orifice controlled the flow through the 210Po ion source with the excess flow being vented to air. Pressures in various regions of the instrument were the same as that for acetate experiments. For the experimental conditions mentioned above, the most abundant ion observed was H3O+·H2O with the next dominant peak being H3O+·(H2O)2. Similar reagent ion distributions have been observed by other researchers, although they used lower ionization region pressures (33 and 53 mbar) (de Gouw et al. Citation2003). The use of a CDC in our instrument alters the detected clusters from the actual distribution present in the IMR and likely contributes to some enhanced proton transfer during the residence time within the CDC though we have not examined this issue in detail. Therefore, the actual cluster distribution in the IMR is likely shifted toward higher cluster sizes (H3O+·(H2O) n , n = 3, 4, 5) than as it appears in our mass spectra. During the experiments described here, the H3O+, H3O+·H2O, and H3O+·(H2O)2 count rates observed were typically, 5 × 104, 1.5 × 106, and 5 × 104 Hz, respectively. It is important to note that the work described here was not aimed at developing an optimized proton transfer method but more to illustrate the capabilities of a MOVI-HRToF-CIMS instrument.
2.3. Interface and HRToFMS
shows a schematic of the instrument assembly. The API and orthogonal extraction, reflectron ToF mass analyzer used here are manufactured by TOFWERK AG (Thun, Switzerland). The differentially pumped API, which was described in detail by Bertram et al. (Citation2011), connects the IMR to the high-vacuum high-resolution ToFMS using three intermediate pumping stages containing ion focusing optics. To simplify the description, below we describe the HRToF-CIMS operation and data analysis procedure only for negative-ion mode.
FIG. 2 Schematic of the complete MOVI-HRToF-CIMS instrument. (Color figure available online.)
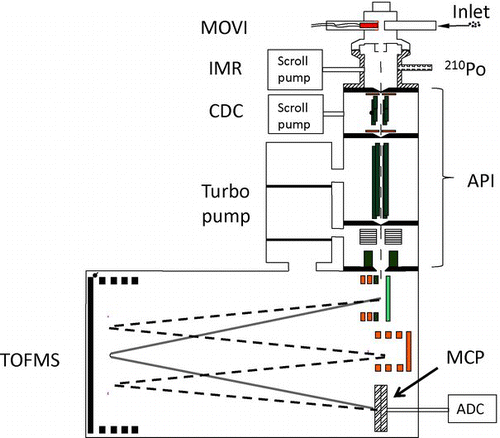
Ions generated in the IMR pass through a 500-μm orifice into the CDC region, which contains a segmented radio frequency (RF-only) quadrupole. The IMR and the orifice are biased at –100 V relative to ground. Pressure in the CDC is maintained at 2.7 mbar using a fine throttle valve and a dry scroll pump (BOC Edwards, Triscroll). The clustered product ions collide with bath gas in the CDC while being guided by the RF and axial fields in the quadrupole. The axial electric field strength across the segments of the CDC quadrupole is adjustable, and was 11.5 V/cm in this work. Bertram et al. (Citation2011) showed that by tuning the electric field strength in the CDC the dissociation of weak ion-molecule clusters can be controlled. Tuning of the CDC and two other vacuum stages of the API are described in detail by Bertram et al. (Citation2011).
The high-resolution ToF can be configured to monitor either positive or negative ions, and ions can traverse the drift region either in a single reflectron (V-mode, nominal drift length = 1.3 m) or double reflectron (W-mode) ion optical configuration (DeCarlo et al. Citation2006). The V-mode has higher sensitivity, while the W-mode has higher mass resolving power. The characterization experiments performed here were generally performed in V-mode with an extraction frequency of 25 kHz resulting in a m/Q range of 5–523 Th. Since both positive and negative ions are discussed in this manuscript, to avoid confusion, negative ion m/Q's will be preceded with a negative (−) sign. HRToF-CIMS data acquisition hardware, data storage procedures, and postprocessing routines used herein are very similar to those described in detail by Bertram et al. (Citation2011). Post processing routines are developed by TOFWERK and use IGOR Pro software (Wavemetrics, Lake Oswego, OR, USA).
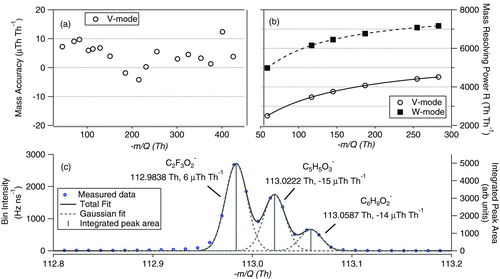
FIG. 3 (a) Relative mass accuracy as a function of m/Q, (b) mass resolving power as a function of m/Q for V and W modes, and (c) expanded view of −113 Th from the α-pinene oxidation experiment. Three ions are marked with their respective mass accuracies in μTh Th−1 (= ppm). (Color figure available online.)
3. METHOD EVALUATION
3.1. Mass Accuracy and Mass Resolving Power
The relatively high mass accuracy and mass resolving power in a field deployable package are important distinguishing characteristics of the HRToF-CIMS. Here, mass accuracy of the instrument is determined by comparing the measured m/Q of specific ions (not used in the calibration) of known elemental composition in a calibrated mass spectrum to the true m/Q of the ions. The m/Q of a recorded ion spectral peak was measured using a custom peak fitting algorithm in IGOR Pro. The peak center of the fitted ion peak was taken as the m/Q of the detected ion and compared to the exact m/Q calculated from the elemental composition. shows the V-mode mass accuracy for a variety of ions with most of the data within ±20 μTh Th−1 (ppm).
The mass resolving power (R) for an isolated peak in a mass spectrum was calculated as the ratio of the m/Q of the peak to its full-width at half-maximum Δ(m/Q)FWHM.
Having a R > 2000 provides the ability to differentiate ions with the same number of nucleons (i.e., integer m/Q) that are separated by a small fraction of a Th in the m/Q scale. An example of this high-resolution ability of the HRToF-CIMS instrument is shown in . This spectrum was obtained during analysis of gas-phase oxidation products from dark ozonolysis of α-pinene in a flow through chamber (discussed further in the method application section). The data clearly shows three distinct ions at −113 Th corresponding to C2F3O− 2, C5H5O− 3, and C6H9O− 2 which all have the same nucleon number, but slightly different mass defects. The fluorinated ion is a persistent internal contaminant due to perfluoro polymers used in seals and tubing upstream of the ion source. Such resolving power is important for rigorous testing and development of chemical mechanisms or emissions inventories.
3.2. Sensitivity
3.2.1. Sensitivity to Gas-Phase Carboxylic Acids Using CH3C(O)O−
To assess the sensitivity of the instrument to gas-phase compounds, calibrations were performed using formic acid with acetate as the reagent ion. Based on previous work, we expect the sensitivity to be a nearly linear function of extraction frequency, a nonlinear function of IMR and CDC pressure (in part due to the RF ion guides), and to depend upon electric field tuning for resolution and declustering as well as the presence of surfaces between the formic acid source and IMR. Therefore, we separate the sensitivity of the instrument that obtained using a high transmission inlet, from that obtained with the MOVI inlet.
Dilute concentrations of formic acid were produced by flowing 10 sccm of N2 over a calibrated permeation tube (Kin-Tek, SRT2, 21.6 ng/min) maintained at 70°C and contained in an insulated aluminum block. The output of the permeation tube was confirmed to be accurate and stable to within 5% via ion chromatography. Formic acid concentrations were varied by increasing or decreasing the carrier flow used to dilute the formic acid and overflow the instrument inlet which for these determinations of optimum sensitivity, consisted of a single fitting bored through with a PFA Teflon tube delivering gas to the critical orifice separating ambient pressure and the IMR.
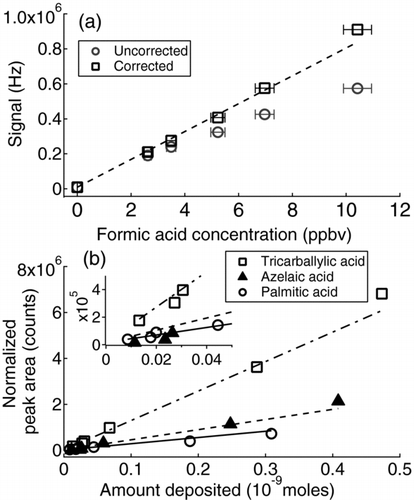
Based on HR analysis, it was confirmed that only the HCOO− ion contributed to the signal at −45 Th, therefore, the instrument response for formic acid was measured using the integrated unit mass resolution (UMR) signal at −45 Th. The formic acid signal was observed to be linear up to 2 ppb above which acetate ion depletions were noticed. At the maximum formic acid concentration tested (10.4 ppb), depletion in acetate ion was measured to be ∼37%. The measured formic acid signal is normalized to depletions in reagent ion using the ratio of the reagent ion signal measured without formic acid to the signal when formic acid is present. Using the normalized counts in response to varying formic acid concentrations, the sensitivity of HRToF-CIMS to formic acid, determined from the slope of the line in is 80 Hz/pptv. This value is consistent with the 338 Hz/pptv obtained by Bertram et al. (Citation2011) using a CToF, taking into consideration (1) the known factor of 4 decrease in ion transmission within the ToF region of the TOFWERK HTOF compared to CTOF (DeCarlo et al. Citation2006) for the same mass range (2) differences in the IMR and CDC pressures and electric fields (135 and 2.6 mbar here, vs. 85 and 2 mbar, respectively, in Bertram et al. [Citation2011]), and (3) different low m/Q transmission efficiency of the RF ion guides and TOFMS tuning. Importantly, a sensitivity of order 80 Hz/pptv for a single gas-phase compound is more than capable of making atmospheric measurements of many gases depending upon the instrument background. Moreover, there are few, if any, field deployable instruments with a better sensitivity coupled to the high-resolution capabilities and duty cycle of the ToF.
FIG. 4 (a) Signal at –45 Th (formic acid) shown in two ways: corrected for variations in the reagent ion (CH3C(O)O−) intensity and uncorrected, as a function of formic acid concentration. (b) Normalized peak areas for (a) palmitic acid, (b) azelaic acid, and (c) tricarballylic acid obtained using the CH3C(O)O− reagent ion. Lines are orthogonal distance regression fits to the data. Inset in Figure 4b shows a smaller range of acid deposited on the MOVI post.
Replacing the gas-phase inlet with a MOVI heated to 200°C and containing an inert coating such as SilcoNert, transmits formic acid to better than 80% of the simpler inlet described above with relatively little loss in sensitivity. However, and not surprisingly, operating the MOVI manifold at 50°C reduces the sensitivity to formic acid by a factor of 5, but only because of inefficient transfer of the formic acid through the MOVI (adsorption in the nozzle tube and on the MOVI surfaces) to the IMR, not because of changes in ion chemistry or ion transmission to the detector. A subsequent article will describe mass closure experiments using the MOVI at various temperatures.
For the above experiments, the ion transmission was optimized for low m/Q compounds. However, for most applications, such as those described below, we are interested in both capturing a large m/Q range, which requires lower extraction frequencies (25 kHz vs. 75 kHz), and operating at the minimum necessary declustering to reduce fragmentation of ions in the CDC. These conditions lower the sensitivity to formic acid by a factor of 4, and operating a MOVI at 50°C lowers the sensitivity to formic acid further as noted above. Therefore, for typical operating conditions employed for the remaining tests discussed below, that is, 50°C MOVI body temperature, 25 kHz extraction frequency and pressures optimized for moderate declustering, we find a sensitivity of formic acid of 3 Hz pptv−1. Using the expression given in Bertram et al. (Citation2011), a background signal of 300 Hz, and a signal-to-noise ratio S/N = 3, we estimate the formic acid detection limit to be 25 pptv in 1 s integrations, which is a factor of 8 lower than that obtained by Bertram et al. (Citation2011) with a high transmission gas-phase inlet and a CToF.
The gas-phase formic acid detection limit reported here makes this instrument suitable for measurements in both clean and polluted regions. Formic acid concentrations range between (lowest and highest values reported) 0.02–19 ppbv in polluted urban air (Chebbi and Carlier Citation1996; Veres et al. Citation2011), 1–40 ppbv in forested region of Sierra Nevada in Southwestern USA (Harrington et al. Citation1993), 0.1–3.8 ppbv in rural and semirural regions across the world (Chebbi and Carlier Citation1996), 0.03–1.7 ppbv and 0.65–0.85 ppbv in remote and pollution impacted marine regions, respectively (Bertram et al. Citation2011), and 0.1–2.9 ppbv in the Amazon forest (Chebbi and Carlier Citation1996).
3.2.2. Sensitivity to Particle-Phase Carboxylic Acids Using CH3C(O)O−
The MOVI-HRToF-CIMS sensitivity for condensed phase organic compounds was measured using submicron polydisperse aerosols generated with a constant output atomizer (TSI 3076, St. Paul, MN, USA). A dilute methanol solution containing approximately 0.12, 0.11, and 0.11 mM of tricarballylic acid (C6H8O6, tri-acid), azelaic acid (C9H16O4, di-acid), and palmitic acid (C16H32O2, mono-acid), respectively, was used as the solution for atomization within 2 days of its preparation. These acids were chosen in order to investigate the ionization efficiency and instrument response to compounds that differed in number of carboxylic acid functionalities and volatility. Other acids (glutaric, benzoic, and decanoic) have also been tested, but fall within a similar range of results as the acids presented here.
The mass collected on the impaction surface was changed by sampling for different times between thermal desorptions. The collected mass was determined from the particle mass concentration, sampling time, and the impactor collection and bounce efficiencies using EquationEquation (1). We assume the dry aerosol particles were more likely amorphous solids rather than liquids. In addition, we assume the components in the aerosol particles retained the same mole fraction as the atomizer solution. The collected particle mass was thermally desorbed into dry N2 and subsequently detected by CI and ToFMS analysis.
The measured signal for each acid (Si , i = tricarballylic, azelaic, palmitic) was normalized by multiplying the respective acid signal by the ratio of the reagent ion signal at the start of desorption cycle (Acetate t = 0) to the reagent ion signal during each time step (Acetate t = t ).
For the acids used here, the corresponding carboxylate anions appear in the mass spectrum at the nominal m/Q of −175, −187, and −255 Th for tricarballylic, azelaic and palmitic acid, respectively. For all these acids minimal signal was observed at the m/Q corresponding to a cluster with acetate ion, and except for tricarballylic acid, the carboxylate anion was the only significant ion observed. For tricarballylic acid, an additional signal was detected at −157 Th, which was highly correlated with −175 Th and was 2–10 times larger. High-resolution analysis of −157 Th peak had an exact mass of −157.0137 Th, which corresponded to the dehydrated TA. At present, it is not known if the dehydration occurred in the solvent used in the atomizer, during thermal vaporization, or after ionization. Further tests are needed to more conclusively determine its cause. For the purpose of the sensitivity calculation it is assumed that the signal at −157 Th is a dehydration product of tricarballylic acid, and therefore the signal at this peak is added to the signal measured at −175 Th to obtain the total tricarballylic acid signal.
shows the tricarballylic, azelaic, and palmitic signals (normalized to acetate signal and integrated ion counts) versus deposited amount in nanomoles (nmol). A linear increase in ion signal is observed with increasing amount deposited for the range studied consistent with the linearity demonstrated previously with the MOVI (Yatavelli and Thornton Citation2010). The sensitivity to each acid is the slope of the linear least squares line shown in and is calculated to be 2.7 ± 0.06 × 106, 4.4 ± 0.1 × 106, and 1.3 ± 0.03 × 107 counts/nmol, for palmitic, azelaic, and tricarballylic acid, respectively. For comparison, the sensitivity to gas-phase formic acid with a hot MOVI is equivalent to 5 × 106 counts/nmol, essentially the same as that for azelaic acid, suggesting a consistency between the sensitivity to gas and particle-phase compounds. Moreover, the sensitivities calculated here correspond to ionization efficiencies of 4.5 × 10−9–2.2 × 10−8 ions/molecule which can be compared to typical ionization efficiencies of 1 × 10−7 ions/molecule for V-mode and 1 × 10−8 ions/molecule for W-mode of the HRToF-AMS using electron-impact ionization (DeCarlo et al. Citation2006). Given the larger volumes sampled by the MOVI-HRToF-CIMS versus the AMS (10 lpm vs. 0.1 lpm, respectively), the detection limits for the MOVI-HRToF-CIMS in terms of ambient concentrations are expected to be similar or better than those of the HR-ToF-AMS.
The sensitivity to carboxylic acid compounds observed using acetate ion chemistry increases with an increasing number of carboxylic acid functionalities per compound and is also consistent with an increase in the relative acidity (ΔG acid) of the analyte with respect to acetic acid. The gas-phase acidity for various mono and dicarboxylic acids (Bartmess Citation1989) suggests an increase in acetate ionization probability with increasing number of acid groups and decreasing carbon number (Figure S2). At some point, ionization will be limited by the ion-molecule collision frequency and residence time in the IMR; increases in relative acidity will not increase sensitivity beyond that point. We hypothesize that the range in sensitivity observed for the carboxylic acids here is due to the range in gas-phase acidities relative to acetate. This conclusion will be the focus of future research by testing more acids and/or modeling the ion-molecule reaction thermodynamics and kinetics. Systematic characterization of the sensitivity to the broad range of compounds detectable by the MOVI-HRToF-CIMS is ultimately necessary to be quantitative for any specific ion detected. Utilizing measured and theoretically determined gas-phase acidities (or proton affinities when operating in proton addition mode) as a function of elemental composition is a likely approach for such a universal sensitivity function. While such a universal sensitivity function is a long-term goal, it is important to note that the relative sensitivity between different compounds (and therefore between detected ions) is likely fixed for constant operating conditions and need not be known to determine and characterize statistical patterns within a time series of mass spectra, as is done for factor analysis.
3.3. Precision and Detection Limits for Particle-Phase Carboxylic Acids
To estimate the precision of the impaction/volatilization method, three replicate calibration experiments were performed for the three organic acids using syringe deposition as described previously (Yatavelli and Thornton Citation2010). These tests suggest a relative measurement precision for palmitic, azelaic, and tricarballylic acid of 8, 16, and 19%, respectively. The overall precision error is larger than the estimated delivery precision. We speculate that matrix effects, such as variable conversion of acids to esters in the ethanol solution are driving the observed variability in this case. Future calibration experiments are planned with different solvents to test this hypothesis. In the absence of independent standards for these acids, it is not possible to calculate the instrument accuracy.
Following Yatavelli and Thornton (Citation2010), we define the detection limit of the MOVI-HRToF-CIMS instrument as being the mass deposited on the impaction post that gives rise to a signal that is three times the precision of the signal, including variations of the background. The uncertainty in the signal peak areas (σ spa), which is the precision mentioned above, and uncertainty in the blank signal peak areas (σ bspa) together with the deposited amount in moles (n) and calibration factor (Cf ) determine the S/N ratio.
These detection limits are suitable for measurements not only in polluted regions but also in cleaner environments. For example, ambient concentrations (lowest and highest values reported) of palmitic and azelaic acids are 35.6–89.1 ng/m3 and 2.6–25.5 ng/m3, respectively, in aerosol collected in Philadelphia, PA, USA (Li et al. Citation2006), 0.95–2.4 ng/m3 and 0.17–4 ng/m3, respectively, in remote marine aerosol (Kawamura and Sakaguchi Citation1999; Mochida et al. Citation2002), and 2.1–12.7 ng/m3 and 0.7–2.5 ng/m3, respectively, in the Amazon forest (Graham et al. Citation2003). Tricarballylic acid has also been measured in various source regions with a range of 0.35–39.1 ng/m3 in urban aerosol collected in Chennai, India (Fu et al. Citation2010), 0.002–3.4 ng/m3 in remote marine aerosol (Fu et al. Citation2011), and 1.5–5.3 ng/m3 in the Amazon forest (Graham et al. Citation2003).
4. METHOD APPLICATION
The capabilities of the technique for broadly characterizing gaseous and particulate oxygenated organic compounds thought to be atmospherically relevant were assessed using OA generated in controlled oxidation experiments. Results presented here are obtained from two such experiments: (1) dark ozonolysis of α-pinene in a flow-through chamber, and (2) photo-oxidation of a cyclic alkane in a large environmental chamber.
4.1. α-Pinene Ozonolysis SOA
SOA was produced by reacting α-pinene with ozone in an ambient pressure, room temperature, entrained aerosol flow reactor. The entrained aerosol flow reactor is similar to that used previously in the Thornton lab and described in detail by Bertram et al. (Citation2009). Details of the experimental setup and conditions are presented in the supplemental information. We only briefly summarize the conditions here. Ozone and α-pinene reacted without seed particles for 8 min (residence time). At the exit of the flow reactor, a fraction of the eluent was sampled by an SMPS, and the rest was drawn through the MOVI inlet (at 40°C) to the HRToF-CIMS. Typically, 76 μg/m3 of SOA were detected at the exit and subsequently diluted by a factor of 5.5 prior to sampling by the MOVI. Total SOA mass collected during the sampling cycle, assuming a density of 1.4 g/cm3 and spherical particles was ∼0.94 μg. These calculations used EquationEquation (1) with an Eb equal to that of solid aerosol-particles (0.65) along with the size dependent CE of the MOVI (Yatavelli and Thornton Citation2010). Acetate was used as the reagent ion during these experiments to detect gas and particle-phase carboxylic acids which are known to be common products of α-pinene ozonolysis.
show average UMR mass spectra obtained during (a) sampling and (b) a temperature-programmed thermal desorption cycle of aerosol material collected during the sampling. All signals related to the reagent ion have been removed for clarity. To provide a means of qualitative comparison, we have assumed that all compounds detected during gas-phase sampling exhibit the same sensitivity as formic acid, while we apply an average of the sensitivities determined for palmitic and azelaic acids to the signals detected during the thermal desorption cycle. Tricarballylic acid sensitivity was not used in the calculation above assuming that the formation of tri-acids during α-pinene ozonolysis is a minor contributor to the overall SOA mass. The total detected organic acid mass evolving during a heating cycle was calculated from the total analyte ion current to be in the range 100–320 ng, where the range arises from using sensitivity equal to that of either azelaic or palmitic acids, respectively. This total acid estimate corresponds to an acid fraction of 11–34% of the total SOA mass. The organic acid fraction estimated above is similar to that reported previously by Yu et al. (Citation1999) for α-pinene ozonolysis in a large environmental chamber (at an SOA concentration of 53 μg m−3). They estimated the major ozonolysis products pinic, norpinic, pinonic, norponinic, hydroxypinonic acid, and an unidentified acid (C10H14O3) accounted for 13.6% (9.5–17.8%) of the total aerosol mass yield formed. Using the estimated mass concentration, shown in , for the same acids, we calculate that ∼12% of the aerosol mass is accounted by these acids; again remarkably similar to the study by Yu et al. (Citation1999).
FIG. 5 Unit mass resolution (UMR) spectra from ozonolysis of α-pinene in low-NOx conditions for (a) sampling and (b) heating cycles. See text for the estimation of mass concentrations from the detected signals.
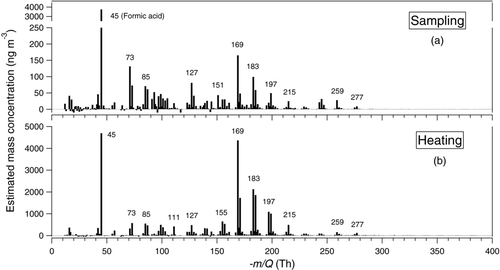
In , the dominant gas-phase analyte ion observed is that of formic acid at −45 Th, the smallest possible gas-phase carboxylic acid presumably formed due to substantial C–C bond breakage during oxidation. Other major well-known products such as pinonic (−183.1026 Th) and norponinic acid (−169.0876 Th), are also observed during sampling, but are much lower in intensity compared to formic acid suggesting that there is little gas-phase abundance of carboxylic acids in the high mass range (>200 Th) under our high OA mass concentrations. Alternately, if they preferentially partition to the gas-phase in the reactor, they may have rapidly partitioned to the reactor walls and/or the walls and collection surface of the sampling inlet. Likely, both effects play a role in these experiments and preserving the gas/particle portioning upon sampling represents a significant analytical challenge for any instrument.
shows the mass spectrum during heating (particle-phase). The spectrum is similar to that observed during sampling but with much higher intensity upon thermal desorption. Some of the notable spectral features that correspond to products reported by other studies include norpinonic acid (−169.0876 Th), terpenylic acid/norpinic acid (−171.0674 Th), pinonic acid (−183.1026 Th), pinic acid (−185.0831 Th), and hydroxy-pinonic acid (−199.0972 Th).
In addition to the known products, the high mass resolving power and high mass accuracy of the HRToF-CIMS, allows us to estimate the elemental composition of a large range of detected organic acids. Exact peak centers are calculated using a custom high-resolution peak fitting algorithm, written at the University of Washington, and exact mass values are input into an online molecular composition calculator (http://www.chemcalc.org), whose output is used to estimate the elemental composition. shows the time-averaged high-resolution mass spectrum obtained during a heating cycle with the possible elemental compositions of several major ions. The mass scale is divided into three different panels; a, b, and c to more clearly show the separation in ion peaks. Table T1 in the supplemental information lists the estimated elemental compositions of all the ion peaks detected during the heating cycle. shows the ion peak at −357.1564 Th and four possible oxygenated compounds that are within ±40 μTh Th−1 of the ion peak center. Since the mass accuracy of the instrument is ±20 μTh Th−1, we can reasonably narrow the possibilities to being some combination of two elemental compositions at this ion peak. The estimated elemental composition can also be used to understand the evolution of oxidation states of the products. This information provides insights on the fragmentation, functionalization, and oligomerization during oxidation of organic species (Kroll et al. Citation2011).
FIG. 6 High-resolution mass spectra marked with likely ion elemental compositions from ozonolysis of α-pinene in low-NOx conditions. (a) Mass range −70–117 Th, (b) mass range −150–210 Th, (c) mass range −319–433 Th, and (d) zoomed in view of ion peak −357 Th with possible oxygenated compounds in ±40 μTh Th−1 range of the ion peak center. (Color figure available online.)
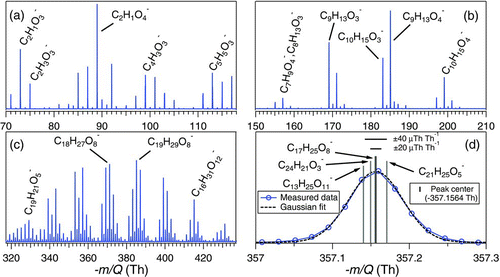
We wish to highlight the large molecular weight products, presumably formed via oligomeric accretion reactions, which are clearly detected using the MOVI-HRToF-CIMS (). Presently, to our knowledge, this is the only instrument that can provide this kind of information at the molecular-level in a field deployable package with a specificity to carboxylic acid containing compounds. The relatively soft nature of the CI coupled with the duty cycle of ToFMS allows detection of these large oligomeric compounds that, to our knowledge, have so far only been detected with extensive offline analyses (Tolocka et al. Citation2004; Reinhardt et al. Citation2007; Walser et al. Citation2008; Heaton et al. Citation2009; Hall and Johnston Citation2011).
The built-in temperature programmed desorption inherent to MOVI analysis provides the ability to monitor the relative volatility of a single compound or of the entire spectrum of compounds. An example of this capability is shown in . The figure shows ion signals, as a function of the desorption temperature (mass thermogram) for m/Q consistent with major α-pinene ozonolysis products having various functionalities. Based on HR analysis of the averaged mass spectra shown in , no interferences were observed for the products shown here, except −155 and −157 Th. Since development of analysis procedures for automated processing of large amounts of HR data is still ongoing, UMR data is used here. For example, pinonic acid (C10H16O3) is a monocarboxylic acid, and thus has a higher equilibrium vapor pressure and desorbs earlier compared to pinic acid (C9H14O4) which is a dicarboxylic acid. The equilibrium vapor pressure of pinic acid is two orders of magnitude lower than pinonic acid (Compernolle et al. Citation2010), resulting in its higher desorption temperature. An interesting feature to note is the double hump structure in the thermogram for −155, −183, and somewhat for −169 Th. This secondary hump is probably thermal decomposition of oligomeric compounds (shown in ) into respective monomers. A recent study by Gao et al. (Citation2010) identified the product at −357 Th as a dimer of pinic acid and terpenylic acid (C8H12O4). Alternately, since the traces shown in are UMR, it is possible that there are other oxidized compounds with lower volatilities at similar nominal masses that are desorbing at higher temperatures.
FIG. 7 Mass spectral signal intensities as a function of MOVI desorption temperature for α-pinene ozonolysis products. Also plotted are signals from two oligomeric compounds (−357 Th and −343 Th) observed during the experiment. (Color figure available online.)
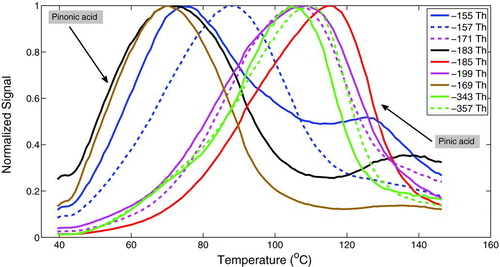
The non zero signal for the products during the start of the heat cycle is possibly due to a combination of evaporation of the compounds from the sample collected on the post and on the manifold walls (during aerosol sampling). During the start of the heating cycle, partitioning of the compounds based on their vapor pressure is in fact visible with pinonic acid, which has a higher vapor pressure compared to pinic acid, and has a correspondingly higher signal than other products. Some of these issues will be addressed with a future MOVI design which has the manifold constantly maintained at 200°C while the collection post is kept at room temperature (by active cooling) thus minimizing adsorption of gas-phase compounds on the walls. This design change will not address the potential evaporation of the compounds from the aerosol particles collected on the post due to the pressure drop across the nozzles. The magnitude of this possible negative bias will ultimately depend upon the volatility and phase-state of the particles collected. The nonzero signal at the end of the heat cycle is possibly due to decomposition of oligomers at high temperatures and the highest temperature reached being lower than what is needed to completely evaporate all the collected material. We are presently testing the new MOVI design with post temperatures reaching around 200°C by the end of the heat cycle with a long soak at 200°C to ensure volatilization of all nonrefractory material from the post during a heating.
4.2. Alkane Photo-oxidation SOA
The MOVI-HRToF-CIMS was recently employed in a study of the photo-oxidation of several C12 alkanes in the Caltech environmental chamber. A detailed analysis of the products and their formation pathways during these experiments is the subject of future articles. Here, we use this dataset to demonstrate the capability of the technique to ascertain time-dependent changes in the gas and aerosol phases. For this purpose we show a full mass spectrum at one time point and the evolution of two products over 18 h of OH oxidation of cyclododecane (C12H24, 10 ppb) in the presence of NOx (initial concentration NO = 377 ppb and NO2 = 359 ppb). The instrument was operated with both acetate and H3O+·(H2O) n clusters over the course of the study with a mass range 5–523 Th. Here, we show results utilizing protonated water clusters as the reagent ion. Other experimental details are provided in the supplemental information.
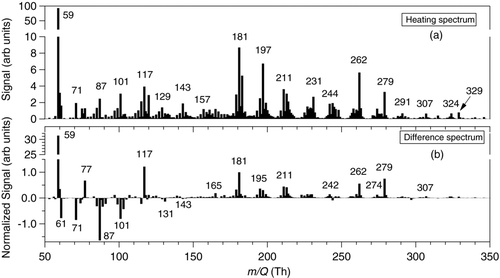
shows an average UMR mass spectrum from a heating cycle after 5 h of oxidation for the post temperature range 125–175°C. The mass spectrum contains numerous peaks with the most intense signals at 59 and 181 Th, which, based on HR analysis, are identified to be either protonated acetone or propionaldehyde (C3H6O·H+, 4 μTh Th−1) and an unknown compound with the likely elemental composition of C12H20O·H+ (1 μTh Th−1). The latter formula implies a cyclic alkenone or an ion after –OH loss. Clusters of peaks every 14 and 16 Th suggest a difference of –CH2, N, and/or O. shows a difference spectrum between the heating and sampling cycles normalized to the signal at 181 Th. The heating spectrum was calculated by integrating the signal during the thermal desorption cycle for each m/Q and then dividing by the sampling time. The sampling spectrum is just an average of the signal at each m/Q measured during the 10 min sampling cycle. There is a clear progression in the difference spectrum from negative to positive signal around 150 Th, indicating the transition from species that are preferentially detected in gas-phase to those predominantly collected on the post (e.g., in the particle-phase) as m/Q increases.
FIG. 8 Unit mass resolution (UMR) spectra after 5 h of cyclododecane oxidation by OH-radicals in the presence of NOx. (a) Average of signal between 125–175°C during a heating cycle, and (b) difference spectrum between heating and sampling cycles.
shows the time evolution of two oxidation products during the 18 h photo-oxidation experiment. A faulty MOVI postheater resulted in incomplete vaporization during some of the heating cycles; therefore, the data corresponding to these times is removed from the plot. Inspection of the high-resolution spectra did not reveal the presence of multiple compounds at the corresponding m/Q for the products discussed below; therefore, UMR signal is used. shows the evolution of the C12H22O·H+ (183 Th), which we presume is the carbonyl product formed from the reaction of an alkoxy radical with O2, and shows the evolution of C12H23NO4·H+ (246 Th), which we presume is the 1,4-hydroxynitrate (1,4-HN) formed from the addition of NO to the hydroxyperoxy radical. Both are first generation products that have been observed previously by other studies (Lim and Ziemann Citation2009; Aschmann et al. Citation2011). Although both of these products form within 1 h of the start of the experiment and partition between gas and particle-phases, they evolve differently during the remaining experiment time indicating differences in their relative production and loss processes. Included for reference is an estimate of the OH exposure during the experiment as well as the NO/NO2 ratio. Both of these quantities demonstrate substantial changes over the course of the experiment and the evolution of the carbonyl and 1,4-HN ions are consistent with the changes in these indicators. In particular, the rapid formation of the first generation carbonyl and 1,4-HN in the first hour of the experiment, followed by their much slower evolution thereafter is consistent with the dramatic loss of the parent hydrocarbon in the first hour followed by much slower loss thereafter (Figures S3b and S4). Moreover, there is a clear change in the 1,4-HN production rate over the first few hours likely due to a strongly increasing NO/NO2 ratio during that time.
FIG. 9 Evolution of cyclododecane photo-oxidation products during sampling and heating cycles. (a) Carbonyl and (b) 1,4-hydroxynitrate. Also shown is estimate of the OH exposure and the measured NO/NO2 ratio during the experiment. (Color figure available online.)
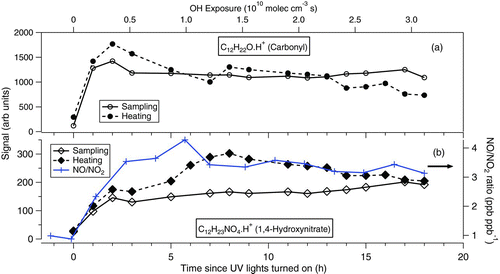
Using the elemental composition and the structure of the carbonyl and 1,4-HN, we can estimate their subcooled liquid vapor pressures, and thus their saturation concentrations,Co i , allowing us to predict their particle-phase fraction (Donahue et al. Citation2006, Citation2011). Note that the Co i reported here does not take into account the activity coefficient of the compounds in a mixture. Subcooled liquid vapor pressures were calculated (Lindsay Yee, personal communication) using the EVAPORATION model (Compernolle et al. Citation2011). For the carbonyl, Co i is estimated to be ∼1.2 × 105 μg/m3, suggesting that it is an intermediate-volatility (300 < Co i < 3 × 106 μg/m3) organic compound. In the case of 1,4-HN, Co i is estimated to be ∼57 μg/m3, indicating that it is a semivolatile (0.3 < Co i < 300 μg/m3) organic compound, and therefore should readily partition between gas and particle-phases compared to the carbonyl. The volatility classes mentioned above are based on Donahue et al. (Citation2012).
Adsorption of gases onto the MOVI manifold and post during sampling cycle, as noted above, is a known artifact for all impaction techniques. Therefore, the actual particle-phase mass loading will be lower than that of implied by the signal measured during the heating cycle for intermediate-volatility and semivolatile organic compounds. This could be one of the reasons for the carbonyl being observed during the heating cycle. Ongoing MOVI design improvements are aimed at further reducing this adsorption artifact. The other possibility for observing the carbonyl during a thermal desorption could be thermal decomposition of oligomeric compounds. Future data analysis and the MOVI passing efficiency for such a carbonyl will investigate this aspect.
5. CONCLUSIONS
The MOVI-HRToF-CIMS instrument is unique in that it provides, within a compact field-deployable package, (1) quantifiable molecular-level information for both gas and particle-phase oxygenated organic species on a semicontinuous timescale across a large range of volatilities, (2) the ability to efficiently probe oxidation and SOA formation mechanisms by estimating the elemental composition of the detected compounds, and (3) relative volatility information of the detected compounds using the programmed thermal desorption information from the MOVI. These three traits make the MOVI-HRToF-CIMS, unique and complementary to other techniques that are presently available for OA and OG analysis. In particular, one of the benefits of our approach apparent from the tests we described is an ability to study highly oxidized organic compounds without temporal limitations or other restrictions induced by preseparation. Chromatographic preseparation has its own advantages, and the desired time response and range of compounds of interest ultimately must be weighed. The prototype instrument described here is in the early stages of application, yet we have demonstrated that it has the sensitivity and precision necessary for making in-situ observations of gas and particle composition in a range of environments. Further improvements in the MOVI collection and desorption approaches are ongoing, aimed at continuing to address the well-known challenges of sampling intermediate and semi volatility compounds while preserving gas/particle partitioning between the atmosphere and instrument. Development and characterization of other CI schemes is also proceeding to further expand the range of detectable compounds.
uast_a_712236_sup_27528072.zip
Download Zip (237.6 KB)Acknowledgments
The MOVI-HRToF-CIMS development was funded by National Science Foundation CAREER award (NSF ATM-0846183) to JAT, Department of Energy SBIR grant to Aerodyne Research Inc. (DE-SC0004577), NSF ATM-0919189, and a CU Innovative Research Program Seed Grant to JLJ and a NASA Earth and Space Science Graduate Fellowship (07-Earth07F-0069) to RLNY. We acknowledge Jill Craven, Christine Loza, and Lindsay Yee of the Caltech chamber group for carrying out the cyclododecane experiments shown here and Prof. John Seinfeld, Prof. Paul Ziemann, Dr. Manjula Canagaratna, and Prof. Nga Lee (Sally) Ng for guidance and discussions during the Caltech chamber experiments.
[Supplementary materials are available for this article. Go to the publisher's online edition of Aerosol Science and Technology to view the free supplementary files.]
REFERENCES
- Aiken , A. C. , DeCarlo , P. F. , Kroll , J. H. , Worsnop , D. R. , Huffman , J. A. Docherty , K. S. 2008 . O/C and OM/OC Ratios of Primary, Secondary, and Ambient Organic Aerosols with High-Resolution Time-of-Flight Mass Spectrometry . Environ. Sci. Technol. , 42 : 4478 – 4485 .
- Aschmann , S. M. , Arey , J. and Atkinson , R. 2011 . Reactions of OH Radicals with C6–C10 Cycloalkanes in the Presence of NO: Isomerization of C7–C10 Cycloalkoxy Radicals . J. Phys. Chem. A , 115 : 14452 – 14461 .
- Bartmess , J. E. 1989 . Gas-phase Equilibrium Affinity Scales and Chemical Ionization Mass Spectrometry . Mass. Spectrom. Rev. , 8 : 297 – 343 .
- Bertram , T. H. , Kimmel , J. R. , Crisp , T. A. , Ryder , O. S. , Yatavelli , R. L. N. Thornton , J. A. 2011 . A Field-Deployable, Chemical Ionization Time-of-Flight Mass Spectrometer . Atmos. Meas. Tech. , 4 : 1471 – 1479 .
- Bertram , T. H. , Thornton , J. A. and Riedel , T. P. 2009 . An Experimental Technique for the Direct Measurement of N2O5 Reactivity on Ambient Particles . Atmos. Meas. Tech. , 2 : 231 – 242 .
- Blake , R. S. , Whyte , C. , Hughes , C. O. , Ellis , A. M. and Monks , P. S. 2004 . Demonstration of Proton-Transfer Reaction Time-of-Flight Mass Spectrometry for Real-Time Analysis of Trace Volatile Organic Compounds . Anal. Chem. , 76 : 3841 – 3845 .
- Chebbi , A. and Carlier , P. 1996 . Carboxylic Acids in the Troposphere, Occurrence, Sources, and Sinks: A Review . Atmos. Environ. , 30 : 4233 – 4249 .
- Chow , J. C. , Doraiswamy , P. , Watson , J. G. , Chen , L. -W. A. , Ho , S. S. H. and Sodeman , D. A. 2008 . Advances in Integrated and Continuous Measurements for Particle Mass and Chemical Composition . JAWMA , 58 : 141 – 163 .
- Compernolle , S. , Ceulemans , K. and Muller , J. T. 2010 . Vapor Pressure Estimation Methods Applied to Secondary Organic Aerosol Constituents from α-pinene Oxidation: An Intercomparison Study . Atmos. Chem. Phys. , 10 : 6271 – 6282 .
- Compernolle , S. , Ceulemans , K. and Muller , J.-F. 2011 . EVAPORATION: A New Vapour Pressure Estimation Method for Organic Molecules Including Non-Additivity and Intramolecular Interactions . Atmos. Chem. Phys. , 11 : 9431 – 9450 .
- de Gouw , J. A. and Warneke , C. 2007 . Measurements of Volatile Organic Compounds in the Earth's Atmosphere Using Proton-Transfer-Reaction Mass Spectrometry . Mass. Spectrom. Rev. , 26 : 223 – 257 .
- de Gouw , J. A. , Warneke , C. , Karl , T. , Eerdekens , G. , Veen , C. and Fall , R. 2003 . Sensitivity and Specificity of Atmospheric Trace Gas Detection by Proton-Transfer-Reaction Mass Spectrometry . Int. J. Mass Spectrom. , 223–224 : 365 – 382 .
- DeCarlo , P. F. , Kimmel , J. R. , Trimborn , A. , Northway , M. J. , Jayne , J. T. Aiken , A. C. 2006 . Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer . Anal. Chem. , 78 : 8281 – 8289 .
- DeCarlo , P. F. , Slowik , J. G. , Worsnop , D. R. , Davidovits , P. and Jimenez , J. L. 2004 . Morphology and Density Characterization by Combined Mobility and Aerodynamic Diameter Measurements. Part 1: Theory . Aerosol Sci. Technol. , 38 : 1185 – 1205 .
- Donahue , N. M. , Epstein , S. A. , Pandis , S. and Robinson , A. L. 2011 . A Two-Dimensional Volatility Basis Set: 1. Organic-Aerosol Mixing Thermodynamics . Atmos. Chem. Phys. , 11 : 3303 – 3318 .
- Donahue , N. M. , Kroll , J. H. , Pandis , S. N. and Robinson , A. L. 2012 . A Two-Dimensional Volatility Basis Set—Part 2: Diagnostics of Organic-Aerosol Evolution . Atmos. Chem. Phys. , 12 : 615 – 634 .
- Donahue , N. M. , Robinson , A. L. , Stanier , C. O. and Pandis , S. N. 2006 . Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics . Environ. Sci. Technol. , 40 : 2635 – 2643 .
- Drewnick , F. , Hings , S. S. , DeCarlo , P. F. , Jayne , J. T. , Gonin , M. Fuhrer , K. 2005 . A New Time-of-Flight Aerosol Mass Spectrometer (TOF-AMS) - Instrument Description and First Field Deployment . Aerosol Sci. Technol. , 39 : 637 – 658 .
- Farmer , D. K. and Jimenez , J. L. 2010 . Real-Time Atmospheric Chemistry Field Instrumentation . Anal. Chem. , 82 : 7879 – 7884 .
- Fu , P. Q. , Kawamura , K. , Pavuluri , C. M. , Swaminathan , T. and Chen , J. 2010 . Molecular Characterization of Urban Organic Aerosol in Tropical India: Contributions of Primary Emissions and Secondary Photooxidation . Atmos. Chem. Phys. , 10 : 2663 – 2689 .
- Fu , P. , Kawamura , K. and Miura , K. 2011 . Molecular Characterization of Marine Organic Aerosols Collected During a Round-the-World Cruise . J. Geophys. Res. , 116 : D13302 doi: 10.1029/2011JD015604
- Gao , Y. , Hall , W. A. and Johnston , M. V. 2010 . Molecular Composition of Monoterpene Secondary Organic Aerosol at Low Mass Loadings . Environ. Sci. Technol. , 44 : 7897 – 7902 .
- Goldstein , A. H. and Galbally , I. E. 2007 . Known and Unexplored Organic Constituents in the Earth's Atmosphere . Environ. Sci. Technol. , 41 : 1515 – 1521 .
- Goldstein , A. H. , Koven , C. D. , Heald , C. L. and Fung , I. Y. 2009 . Biogenic Carbon and Anthropogenic Pollutants Combine to form a Cooling Haze Over the Southeastern United States . PNAS , 106 : 8835 – 8840 .
- Graham , B. , Guyon , P. , Taylor , P. E. , Artaxo , P. , Maenhaut , W. Glovsky , M. M. 2003 . Organic Compounds Present in the Natural Amazonia Aerosol: Characterization by Gas Chromatography - Mass Spectrometry . J. Geophys. Res. , 108 : 4766 – 4779 .
- Graus , M. , Muller , M. and Hansel , A. 2010 . High Resolution PTR-TOF: Quantification and Formula Confirmation of VOC in Real Time . J. Am. Soc. Mass Spectrom. , 21 : 1037 – 1044 .
- Hall , W. A. and Johnston , M. V. 2011 . Oligomeric Content of α-Pinene Secondary Organic Aerosol . Aerosol Sci. Technol. , 45 : 37 – 45 .
- Hallquist , M. , Wenger , J. C. , Baltensperger , U. , Rudich , Y. , Simpson , D. Claeys , M. 2009 . The Formation, Properties and Impacts of Secondary Organic Aerosol: Current and Emerging Issues . Atmos. Chem. Phys. , 9 : 5155 – 5236 .
- Harrington , R. F. , Gertler , A. W. , Grosjean , D. and Amar , P. 1993 . Formic Acid and Acetic Acid in the Western Sierra Nevada, California . Atmos. Environ. , 27A : 1843 – 1849 .
- Heald , C. L. , Ridley , D. A. , Kreidenweis , S. M. and Drury , E. E. 2010 . Satellite Observations Cap the Atmospheric Organic Aerosol Budget . Geophys. Res. Lett. , 37 : L24808 doi: 10.1029/2010GL045095
- Hearn , J. D. and Smith , G. D. 2004 . A Chemical Ionization Mass Spectrometry Method for the Online Analysis of Organic Aerosols . Anal. Chem. , 76 : 2820 – 2826 .
- Heaton , K. J. , Sleighter , R. L. , Hatcher , P. G. , Hall , W. A. and Johnston , M. V. 2009 . Composition Domains in Monoterpene Secondary Organic Aerosol . Environ. Sci. Technol. , 43 : 7797 – 7802 .
- Holzinger , R. , Kasper-Giebl , A. , Staudinger , M. , Schauer , G. and Rockmann , T. 2010 . Analysis of the Chemical Composition of Organic Aerosol at the Mt. Sonnblick Observatory Using a Novel High Mass Resolution Thermal-Desorption Proton-Transfer-Reaction Mass-Spectrometer (hr-TD-PTR-MS) . Atmos. Chem. Phys. , 10 : 10111 – 10128 .
- Jimenez , J. L. , Canagaratna , M. R. , Donahue , N. M. , Prevot , A. S. H. , Zhang , Q. Kroll , J. H. 2009 . Evolution of Organic Aerosols in the Atmosphere . Science , 326 : 1525 – 1529 .
- Jordan , A. , Haidacher , S. , Hanel , G. , Hartungen , E. , Mark , L. Seehauser , H. 2009 . A High Resolution and Highsensitivity Proton-Transfer Time-of-Flight Mass Spectrometer (PTR-TOF-MS) . Internation Journal of Mass Spectrometry , 286 : 122 – 128 .
- Junninen , H. , Ehn , M. , Petaja , T. , Luosujarvi , L. , Kotiaho , T. Kostiainen , R. 2010 . A High-Resolution Mass Spectrometer to Measure Atmospheric Ion Composition . Atmos. Meas. Tech. , 3 : 1039 – 1053 .
- Kawamura , K. and Sakaguchi , F. 1999 . Molecular Distributions of Water Soluble Dicarboxylic Acids in Marine Aerosols Over the Pacific Ocean Including Tropics . Journal of Geophysical Research-Atmospheres , 104 : 3501 – 3509 .
- Kimmel , J. R. , Farmer , D. K. , Cubison , M. J. , Sueper , D. , Tanner , C. Nemitz , E. 2010 . Real-Time Aerosol Mass Spectrometry with Millisecond Resolution . Int. J. Mass Spectrom. , 303 : 15 – 26 .
- Koop , T. , Bookhold , J. , Shiraiwa , M. and Poschl , U. 2011 . Glass Transition and Phase State of Organic Compounds: Dependency on Molecular Properties and Implication for Secondary Organic Aerosols in the Atmosphere . Phys. Chem. Chem. Phys. , 13 : 19238 – 19255 .
- Kroll , J. H. , Donahue , N. M. , Jimenez , J. L. , Kessler , S. H. , Canagaratna , M. R. Wilson , K. R. 2011 . Carbon Oxidation State as a Metric for Describing the Chemistry of Atmospheric Orgnaic Aerosol . Nature Chemistry , 3 : 133 – 139 .
- Li , M. , McDow , S. R. , Tollerud , D. J. and Mazurek , M. A. 2006 . Seasonal Abundance of Organic Molecular Markers in Urban Particulate Matter from Philadelphia, PA . Atmos. Environ. , 40 : 2260 – 2273 .
- Lim , Y. B. and Ziemann , P. J. 2009 . Chemistry and Secondary Organic Aerosol Formation From OH Radical-Initiated Reactions of Linear, Branched, and Cyclic Alkanes in the Presence of NOx . Aerosol Sci. Technol. , 43 : 604 – 619 .
- Lindinger , W. , Hansel , A. and Jordan , A. 1998 . On-Line Monitoring of Volatile Organic Compounds at pptv Levels by Means of Proton-Transfer-Reaction Mass Spectrometry (PTR-MS); Medical Applications, Food Control, and Environmental Research (1998) . Int. J. Mass. Spectrom. Ion Proc. , 173 : 191 – 241 .
- Mochida , M. , Kitamori , Y. , Kawamura , K. , Nojiri , Y. and Suzuki , K. 2002 . Fatty Acids in the Marine Atmosphere: Factors Governing Their Concentrations and Evaluations of Organic Films on Sea-Salt Particles . J. Geophys. Res. , 107 : 4325 – 4334 .
- Mochida , M. , Miyakawa , T. , Takegawa , N. , Morino , Y. , Kawamura , K. and Kondo , Y. 2008 . Significant Alteration in the Hygroscopic Properties of Urban Aerosol Particles by the Secondary Formation of Organics . Geophys. Res. Lett. , 35 : L02804 doi: 10.1029/2007GL031310
- Murphy , D. M. and Thomson , D. S. 1995 . Laser Ionization Mass Spectroscopy of Single Aerosol Particles . Aerosol Sci. Technol. , 22 : 237 – 249 .
- Nash , D. G. , Baer , T. and Johnston , M. V. 2006 . Aerosol Mass Spectrometry: An Introductory Review . Int. J. Mass Spectrom. , 258 : 2 – 12 .
- Ng , N. L. , Canagaratna , M. R. , Jimenez , J. L. , Chhabra , P. S. , Seinfeld , J. H. and Worsnop , D. R. 2011 . Changes in Organic Aerosol Composition with Aging Inferred from Aerosol Mass Spectra . Atmos. Chem. Phys. , 11 : 6465 – 6474 .
- Novakov , T. and Penner , J. E. 1993 . Large Contribution of Organic Aerosols to Cloud-Condensation Nuclei Concentrations . Nature , 365 : 823 – 826 .
- Oktem , B. , Tolocka , M. P. and Johnston , M. V. 2004 . On-Line Analysis of Organic Components in Fine and Ultrafine Particles by Photoionization Aerosol Mass Spectrometry . Anal. Chem. , 76 : 253 – 261 .
- Paulot , F. , Crounse , J. D. , Kjaergaard , H. G. , Kurten , A. St. , Clair , J. M. Seinfeld , J. H. 2009 . Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene . Science , 325 : 730 – 733 .
- Perraud , V. , Burns , E. A. , Ezell , M. J. , Johnson , S. N. , Yu , Y. Alexander , M. L. 2012 . Nonequilibrium Atmomspheric Secondary Organic Aerosol Formation and Growth . PNAS , 109 : 2836 – 2841 .
- Pope , A. C. , Ezzati , M. and Dockery , D. W. 2009 . Fine-Particulate Air Pollution and Life Expectancy in the United States . N. Engl. J. Med. , 360 : 376 – 386 .
- Prather , K. A. , Nordmeyer , T. and Salt , K. 1994 . Real-Time Characterization of Individual Aerosol Particles Using Time-of-Flight Mass Spectrometry . Anal. Chem. , 66 : 1403 – 1407 .
- Quinn , P. K. , Bates , T. S. , Baynard , T. , Clarke , A. D. , Onasch , T. B. Wang , W. 2005 . Impact of Particulate Organic Matter on the Relative Humidity Dependence of Light Scattering: A Simplified Parameterization . Geophys. Res. Lett. , 32 : L22809 doi: 10.1029/2005GL024322
- Reinhardt , A. , Emmenegger , C. , Gerrits , B. , Panse , C. , Dommen , J. Baltensperger , U. 2007 . Ultrahigh Mass Resolution and Accurate Mass Measurements as a Tool to Characterize Oligomers in Secondary Organic Aerosols . Anal. Chem. , 79 : 4074 – 4082 .
- Roberts , J. M. , Veres , P. , Werneke , C. , Neuman , J. A. , Washenfelder , R. Brown , S. S. 2010 . Measurement of HONO, HNCO, and Other Inorganic Acids by Negative-Ion Proton-Transfer Chemical-Ionization Mass Spectrometry (NI-PT-CIMS): Application to Biomass Burning Emissions . Atmos. Meas. Tech. , 3 : 981 – 990 .
- Roberts , J. M. , Veres , P. R. , Cochran , A. K. , Warneke , C. , Burling , I. R. Yokelson , R. J. 2011 . Isocyanic Acid in the Atmosphere and its Possible Link to Smoke-related Health Effects . PNAS , 108 : 8966 – 8971 .
- Robinson , A. L. , Donahue , N. M. , Shrivastava , M. K. , Weitkamp , E. A. , Sage , A. M. Grieshop , A. P. 2007 . Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging . Science , 315 : 1259 – 1262 .
- Sorooshian , A. , Feingold , G. , Lebsock , M. D. , Jiang , H. and Stephens , G. 2009 . On the Precipitation Susceptibility of Clouds to Aerosol Perturbations . Geophys. Res. Lett. , 36 : L13803 doi: 10.1029/2009GL038993
- Spanel , P. and Smith , D. 1995 . Reactions of Hydrated Hydronium Ions and Hydrated Hydroxide Ions with Some Hydrocarbons and Oxygen-Bearing Organic Molecules . J. Phys. Chem. A , 99 : 15551 – 15556 .
- Sullivan , R. C. and Prather , K. A. 2005 . Recent Advances in Our Understanding of Atmospheric Chemistry and Climate Made Possible by On-Line Aerosol Analysis Instrumentation . Anal. Chem. , 77 : 3861 – 3886 .
- Tolocka , M. P. , Jang , M. , Ginter , M. J. , Cox , F. J. , Kamens , R. M. and Johnston , M. V. 2004 . Formation of Oligomers in Secondary Organic Aerosols . Environ. Sci. Technol. , 38 : 1428 – 1434 .
- Vaden , T. D. , Imre , D. , Beranek , J. , Shrivastava , M. and Zelenyuk , A. 2011 . Evaporation Kinetics and Phase of Laboratory and Ambient Secondary Orgnaic Aerosol . PNAS , 108 : 2190 – 2195 .
- Vaden , T. D. , Song , C. , Zaveri , R. A. , Imre , D. and Zelenyuk , A. 2010 . Morphology of Mixed Primary and Secondary Organic Particles and the Adsorption of Spectator Organic Gases During Aerosol Formation . PNAS , 107 : 6658 – 6663 .
- Veres , P. R. , Roberts , J. M. , Cochran , A. K. , Gillman , J. B. , Kuster , W. C. Holloway , J. S. 2011 . Evidence of Rapid Production of Organic Acids in an Urban Air Mass . Geophys. Res. Lett. , 38 : L17807 doi: 10.1029/2011GL048420
- Veres , P. R. , Roberts , J. M. , Warneke , C. , Welsh-Bon , D. , Zahniser , M. Herndon , S. 2008 . Development of Negative-Ion Proton-Transfer Chemical-Ionization Mass Spectrometry (NI-PT-CIMS) for the Measurement of Gas-Phase Organic Acids in the Atmosphere . Int. J. Mass Spectrom. , 274 : 48 – 55 .
- Virtanen , A. , Joutsensaari , J. , Koop , T. , Kannosto , J. , Yli-Pirila , P. Leskinen , J. 2010 . An Amorphous Solid State of Biogenic Secondary Organic Aerosol Particles . Nature , 467 : 824 – 827 .
- Walser , M. L. , Desyaterik , Y. , Laskin , J. , Laskin , A. and Nizkorodov , S. A. 2008 . High-Resolution Mass Spectrometric Analysis of Secondary Organic Aerosol Produced by Ozonation of Limonene . Phys. Chem. Chem. Phys. , 10 : 1009 – 1022 .
- Yatavelli , R. L. N. and Thornton , J. A. 2010 . Particulate Organic Matter Detection Using a Micro-Orifice Volatilization Impactor Coupled to a Chemical Ionization Mass Spectrometer (MOVI-CIMS) . Aerosol Sci. Technol. , 44 : 61 – 74 .
- Yu , J. , Crocker , D. R. III , Griffin , R. J. , Flagan , R. C. and Seinfeld , J. H. 1999 . Gas-Phase Ozone Oxidation of Monoterpenes: Gaseous and Particulate Products . J. Atmos. Chem. , 34 : 207 – 258 .
- Zelenyuk , A. and Imre , D. 2005 . Single Particle Laser Ablation Time-of-Flight Mass Spectrometer: An Introduction to SPLAT . Aerosol Sci. Technol. , 39 : 554 – 568 .
- Zhang , Q. , Jimenez , J. L. , Canagaratna , M. R. , Ulbrich , I. M. , Ng , N. L. Worsnop , D. R. 2011 . Understanding Atmospheric Organic Aerosols Via Factor Analysis of Aerosol Mass Spectrometry: A Review . Analytical and Bioanalytical Chemistry , 401 : 3045 – 3067 .