Abstract
Organonitrate (ON) groups are thought to be important substituents in secondary organic aerosols (SOAs). Model simulations and laboratory studies indicate a large fraction of ON groups in aerosol particles, but much lower quantities are observed in the atmosphere. Hydrolysis of ON groups in aerosol particles has been proposed recently to account for this discrepancy. To test this hypothesis, we simulated formation of ON molecules in a reaction chamber under a wide range of relative humidity (RH) (0 to 90%). The mass fraction of ON groups (5 to 20% for high-NOx experiments) consistently decreased with increasing RH, which was best explained by hydrolysis of ON groups at a rate of 4 day−1 (lifetime of 6 h) for reactions under RH greater than 20%. In addition, we found that secondary nitrogen-containing molecules absorb light, with greater absorption under dry and high-NOx conditions. This work provides the first evidence for particle-phase hydrolysis of ON groups, a process that could substantially reduce ON group concentration in atmospheric SOAs.
Copyright 2012 American Association for Aerosol Research
1. INTRODUCTION
Atmospheric organonitrate (ON) molecules are produced by oxidation of volatile organic compounds (VOCs) in the presence of NOx. Major oxidants in the atmosphere are OH radicals, ozone (O3), and NO3 radicals, and the VOCs responsible for secondary organic aerosol (SOA) formation are largely composed of aromatics, alkanes, and alkenes. The most important step during photochemical oxidation in which ON groups (ONO2) form is the reaction of peroxy radicals (RO2) with NO (Roberts Citation1990):
Despite these model predictions, direct measurements of particle-phase ON concentrations in both laboratory and ambient studies are scarce due to limitations of measuring techniques and ON concentrations often must be inferred. For example, Zaveri et al. (Citation2010) inferred up to ∼0.4 μg m−3 of particulate ON (corresponding to <5% of total particulate mass) in a nocturnal power plant plume based on particle acidity and gas-particle equilibrium analysis of Aerodyne quadrupole aerosol mass spectrometer (Q-AMS) measurements. While aerosol mass spectroscopy is the most commonly employed technique for measuring the chemical composition of OM, it is not ideal for directly measuring ON concentrations (Farmer et al. Citation2010). Bruns et al. (Citation2010) suggest that ON molecules can only be unambiguously identified by high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) when the ratio of ON groups to inorganic nitrate is larger than 3:20 to 3:5, which is 2 to 7 times higher than reported ratio of organic-to-inorganic nitrate measured in the atmosphere (Day et al. Citation2010). An alternative method of measuring the OM chemical composition is Fourier transform infrared (FTIR) spectroscopy (Maria et al. Citation2002). Measuring particle-phase ON groups using FTIR can be traced back to the 1990s (Allen et al. Citation1994), but quantification of ON group mass was not possible until recently (Day et al. Citation2010). Using the FTIR technique, ON groups were quantified as 3% of OM in fine particles in coastal Southern California (Day et al. Citation2010; Liu et al. Citation2011) and below detection limit (DL) in most other regions. The difference between observations and model predictions suggests a significant sink of ON exists that operates on timescales of several hours. One possibility proposed by Day et al. (Citation2010) and Russell et al. (Citation2011) is that ON groups hydrolyze under ambient conditions to give alcohols and nitric acid:
In this study, we simulated formation of SOA that contains ON groups in a reaction chamber under controlled conditions. We used an aromatic hydrocarbon, an important compound class that typically accounts for 20 to 30% of VOCs in urban areas (Lim and Ziemann Citation2005), as the SOA precursor. Specifically, 1,2,4-trimethylbennzene (TMB) was selected as the model precursor because of its abundance in aromatic hydrocarbons, for example, 1,2,4-TMB accounts for ∼10% of the measured light aromatic hydrocarbons in a Los Angeles roadway tunnel (Fraser et al. Citation1998). We focused on effects of RH and VOC-to-NOx ratio, the two environmentally relevant variables, on formation and loss of ON groups. To probe for possible formation of “brown carbon” in our experiments, we investigate optical properties of nitrogen-containing SOA components. These measurements are built on complementary particle- and gas-phase measurements described below.
2. EXPERIMENTAL
2.1. Experimental Procedures
SOAs were produced in a temperature-controlled (25°C), 10.6 m3 Teflon reaction chamber at Pacific Northwest National Laboratory (Richland, WA, USA). The chamber is surrounded by 104 UV blacklights (Q-labs, UV-340) with their output centered at 340 nm. A custom Teflon-coated stainless steel manifold is used to inject reactants and pure air into the chamber and to sample aerosol and gas-phase species. Teflon lines were used for the sampling of ozone and NOx, stainless steel lines were used to sample aerosol particles, and a PEEK line was used to sample VOCs with the proton transfer reaction mass spectrometer (PTR-MS). HONO was used as the NOx and OH radical initiator and was generated by reaction of NaNO2 (Sigma-Aldrich, ≥ 97%) with excess H2SO4 (Sigma-Aldrich, 99.999%) in a glass bulb at room temperature. The resultant HONO was transferred to the chamber by flushing the bulb with 1.0 lpm of pure air. For low-NOx experiments, H2O2 was added to keep the OH radical concentrations generated in each experiment approximately equal. The 1,2,4-TMB (Sigma-Aldrich, 98%), the VOC precursor, was introduced into the chamber by injecting the desired volume into a gently warmed glass bulb and flushing 1.0 lpm of pure air through the bulb for 20 min. After the injection, we allow 60 to 90 min for the HONO and TMB to become homogenously mixed in the chamber, as observed by the NOx analyzer and PTR-MS. The reaction was initiated by turning on all 104 lights, which photolyzed the HONO and/or H2O2 to generate OH radicals that reacted with TMB. The pure air was generated with an AADCO pure air generator. For experiments with relative humidity (RH) greater than 0%, the chamber was conditioned with humid air, which was produced by mixing pure air (RH = 0%) with water vapor-saturated air (RH = 100%) generated from a heated water bubbler (18 MΩ cm). By adjusting flow rates of the dry and saturated air, desired chamber RH can be achieved. A HEPA filter was placed downstream of the water bubbler to eliminate particles. No seed aerosol was used in this study. The chamber was continuously flushed with pure air between all experiments.
Experimental conditions are summarized in . Two sets of experiments were conducted to study the effects of (1) RH and (2) NOx on formation and loss of ON groups. The first set of experiments (12–28 March) were conducted under high-NOx conditions (TMB:NOx ∼0.2) with RH being the only changing variable (varied from 0 to 90%). In the second set of experiments (April 2–7), SOA was produced with intermediate-NOx (TMB:NOx ∼2) and low-NOx (TMB:NOx ∼10) levels under dry (∼0%) and intermediate RH (∼50%).
TABLE 1 Experimental conditions including initial mixing ratios of TMB and NOx, RH, total organic loading (M org), mass fraction of ON groups (FON), yield of OM (Y OM), molar ratio of alkane and ON groups (R mole) in SOA, and availability of PSAP measurements.
2.2. Particle- and Gas-Phase Measurements
A suite of particle- and gas-phase instruments was deployed around the chamber to measure chemical composition of SOA components. One filter (Teflo, Pall Inc., Ann Arbor, MI, USA) sample was collected for each experiment to quantify organic functional group mass using a Bruker Tensor 37 FTIR (Bruker, Waltham, MA, USA). The filter sample collection began ∼1 h after the lights were turned on, when SOA concentration reached its maximum, and typically lasted 3–4 h. The filter samples were measured immediately after sample collection with FTIR. Before each IR scan, the FTIR sample chamber (with sample in it) was purged with pure N2 for 3 min to eliminate interference of CO2 and gas and liquid-phase water. Organic functional groups, including alkane, hydroxyl, carboxylic acid, nonacid carbonyl, and ON groups, were quantified using algorithms described previously (Gilardoni et al. Citation2009; Liu et al. Citation2009, Citation2011; Russell et al. Citation2009; Day et al. Citation2010). The total FTIR-measured OM mass correlated well to the mass quantified by a scanning mobility particle sizer (SMPS, TSI 3080) (assuming a density of 1.4 g cm−3 for SOA particles) with an R of 0.90 and a slope of 1.05, well within the uncertainties of FTIR and SMPS.
In addition to the sample collected for FTIR analysis, one separate sample from each experiment was collected for X-ray analysis. This sample collection started ∼1 h after the lights were on and lasted ∼30 min. Single-particle X-ray absorption spectra were acquired using scanning transmission X-ray microscopy (STXM) and near-edge X-ray absorption fine structure (NEXAFS) at the Advanced Light Source (Lawrence Berkeley National Laboratory, CA, USA). Single-particle functional groups, including alkane, carboxylic acid, hydroxyl, ketone, and alkene groups were measured. Detailed analysis procedures are described in Takahama et al. (Citation2010).
An Aerodyne HR-ToF-AMS (Billerica, MA, USA) was used to measure SOA mass loadings with ∼10 min time resolution. In this instrument, particles focused by an aerodynamic lens impact on a heated surface (600°C). Nonrefractory components flash vaporize and are ionized and fragmented by electron impaction. The fragments are analyzed by a time-of-flight mass spectrometer (Tofwerk AG, Thun, Switzerland). In addition, particle size is measured by particle time-of-flight between a rotating chopper, which modulates the particle beam, and the vaporizer (Jayne et al. Citation2000), providing time-resolved size distribution measurements of particle fragments. A detailed description of HR-ToF-AMS is provided in DeCarlo et al. (Citation2006).
Light absorption of SOA particles was continuously measured using a particle/soot absorption photometer (PSAP, Radiance Research, Seattle, WA, USA). The absorption coefficient was quantified by measuring the change in transmittance across a filter on which SOA particles impacted. In this instrument, absorption at three wavelengths was monitored, representing blue (467 nm), green (530 nm), and red (660 nm) light. The absorption coefficient retrieved from the PSAP measurements was corrected for flowrate, spot size, and light scattering interferences following the procedures described by Bond et al. (Citation1999) and Ogren (Citation2010). The light scattering coefficient was calculated from the SMPS-derived particle size distribution and an assumed refractive index of 1.45 + 0i (Kim et al. Citation2010) for the 30 min average around the peak absorption. The scattering calculations were performed using conventional Mie theory, which assumes the particles to be spherical. Note that the corrected absorption coefficient appeared slightly negative for experiments conducted on 5 and 7 April when the raw (uncorrected) absorption coefficient of SOA was low, indicating that the light scattering coefficient might be slightly overestimated. However, we focus on the relative change in absorption coefficient to study the link between SOA chemical composition and light absorbing properties.
Additional measurements include: (1) O3 measured by an ultraviolet photometric O3 analyzer (Thermo Electron Instruments, Model 49C), (2) NO and NOx monitored by a NO–NO2–NOx Analyzer (Thermo Environmental Instruments, 42C), (3) 1,2,4-TMB mixing ratio recorded by a PTR-MS (Ionicon; Lindinger et al. Citation1998), and (4) RH and temperature were monitored by a Rotronic hygroclip (SC05) sensor.
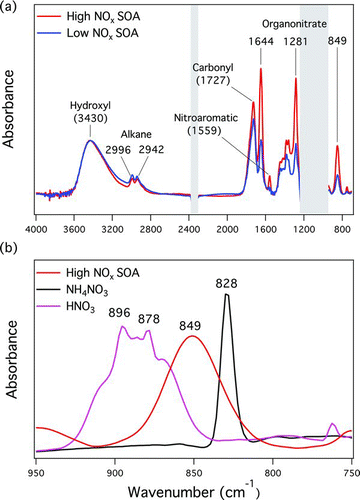
FIG. 1 (a) Representative FTIR spectra for SOA produced under high-NOx (23 March) and low-NOx conditions (2 April). Absorptions near 1100 and 2360 cm−1 (grey areas) are not shown due to interferences by Teflon substrates and CO2, respectively. (b) Comparison of IR spectra in the 750 to 950 cm−1 region for high-NOx SOA, ammonium nitrate, and condensed-phase nitric acid. Spectra are normalized in order to show differences. (Color figure available online.)
2.3. Identification and Quantification of ON Groups
ON groups were observed in all SOA samples, as indicated by peaks at 1644, 1281, and 849 cm−1 in the IR spectra () (Roberts Citation1990). To confirm that there was no interference from inorganic nitrates, most likely ammonium nitrate, an IR spectrum of ammonium nitrate was compared to the spectra of SOA samples (). The ammonium nitrate sample was acquired by atomizing ammonium nitrate solution followed by particle collection on Teflon filter at a sampling rate of ∼4 L min−1, the same rate used for SOA sample collection. A sharp peak at 828 cm−1 was observed, consistent with documented peak locations for inorganic nitrate ions (Miller and Wilkins Citation1952). The distinct IR spectra of ON groups and inorganic nitrate exclude the existence of inorganic nitrate salts, which is expected since ammonia was not present in the reaction system. A sample of condensed-phase nitric acid was prepared by applying a droplet of nitric acid to the Teflon filter. The observed vibration peaks between 860 and 900 cm−1 of nitric acid () were consistent with reported peak assignments (McGraw et al. Citation1965; Maki and Wells Citation1992; Sporleder and Ewing Citation2001). These peaks differ significantly from the 849 cm−1 peak of ON groups and were not observed in SOA samples, suggesting that nitric acid, a terminal sink of NOx, was either not present in the particle phase or below DL of the FTIR analysis (the DL of HNO3 is not quantified, but is estimated at 0.01 μg m−3 based on the DL of ON groups). Assuming all the NOx in the chamber is converted to nitric acid, the maximum possible concentration of nitric acid in the chamber is 0.2 Pa (for 2000 ppb NOx), which is well below its vapor pressure of 8000 Pa (Duisman and Stern, 1969), suggesting that nitric acid was too volatile to condense on the particles. Handley et al. (Citation2007) observed that gas-phase nitric acid adsorbs and dissociates into a proton and a nitrate anion on dry, hydrophobic organic films at partial pressures below the saturation pressure of nitric acid, which suggests uptake of nitric acid on dry organic particles may occur. However, Zhou et al. (Citation2003) and Handley et al. (Citation2007) also demonstrated that the proton and nitrate ion are photolyzed under actinic radiation and evaporate to the gas phase. Therefore, the adsorbed nitric acid on dry particles is unlikely to be significant in these photo-oxidation experiments. In addition, the nitrate temporal profiles discussed below suggest that the observed nitrates were formed on the same timescales as the TMB oxidation, while generation of nitric acid is delayed in time. By excluding interference of inorganic nitrates (ammonium nitrate and nitric acid), the IR peak at 849 cm−1 was unambiguously assigned to ON group absorption. This peak was used to quantify ON group mass, using an absorptivity of 9.3 cm−1 μmol−1 derived by Day et al. (Citation2010).
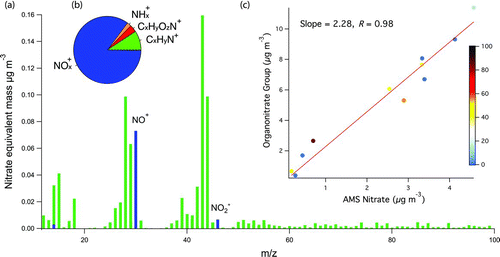
In addition to FTIR, ON groups were measured by the HR-ToF-AMS. Since ammonium was not observed from the AMS measurements and nitric acid was excluded in the particle phase by the FTIR measurements, NO+ and NO2 +, which typically accounted for more than 75% of nitrogen-containing fragments (), most likely originated from ON group parents. The ratio of NO+ to NO2 +, 8.8 ± 1.9 (), was dramatically different from the ratio for NH4NO3 (2.4) and NaNO3 (80) but consistent with the ratio for ON molecules (5–11) reported previously (Fry et al. Citation2009; Bruns et al. Citation2010). The FTIR-measured ON groups (with the formula of ONO2) and AMS-measured nitrate (derived from NO+ and NO2 +) correlated well (R = 0.98), with the former 2.28 times higher than the latter (). Possible explanation for the large slope include: (1) ON groups are fragmented by electron impact ionization and do not have a uniform probability of carrying the positive charge necessary for detection (Bruns et al. Citation2010), or the related point (2) the true relative ionization efficiency of ON molecules is lower than the value of 1.1 used to calculate nitrate mass, or (3) ON groups dissociated (during ionization processes) to form other nitrogen-containing fragments (e.g., CxHyOzN+ in ) that were small (compared to NO+ and NO2 +) and caused the scatter in .
FIG. 2 (a) Typical mass spectrum for SOA generated under high-NOx conditions (23 March). (b) Mass fraction of nitrogen-containing fragments (on 23 March). (c) Comparison of FTIR-measured ON groups (ONO2) and AMS-measured nitrate for all experiments. The RH (%) is indicated by the vertical scale. (Color figure available online.)
3. RESULTS AND DISCUSSION
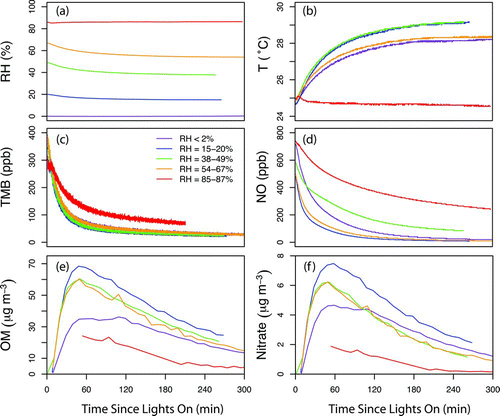
The time dependence of AMS-measured OM and nitrate is shown in and , respectively. Concentrations of OM and nitrate are highly correlated (R = 0.99), suggesting that both participated in particle growth. Size distributions of OM and nitrate showed the same peak locations (), and their ratios were independent of particle size, suggesting that ON molecules were internally mixed with other SOA components, and processing of ON molecules (if any) occurred within the particles rather than on their surface. The concentration of nitrate increased rapidly at the beginning of the reaction and reached a maximum after ∼1 h. During the first 2 h of reaction, TMB and NO decayed exponentially ( and ), suggesting that the majority of ON molecules were formed within 2 h of the start of the reaction. Decay rates of TMB and NO for dry and humid experiments were similar (except for the lower decay rates observed on 28 March with RH of ∼85%, for which the reason is not clear), which is consistent with the fact that water vapor has no effects on OH radical formation (Atkinson et al. Citation1992) at the wavelengths of light (centered at 340 nm) employed in this study. In addition, it has been shown that the reaction of RO2 and NO (reaction (1)) is not affected by RH (Matsunaga and Ziemann, Citation2009). Thus, evidence suggests that the effects of water vapor on gas-phase reactions, including formation of ON groups, were likely negligible.
FIG. 3 Time-dependence of (a) RH, (b) temperature, (c) TMB, (d) NO, (e) AMS-measured OM, and (f) AMS-measured nitrate. The AMS measurements on 28 March (RH ∼85%) were only available 1 h after the lights were on due to improper experimental setup during the first hour. (Color figure available online.)
3.1. ON Group Mass Fraction
The mass fraction of ON groups in high-NOx experiments ranged from 4.8 to 19.9% (), with higher mass fraction observed under drier conditions (). The mass fraction (4.8%) of ON groups under high-NOx and high-RH conditions (28 March) matched the mass fraction (3.2 ± 2.0%) measured at coastal Southern California (Day et al. Citation2010), a site that was significantly influenced by anthropogenic emissions (from the Los Angeles Basin) and associated with high RH. The OM yield, quantified by FTIR and defined as the ratio of OM to the mass of reacted TMB (Odum et al. Citation1996), spanned from 1.7 to 5.9% in agreement with reported SOA yields for TMB (Odum et al. Citation1996; Cocker et al. Citation2001; Wyche et al. Citation2009).
The mass fraction of ON groups decreased with increasing TMB-to-NOx ratio. The decrease is likely due to the competitive chemistry of RO2, which reacts with both HO2 and NO. Under low-NOx conditions, reaction of RO2 with HO2 begins to compete with reaction with NO (Kroll and Seinfeld Citation2008), yielding smaller amounts of ON groups.
FIG. 4 Example (15 March) time-dependence of size distributions of AMS-measured OM and nitrate. (Color figure available online.)
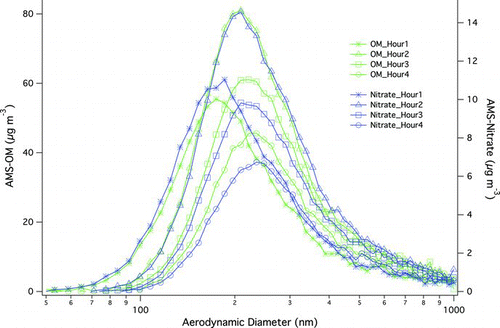
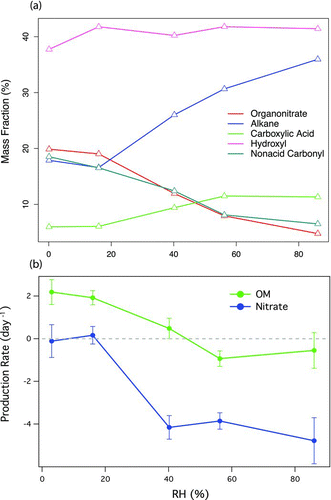
FIG. 5 (a) RH dependence of organic functional group mass fraction for the high-NOx experiments (12–28 March). The mass fractions are derived from the FTIR measurements. (b) Production rate of AMS-measured OM and nitrate. (Color figure available online.)
3.2. Hydrolysis of ON Groups
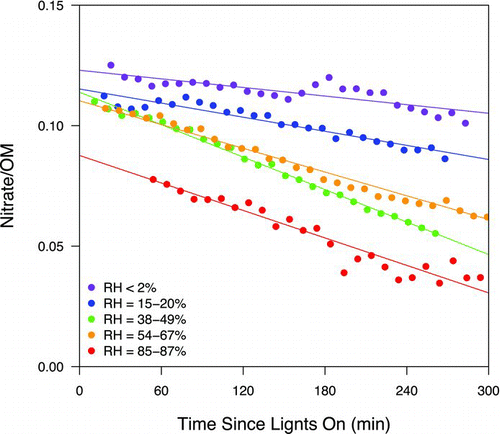
The average ON group mass fraction measured by FTIR consistently decreased with increasing RH (), especially when RH was greater than 20%. Similar reduction of ON groups in humid conditions was reported in four reaction chamber studies. Baltensperger et al. (Citation2005) and Sax et al. (Citation2005) measured the oxidation products of 1,3,5-TMB formed at 50 to 60% RH using FTIR. Both studies showed that the abundance of ON groups decreased with time, but explanations were not given. Zhang et al. (Citation2011) and Nguyen et al. (Citation2011) studied isoprene SOA formation under dry (0–40%) and humid (40–90%) conditions in the presence of high-NOx concentrations. In Zhang et al. (Citation2011), the concentrations of the ON monomer (similar to 2-methylglyceric acid (2-MG) except one of the OH groups is replaced with a tertiary ON group) and its oligoester identified in the SOA were significantly lower under humid conditions. While the decreased oligoester concentrations could be explained by constrained particle-phase esterification under humid conditions, the reduced ON monomer concentrations under elevated RH remains unresolved. In Nguyen et al. (Citation2011), the relative abundance of nitrogen-containing compounds (measured by mass spectrometry and not assigned to specific compound classes) decreased by 40% in the humid experiments. The explanation proposed by Nguyen et al. (Citation2011) was that particle-phase water suppressed condensation-type reactions that generated water. The condensation-type processes may include (1) esterification of hydroxyl groups and nitric acid to form ON groups, and (2) oligomer formation from monomers containing ON groups. Oligomers are more ionizable (than monomers) so they are more likely to be detected by the mass spectral technique used in Nguyen et al. (Citation2011). Explanation (1) was excluded in this study because significant quantities of particle-phase nitric acid—required for establishing equilibrium in esterification—was not observed, as discussed in the experimental section. Explanation (2) was not applicable to this study since quantification of ON groups by FTIR does not require ionizing organic molecules. Finally, the temporal profiles () show that SOA and ON production occurred simultaneously; if oligomerization reactions were a significant source of ON, we would expect that the nitrate signal would be delayed relative to the production of total OM.
FIG. 6 Time-dependence of nitrate-to-OM ratio (measured by the AMS). The lines represent linear fits of the measurements, with slopes of 7.1 × 10−5, 1.0 × 10−4, 2.2 × 10−4, 1.7 × 10−4, and 2.0 × 10−4 for RH < 2%, RH = 15–20%, RH = 38–49%, RH = 54–67%, and RH = 85–87% experiments, respectively. (Color figure available online.)
The decrease of ON group mass fraction (as a function of RH), an indication of ON group loss, was supported by real-time measurements of ON group loss under humid conditions (RH > 20%). To quantify the loss rate, particle wall loss, and dilution of ON group concentrations by condensation of non-nitrate SOA need to be taken into account. The wall loss rate was determined by fitting the decay curve of the integrated particle number concentration (measured by the SMPS) to an exponential decay function (Paulsen et al. Citation2005):
Based on the following observations: (1) the only changing variable was RH (for the first set of experiments), (2) water vapor did not affect gas-phase chemistry, (3) condensed-phase water increased with increasing RH (Seinfeld et al. Citation2001), and (4) the observed loss of ON group mass under humid conditions, particle-phase hydrolysis of ON groups via reaction (2) provides the most consistent explanation for the observations. According to Wyche et al. (Citation2009), one likely product of 1,2,4-TMB and OH reaction system is molecule A (), which contains a tertiary ON group and is sufficiently nonvolatile to partition into particle phase (Johnson et al. Citation2004, Citation2005; Wyche et al. Citation2009).
FIG. 7 Structure of molecule A—a likely product of 1,2,4-TMB and OH reaction system.
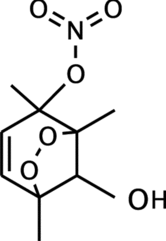
Each molecule of A has 10 alkane groups and 1 ON group, consistent with the observed molar ratio of alkane to ON groups in SOA particles () in dry conditions but much lower than the ratios for high-RH SOA. These observations suggest that product A likely hydrolyzed via reaction Equation(2) under high-RH conditions where the amounts of condensed-phase water was greatly enhanced. Assuming only tertiary ON molecules hydrolyze in aerosols based on the kinetic studies in bulk solutions (Hu et al. Citation2011), our measured particle-phase tertiary ON hydrolysis rate of 4 day−1 at RH greater than ∼20% is 200 times slower than the measured hydrolysis rate of tertiary ON molecules in dilute solutions (Hu et al., Citation2011). While the relative importance of tertiary ON groups versus primary or secondary ON groups in TMB oxidation products has not been reported, it is expected that the tertiary ON groups are produced in the alkyl group-substituted benzene carbons (e.g., product A). Since 1,2,4-TMB has three –CH3 substituted benzene carbons, we estimate that tertiary ON groups account for approximately 50% of the total ON groups in 1,2,4-TMB SOA, which is quantitatively consistent with the ∼50% decrease of ON group mass fraction after ∼5-h reaction for the RH > 20% experiments (). The slower particle-phase hydrolysis rate likely resulted from a combination of interrelated factors: limited availability of condensed-phase water and particle phase.
First, molecular diffusion coefficients for ON and water molecules in particles may be slower than in solutions, potentially reducing the hydrolysis rate. Recent studies suggest that laboratory and ambient SOA (both anthropogenic and biogenic) exist in an amorphous solid (glassy) state when RH < 30% (Virtanen et al. Citation2010; Vaden et al. Citation2011; Saukko et al. Citation2012). One likely consequence of this phase state is that molecular diffusion through the particle would be hindered. A moisture-induced phase transition of pure SOA from a solid state to a semi-solid or liquid state is suggested to occur at RH > 40% (Shiraiwa et al. Citation2011; Saukko et al. Citation2012). In the semisolid or liquid state, organic would diffuse to the organic interface much more rapidly (Vaden et al. Citation2011). The observed particle-phase ON group hydrolysis rate () is consistent with the phase transition of SOA from solid to semisolid/liquid particles: The hydrolysis rate was ∼0 and 4 day−1 (corresponding to a lifetime of 6 h) for RH < 20% and > 40% experiments, respectively. The pronounced increase of hydrolysis rate at 40% RH also agrees with the hydrolysis loss rate of N2O5 on organic particles, which is dependent on water concentration when RH < 50% and appears to be independent of water when RH is greater than 50% (Thornton et al. Citation2003).
However, even after the SOA phase transition, two separate phases, an organic-rich phase and an aqueous phase, may form as the particle continues to hydrate. This phase separation has been observed for similar laboratory SOA samples and for ambient samples (You et al. Citation2012). The separation of the organic and aqueous phase would confine the interaction of water and ON molecules to the interface of the two phases, significantly lowering the rate of hydrolysis reactions.
Second, although particle-phase hydrolysis occurs at RH > 20%, water is likely the rate-limiting reagent in our experiments. In dilute solutions, the number of water molecules consumed by hydrolysis is negligible compared to the total number of water molecules available; hydrolysis of ON molecule therefore follows a pseudo-first order rate law, where ON molecules are the rate-limiting reagent (Hu et al. Citation2011). Such an approximation is not applicable to particle-phase hydrolysis because of the limited amount of water in the particles. Assuming hygroscopic growth factors of 1.01 (20% RH), 1.02 (40% RH), 1.04 (60% RH), and 1.08 (85% RH) (Baltensperger et al. Citation2005), the molar ratio of water to ON groups is 1, 2, 5, and 21 for reactions at 20, 40, 60, and 85% RH, respectively. The comparable molar concentration of ON groups and water suggests that the hydrolysis reaction consumes significant fraction of condensed-phase water molecules and its rate is likely limited by both water and ON molecules. As a result, the establishment of hydrolysis equilibrium is expected to be slower than in dilute solutions.
The limited amount of particle-phase water may also limit dissolution of ON molecules, which likely slows down the hydrolysis process. In contrast, ON molecules will fully dissolve in dilute solutions, since the ON molecules are of intermediate solubility. For example, alkyl nitrates have Henry's law constants (H) of 1–4 M atm−1 (Roberts Citation1990), comparable to some aldehyde compounds, such as heptanal (H = 2.3 M atm−1; Yaws and Yang Citation1992) and octanal (H = 2.1 M atm−1; Zhou and Mopper Citation1990).
The hydrolysis mechanism could potentially explain the decrease of ON groups under high-RH conditions observed in previous chamber studies (Nguyen et al. Citation2011; Zhang et al. Citation2011). For example, the ON monomer identified in isoprene SOA by Zhang et al. (Citation2011) could hydrolyze to form 2-MG, resulting in greater decrease of the monomer than the decrease of 2-MG under high-RH conditions, which is consistent with the reduction of ON monomer and 2-MG (compared to dry conditions) by factors of 2.9 and 1.8, respectively.
Similar to the ON groups, the mass fraction of nonacid carbonyl groups decreased as RH increased—likely resulting from hydration of nonacid carbonyl groups (ketones or aldehydes) to form acetals or hemiacetals, which could be catalyzed by secondary carboxylic acids (Jang et al. Citation2002; Kalberer et al. Citation2004) or by small, undetected amounts of adsorbed nitric acid. As a result of ON group hydrolysis and carbonyl group hydration, the absolute mass of hydroxyl groups increased with increasing RH. However, the mass fraction of hydroxyl groups appeared independent of RH due to the enhancement of alkane and carboxylic acid group mass with increasing RH (). The alkane and carboxylic groups correlated with R of 0.95 and 0.92 in ensemble particles (measured by FTIR) and single particles (measured by STXM-NEXAFS), respectively, suggesting that they likely formed in the same molecules and condensed simultaneously on the same particles.
3.3. Light-Absorbing Properties of Nitrogen-Containing SOA
SOA formed under high-NOx and low-NOx conditions had different optical properties. The high-NOx SOA appeared yellow by eye, while the low-NOx SOA was white. The mass absorption coefficient (MAC) at 467 nm of the high-NOx SOA was more than three times higher than that of the low-NOx SOA for the dry and intermediate-RH experiments (), consistent with the observed yellow color of the high-NOx SOA. Absorption of blue light by the high-NOx SOA can be partially explained by the formation of nitroaromatic groups, which are naturally yellow (Booth Citation2007). Nitroaromatic groups form from the reaction of OH-TMB adduct (the aromatic radical produced from hydrogen abstraction of TMB by OH radicals) and NO2 and are therefore more likely to form under high-NOx conditions (). Under high-NOx conditions, the OH-TMB adduct may react with NO2 or O2, forming ON groups as well as nitroaromatic groups (Nishino et al. Citation2008, Citation2010), which are low-volatility compounds and readily partition into particle phase. In contrast, the OH-TMB adduct reacts predominately with O2 when NOx concentration is low and nitroaromatic groups do not form. Benzenenitroaromatic compounds are typically identified in highly-polluted, urban areas with high NOx emissions, such as Los Angeles (Allen et al. Citation1994). Light absorption by nitroaromatic groups is consistent with studies that suggest C–N bonds in SOA can absorb light (Galloway et al. Citation2009; Shapiro et al. Citation2009; Zhong and Jang Citation2011).
FIG. 8 Mass absorption coefficient (MAC) (left axis), ON group mass fraction (right axis), and normalized (by OM) nitroaromatic group peak area (scale is not shown) for low-RH (<2%) and intermediate-RH (∼50%) experiments (dates are shown in the parentheses), respectively. The TMB-to-NOx ratio is indicated by the vertical scale. The ON group mass fraction was determined for the 30 min average around the peak absorption for each experiment using the scaled AMS nitrate mass fraction. The scaling factor was derived by linear regression (slope = 0.60; R = 0.83) of FTIR ON group mass fraction and AMS nitrate mass fraction. (Color figure available online.)
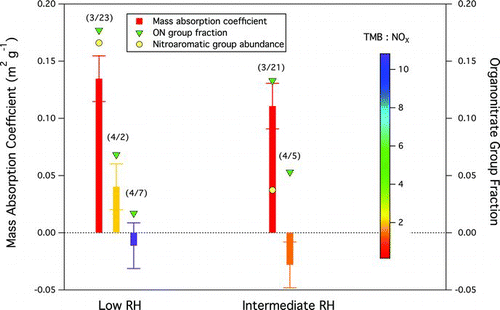
However, the difference of MAC for low-RH and intermediate-RH SOA produced under low-NOx conditions cannot be solely accounted for by nitroaromatic groups because nitroaromatic groups were not observed in the FTIR spectra when the TMB-to-NOx ratio was greater than 1 (i.e., under the conditions employed in the low-NOx experiments). Under low-NOx conditions, the RH-induced differences in absorption were consistent with mass fractions of ON groups, that is, higher MAC corresponded to greater ON group mass fraction (), suggesting that ON groups also play a role in absorbance of the 467 nm light. Further studies are needed to validate this connection.
4. CONCLUSIONS
In summary, this study demonstrates that ON groups may comprise up to 20% of the overall OM yield, but hydrolysis of ON groups in aerosol particles significantly reduces ON group mass fraction (2–4 times) under typical environmental conditions (RH > 50%). We derive a hydrolysis rate of 4 day−1 (lifetime of 6 h) from our measurements, which potentially explains the low-ON group mass fraction measured in ambient air. Future work exploring the hydrolysis of ON molecules produced from precursor VOCs without alkyl-group substitutions (e.g., benzene, linear alkanes) would potentially clarify the atmospheric relevance of hydrolysis reactions. Such precursor compounds are not expected to form tertiary ON groups (i.e., only primary and secondary ON groups are produced), and therefore, based on studies in bulk solutions, hydrolysis of these ON molecules would not be expected to occur on atmospheric-relevant time scales. In light of these observations, model predictions should take hydrolysis into account, especially in coastal regions with high-RH and high-ON group production. In addition, nitrogen-containing SOA particles absorb visible light, suggesting that these compounds are potentially important for aerosols’ effects on the energy balance of the atmosphere.
Acknowledgments
Funding for this work was provided by the Pacific Northwest National Laboratory Aerosol Climate Initiative and the National Science Foundation under grant ATM-0904203. PNNL is operated for the U.S. Department of Energy by Battelle Memorial Institute under contract DE-AC06–76RLO 1830. The authors are grateful to the Lawrence Berkeley National Laboratory Advanced Light Source Beamline 5.3.2 for providing facilities for the STXM analysis and beamline scientists David Kilcoyne and Tolek Tyliszczak for their generous help during sample analysis.
REFERENCES
- Allen , D. T. , Palen , E. J. , Haimov , M. I. , Hering , S. V. and Young , J. R. 1994 . Fourier Transform Infrared Spectroscopy of Aerosol Collected in a Low Pressure Impactor (LPI/FTIR): Method Development and Field Calibration . Aerosol Sci. Technol. , 21 : 325 – 342 .
- Atkinson , R. and Arey , J. 2003 . Atmospheric Degradation of Volatile Organic Compounds . Chem. Rev. , 103 : 4605 – 4638 .
- Atkinson , R. , Aschmann , S. M. , Arey , J. and Shorees , B. 1992 . Formation of Radicals in the Gas-Phase Reactions of O3 with a Series of Terpenes . J. Geoph. Res.-Atmosp. , 97 : 6065 – 6073 .
- Baker , J. W. and Easty , D. M. 1950 . Hydrolysis of Organic Nitrates . Nature , 166 : 156 – 156 .
- Baker , J. W. and Easty , D. M. 1952 . Hydrolytic Decomposition of Esters of Nitric Acid. Part I. General Experimental Techniques. Alkaline Hydrolysis and Neutral Solvolysis of Methyl, Ethyl, Isopropyl, and Tert-butyl Nitrates in Aqueous Alcohol . J. Chem. Soc. , 1952 : 1193 – 1207 .
- Baltensperger , U. , Kalberer , M. , Dommen , J. , Paulsen , D. , Alfarra , M. R. Coe , H. 2005 . Secondary Organic Aerosols from Anthropogenic and Biogenic Precursors . Faraday Disc. , 130 : 265 – 278 .
- Barket , D. J. , Grossenbacher , J. W. , Hurst , J. M. , Shepson , P. B. , Olszyna , K. Thornberry , T. 2004 . A study of the NOx Dependence of Isoprene Oxidation . J. Geoph. Res.-Atmospheres , 109 : D11310
- Bond , T. C. , Anderson , T. L. and Campbell , D. 1999 . Calibration and Intercomparison of Filter-based Measurements of Visible Light Absorption by Aerosols . Aerosol Sci. and Techn. , 30 : 582 – 600 .
- Booth , G. 2007 . “ Nitro Compounds, Aromatic ” . In Ullmann's Encyclopedia of Industrial Chemistry , Edited by: Booth , G. 301 – 349 . New York : John Wiley & Sons .
- Boschan , R. , Merrow , R. T. and Vandolah , R. W. 1955 . The Chemistry of Nitrate Esters . Chem. Rev. , 55 : 485 – 510 .
- Bruns , E. A. , Perraud , V. , Zelenyuk , A. , Ezell , M. J. , Johnson , S. N. Yu , Y. 2010 . Comparison of FTIR and Particle Mass Spectrometry for the Measurement of Particulate Organic Nitrates . Environ. Sci. & Techn. , 44 : 1056 – 1061 .
- Camredon , M. , Aumont , B. , Lee-Taylor , J. and Madronich , S. 2007 . The SOA/VOC/NOx System: An Explicit Model of Secondary Organic Aerosol Formation . Atmosph. Chem. and Phys. , 7 : 5599 – 5610 .
- Cocker , D. R. , Mader , B. T. , Kalberer , M. , Flagan , R. C. and Seinfeld , J. H. 2001 . The Effect of Water on Gas-particle Partitioning of Secondary Organic aerosol: II. m-xylene and 1,3,5-trimethylbenzene Photooxidation Systems . Atmosph. Environ. , 35 : 6073 – 6085 .
- Darer , A. I. , Cole-Filipiak , N. C. , O’Connor , A. E. and Elrod , M. J. 2011 . Formation and Stability of Atmospherically Relevant Isoprene-Derived Organosulfates and Organonitrates . Environ. Sci. & Technol. , 45 : 1895 – 1902 .
- Day , D. A. , Liu , S. , Russell , L. M. and Ziemann , P. J. 2010 . Organonitrate Group Concentrations in Submicron Particles with High Nitrate and Organic Fractions in Coastal Southern California . Atmosph. Environ. , 44 : 1970 – 1979 .
- DeCarlo , P. F. , Kimmel , J. R. , Trimborn , A. , Northway , M. J. , Jayne , J. T. Aiken , A. C. 2006 . Field-deployable, High-resolution, Time-of-flight Aerosol Mass Spectrometer . Analyt. Chem. , 78 : 8281 – 8289 .
- Duisman , J. A. and Stern , S. A. 1969 . Vapor Pressure and Boiling Point of Pure Nitric Acid . Journal of Chemical and Engineering Data , 14 ( 4 ) : 457 – 459 .
- Farmer , D. K. , Matsunaga , A. , Docherty , K. S. , Surratt , J. D. , Seinfeld , J. H. Ziemann , P. J. 2010 . Response of an Aerosol Mass Spectrometer to Organonitrates and Organosulfates and Implications for Atmospheric Chemistry . Proc. Nat. Acad. Sci. USA , 107 : 6670 – 6675 .
- Fraser , M. P. , Cass , G. R. and Simoneit , B. R. T. 1998 . Gas-Phase and Particle-phase Organic Compounds Emitted from Motor Vehicle Traffic in a Los Angeles Roadway Tunnel . Environ. Sci. & Techn. , 32 : 2051 – 2060 .
- Fry , J. L. , Kiendler-Scharr , A. , Rollins , A. W. , Wooldridge , P. J. , Brown , S. S. Fuchs , H. 2009 . Organic Nitrate and Secondary Organic Aerosol yield from NO(3) Oxidation of Beta-pinene Evaluated Using a Gas-phase Kinetics/aerosol Partitioning Model . Atmos. Chem. and Phys. , 9 : 1431 – 1449 .
- Galloway , M. M. , Chhabra , P. S. , Chan , A. W. H. , Surratt , J. D. , Flagan , R. C. Seinfeld , J. H. 2009 . Glyoxal Uptake on Ammonium Sulphate Seed Aerosol: Reaction Products and Reversibility of Uptake Under Dark and Irradiated Conditions . Atmosph. Chem. Phys. , 9 : 3331 – 3345 .
- Gilardoni , S. , Liu , S. , Takahama , S. , Russell , L. M. , Allan , J. D. Steinbrecher , R. 2009 . Characterization of Organic Ambient Aerosol During MIRAGE 2006 on Three Platforms . Atmosph. Chem. Phys. , 9 : 5417 – 5432 .
- Hallquist , M. , Wangberg , I. , Ljungstrom , E. , Barnes , I. and Becker , K. H. 1999 . Aerosol and Product Yields from NO3 Radical-Initiated Oxidation of Selected Monoterpenes . Environ. Sci. & Technol. , 33 : 553 – 559 .
- Handley , S. R. , Clifford , D. and Donaldson , D. J. 2007 . Photochemical Loss of Nitric Acid on Organic Films: A Possible Recycling Mechanism for NOx . Environ. Sci. & Technol. , 41 : 3898 – 3903 .
- Hu , K. S. , Darer , A. I. and Elrod , M. J. 2011 . Thermodynamics And Kinetics Of The Hydrolysis Of Atmospherically Relevant Organonitrates And Organosulfates . Atmosph. Chem. Phys. , 11 : 8307 – 8320 .
- Jang , M. S. , Czoschke , N. M. , Lee , S. and Kamens , R. M. 2002 . Heterogeneous Atmospheric Aerosol Production by Acid-Catalyzed Particle-Phase Reactions . Science , 298 : 814 – 817 .
- Jang , M. , Czoschke , N. M. and Northcross , A. L. 2004 . Atmospheric Organic Aerosol Production by Heterogeneous Acid-Catalyzed Reactions . Chemphyschem , 5 : 1647 – 1661 .
- Jayne , J. T. , Leard , D. C. , Zhang , X. F. , Davidovits , P. , Smith , K. A. Kolb , C. E. 2000 . Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles . Aerosol Sci. Technol. , 33 : 49 – 70 .
- Johnson , D. , Jenkin , M. E. , Wirtz , K. and Martin-Reviejo , M. 2004 . Simulating the Formation of Secondary Organic Aerosol from the Photooxidation of Toluene . Environ. Chem. , 1 : 150 – 165 .
- Johnson , K. S. , Zuberi , B. , Molina , L. T. , Molina , M. J. , Iedema , M. J. Cowin , J. P. 2005 . Processing of Soot in an Urban Environment: Case Study from the Mexico City Metropolitan Area . Atmosph. Chem. Phys. , 5 : 3033 – 3043 .
- Kalberer , M. , Paulsen , D. , Sax , M. , Steinbacher , M. , Dommen , J. Prevot , A. S. H. 2004 . Identification of Polymers as Major Components of Atmospheric Organic Aerosols . Science , 303 : 1659 – 1662 .
- Kim , H. , Barkey , B. and Paulson , S. E. 2010 . Real Refractive Indices of alpha- and beta-pinene and Toluene Secondary Organic Aerosols Generated from Ozonolysis and Photo-Oxidation . J. Geoph. Res.-Atmosph. , 115 : D24212
- Kroll , J. H. and Seinfeld , J. H. 2008 . Chemistry of Secondary Organic Aerosol: Formation and Evolution of Low-volatility Organics in the Atmosphere . Atmosph. Environ. , 42 : 3593 – 3624 .
- Lindinger , W. , Hansel , A. and Jordan , A. 1998 . On-line Monitoring of Volatile Organic Compounds at pptv Levels by Means of Proton-Transfer-Reaction Mass Spectrometry (PTR-MS) - Medical Applications, food Control and Environmental research . Int. J. Mass Spectrom. , 173 : 191 – 241 .
- Lim , Y. B. and Ziemann , P. J. 2005 . Products and Mechanism of Secondary Organic Aerosol Formation from Reactions of n-alkanes with OH Radicals in the Presence of NOx . Environ. Sci. & Technol. , 39 : 9229 – 9236 .
- Liu , S. , Day , D. A. , Shields , J. E. and Russell , L. M. 2011 . Ozone-Driven Daytime Formation of Secondary Organic Aerosol Containing Carboxylic Acid Groups and Alkane Groups . Atmosph. Chem. Phys. , 11 : 8321 – 8341 .
- Liu , S. , Takahama , S. , Russell , L. M. , Gilardoni , S. and Baumgardner , D. 2009 . Oxygenated Organic Functional Groups and Their Sources in Single and Submicron Organic Particles in MILAGRO 2006 Campaign . Atmosph. Chem. Phys. , 9 : 6849 – 6863 .
- Lockwood , A. L. , Shepson , P. B. , Fiddler , M. N. and Alaghmand , M. 2010 . Isoprene Nitrates: Preparation, Separation, Identification, Yields, and Atmospheric Chemistry . Atmosph. Chem. Phys. , 10 : 6169 – 6178 .
- Maki , A. G. and Wells , J. S. 1992 . Measurement And Analysis Of The Fermi Resonance Between Nu-5 And 2-Nu-9 Of Nitric-Acid . J. Mol. Spect. , 152 : 69 – 79 .
- Maria , S. F. , Russell , L. M. , Turpin , B. J. and Porcja , R. J. 2002 . FTIR Measurements of Functional Groups and Organic Mass in Aerosol Samples Over the Caribbean . Atmosph. Envir. , 36 : 5185 – 5196 .
- Matsunaga , A. and Ziemann , P. J. 2009 . Yields of beta-Hydroxynitrates and Dihydroxynitrates in Aerosol Formed from OH Radical-Initiated Reactions of Linear Alkenes in the Presence of NO(x) . J. Phys. Chem. A , 113 : 599 – 606 .
- McGraw , G. E. , Bernitt , D. L. and Ic , Hisatsun. 1965 . Vibrational Spectra of Isotopic Nitric Acids . J. Chem. Phys. , 42 : 237 – 244 .
- Miller , F. A. and Wilkins , C. H. 1952 . Infrared Spectra and Characteristic Frequences of Inorganic Ions-thier Use in Quantitative Analysis . Anal. Chem. , 24 : 1253 – 1294 .
- Nguyen , T. B. , Roach , P. J. , Laskin , J. , Laskin , A. and Nizkorodov , S. A. 2011 . Effect of Humidity on the Composition of Isoprene Photooxidation Secondary Organic Aerosol . Atmosph. Chem. Phys. , 11 : 6931 – 6944 .
- Nishino , N. , Arey , J. and Atkinson , R. 2010 . Formation Yields of Glyoxal and Methylglyoxal from the Gas-Phase OH Radical-Initiated Reactions of Toluene, Xylenes, and Trimethylbenzenes as a Function of NO(2) Concentration . J. Phys. Chem. A , 114 : 10140 – 10147 .
- Nishino , N. , Atkinson , R. and Arey , J. 2008 . Formation of Nitro Products from the Gas-Phase OH Radical-Initiated Reactions of Toluene, Naphthalene, and Biphenyl: Effect of NO(2) Concentration . Environ. Sci. & Technol. , 42 : 9203 – 9209 .
- Odum , J. R. , Hoffmann , T. , Bowman , F. , Collins , D. , Flagan , R. C. and Seinfeld , J. H. 1996 . Gas/particle Partitioning and Secondary Organic Aerosol Yields . Environ. Sci. Techn. , 30 : 2580 – 2585 .
- Ogren , J. A. 2010 . Comment on oCalibration and Intercomparison of Filter-Based Measurements of Visible Light Absorption by Aerosolso . Aerosol Sci. Technol. , 44 : 589 – 591 .
- Paulot , F. , Crounse , J. D. , Kjaergaard , H. G. , Kroll , J. H. , Seinfeld , J. H. and Wennberg , P. O. 2009 . Isoprene Photooxidation: New Insights into the Production of Acids and Organic Nitrates . Atmosph. Chem. Phys. , 9 : 1479 – 1501 .
- Paulsen , D. , Dommen , J. , Kalberer , M. , Prevot , A. S. H. , Richter , R. Sax , M. 2005 . Secondary Organic Aerosol Formation by Irradiation of 1,3,5-trimethylbenzene-NOx-H2O in a New Reaction Chamber for Atmospheric Chemistry and Physics . Environ. Sci. Technol. , 39 : 2668 – 2678 .
- Roberts , J. M. 1990 . The Atmospheric Chemistry of Organic Nitrates . Atmosph. Environ. Part a-Gen. Topics , 24 : 243 – 287 .
- Robinson , A. L. , Donahue , N. M. , Shrivastava , M. K. , Weitkamp , E. A. , Sage , A. M. Grieshop , A. P. 2007 . Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging . Science , 315 : 1259 – 1262 .
- Russell , L. M. , Bahadur , R. and Ziemann , P. J. 2011 . Identifying Organic Aerosol Sources by Comparing Functional Group Composition in Chamber and Atmospheric Particles . Proc. Nat. Acad. Sci. USA , 108 : 3516 – 3521 .
- Russell , L. M. , Takahama , S. , Liu , S. , Hawkins , L. N. , Covert , D. S. Quinn , P. K. 2009 . Oxygenated Fraction and Mass of Organic Aerosol from Direct Emission and Atmospheric Processing Measured on the R/V Ronald Brown During TEXAQS/GoMACCS 2006 . J. Geoph. Res.-Atmosph. , 114 : D00F05
- Saukko , E. , Lambe , A. T. , Massoli , P. , Koop , T. , Wright , J. P. Croasdale , D. R. 2012 . Humidity-dependent Phase State of SOA Particles from Biogenic and Anthropogenic Precursors . Atmosph. Chem. Phy. Dis. , 12 : 4447 – 4476 .
- Sax , M. , Zenobi , R. , Baltensperger , U. and Kalberer , M. 2005 . Time Resolved Infrared Spectroscopic Analysis of Aerosol Formed by photo-oxidation of 1,3,5-trimethylbenzene and Alpha-pinene . Aerosol Sci. Techn. , 39 : 822 – 830 .
- Seinfeld , J. H. , Erdakos , G. B. , Asher , W. E. and Pankow , J. F. 2001 . Modeling the Formation of Secondary Organic Aerosol (SOA). 2. The predicted Effects of Relative Humidity on Aerosol Formation in the Alpha-pinene-, Beta-pinene-, sabinene-, Delta(3)-Carene-, and Cyclohexene-ozone Systems . Environ. Sci. Technol. , 35 : 1806 – 1817 .
- Shapiro , E. L. , Szprengiel , J. , Sareen , N. , Jen , C. N. , Giordano , M. R. and McNeill , V. F. 2009 . Light-absorbing Secondary Organic Material Formed by Glyoxal in Aqueous Aerosol Mimics . Atmosph. Chem. Phys. , 9 : 2289 – 2300 .
- Shiraiwa , M. , Ammann , M. , Koop , T. and Poeschl , U. 2011 . Gas uptake and Chemical Aging of Semisolid Organic Aerosol Particles . Proc. Nat. Acad. Sci. USA , 108 : 11003 – 11008 .
- Sporleder , D. and Ewing , G. E. 2001 . Infrared Spectroscopic Investigation of the Heterogeneous Reaction of HNO3 and NaCl(100) . J. Phys. Chem. A , 105 : 1838 – 1846 .
- Takahama , S. , Liu , S. and Russell , L. M. 2010 . Coatings and Clusters of Carboxylic Acids in Carbon-Containing Atmospheric Particles from Spectromicroscopy and Their Implications for Cloud-Nucleating and Optical Properties . J. Geophys. Res.-Atmos. , : 115 doi: 10.1029/2009JD012622
- Thornton , J. A. , Braban , C. F. and Abbatt , J. P. D. 2003 . N2O5 Hydrolysis on Sub-Micron Organic Aerosols: The Effect of Relative Humidity, Particle Phase, and Particle Size . Phys. Chem. Chem. Phys. , 5 : 4593 – 4603 .
- Vaden , T. D. , Imre , D. , Beranek , J. , Shrivastava , M. and Zelenyuk , A. 2011 . Evaporation Kinetics and Phase of Laboratory and Ambient Secondary Organic Aerosol . Proc. Nat. Acad. Sci. USA , 108 : 2190 – 2195 .
- Virtanen , A. , Joutsensaari , J. , Koop , T. , Kannosto , J. , Yli-Pirila , P. Leskinen , J. 2010 . An Amorphous Solid State of Biogenic Secondary Organic Aerosol Particles . Nature , 467 : 824 – 827 .
- Wangberg , I. , Barnes , I. and Becker , K. H. 1997 . Product and Mechanistic Study of the Reaction of NO3 Radicals with Alpha-Pinene . Environ. Sci. & Technol. , 31 : 2130 – 2135 .
- Wyche , K. P. , Monks , P. S. , Ellis , A. M. , Cordell , R. L. , Parker , A. E. Whyte , C. 2009 . Gas Phase Precursors to Anthropogenic Secondary Organic Aerosol: Detailed Observations of 1,3,5-trimethylbenzene Photooxidation . Atmospheric Chem. and Phys. , 9 : 635 – 665 .
- Yaws , C. L. and Yang , H.-C. 1992 . “ Henry's Law Constant for Compound in Water ” . In Thermodynamic and Physical Property Data , Edited by: Yaws , C. L. Houston : Gulf Publishing Company .
- You , Y. , Renbaum-Wolff , L. , Carreras-Sospedra , M. , Hanna , S. J. , Hiranuma , N. Kamal , S. 2012 . Images Reveal that Atmospheric Particles Can Undergo Liquid-Liquid Phase Separations . Proc. of the Nat. Acad. Sci. USA , in press doi: 10.1073/pnas.1206414109
- Zaveri , R. A. , Berkowitz , C. M. , Brechtel , F. J. , Gilles , M. K. , Hubbe , J. M. Jayne , J. T. 2010 . Nighttime Chemical Evolution of Aerosol and Trace Gases in a Power Plant Plume: IMPLICATIONS for Secondary Organic Nitrate and Organosulfate Aerosol Formation, NO3 Radical Chemistry, and N2O5 Heterogeneous Hydrolysis . J. Geoph. Res.-Atmosp. , 115 : D12304
- Zhang , H. , Surratt , J. D. , Lin , Y. H. , Bapat , J. and Kamens , R. M. 2011 . Effect of Relative Humidity on SOA Formation from Isoprene/NO Photooxidation: Enhancement of 2-methylglyceric Acid and its Corresponding Oligoesters Under Dry Conditions . Atmospheric Chem. and Phys. , 11 : 6411 – 6424 .
- Zhong , M. and Jang , M. 2011 . Light absorption Coefficient Measurement of SOA Using a UV-Visible specTrometer Connected with an Integrating Sphere . Atmospheric Environment , 45 : 4263 – 4271 .
- Zhou , X. L. , Gao , H. L. , He , Y. , Huang , G. , Bertman , S. B. Civerolo , K. 2003 . Nitric Acid Photolysis on Ssurfaces in Low-NOx Eenvironments: Significant Atmospheric Implications . Geophysical Res. Lett. , 30 ( 23 ) : 2217
- Zhou , X. L. and Mopper , K. 1990 . Apparent Partition-Ccoefficients of 15 Ccarbonyl Compounds Between air and Ssea Wwater and Between Aair and Fresh Water-Implications for Aair Sea Exchange . Environ. Sci. Technol. , 24 : 1864 – 1869 .