Abstract
Semi-volatile organic compounds (SVOCs) play a significant role in the formation of secondary organic aerosol, but their atmospheric abundance and chemical composition are poorly understood. We have developed a new system for the thermal desorption aerosol gas chromatograph (TAG) that extends its capability to quantitatively speciate SVOCs (semi-volatile TAG) having vapor pressures lower than tetradecane (C14) with hourly time resolution. The key component is a passivated stainless steel fiber filter that quantitatively collects both gas and particle phase organic compounds. A separation between gas and particle phase collection is determined through a difference method by periodically sampling ambient air through a multichannel charcoal denuder that efficiently removes gas-phase compounds. Measurements made with this new instrument will provide constraints on the abundance, chemical composition, and gas-to-particle partitioning of atmospheric SVOCs and provide opportunities to improve our understanding of secondary organic aerosol formation in the atmosphere.
Copyright 2013 American Association for Aerosol Research
INTRODUCTION
Directly emitted, intermediate volatility organic compounds (IVOCs, defined as having an effective saturation concentration C* of 103 to 106 μg/m3) and semi-volatile organic compounds (SVOCs, C* of 10−1 to 103 μg/m3) have been proposed as a substantial, yet unaccounted for source of secondary organic aerosol (SOA) precursors (Robinson et al. Citation2007; Weitkamp et al. Citation2007). Estimated atmospheric abundances of directly emitted I/SVOCs can be an order of magnitude larger than primary organic aerosol (POA; Robinson et al. Citation2007). Chamber experiments and field observations show that oxidation of compounds in the I/SVOC range produces SOA efficiently (Chan et al. Citation2009; Lim and Ziemann Citation2009; Presto et al. Citation2010; de Gouw et al. Citation2011). The importance of I/SVOCs to SOA formation is further supported by recent modeling work, showing that the inclusion of estimated I/SVOC emissions by means of the volatility basis-set approach (Robinson et al. Citation2007; Shrivastava et al. Citation2008; Grieshop et al. Citation2009) resulted in better agreement between measured and modeled SOA (Dzepina et al. Citation2009; Hodzic et al. Citation2010; Tsimpidi et al. Citation2010) than models that did not include I/SVOCs (Volkamer et al. Citation2006; Hodzic et al. Citation2010). However, these newer models cannot reproduce both the SOA mass and the oxygen to carbon ratio of the bulk aerosol (Hodzic et al. Citation2010), which highlight substantial deficiencies in the understanding of I/SVOC emissions and oxidation mechanisms.
The volatility basis-set approach used in these newer models estimates primary I/SVOCs by multiplying concentrations of POA by a scaling factor derived from dilution measurements of emissions from two primary sources: diesel exhaust and wood burning (Robinson et al. Citation2007; Shrivastava et al. Citation2008; Grieshop et al. Citation2009). The corresponding uncertainty in the estimated IVOC abundance could be as much as three times the magnitude of POA emissions (Robinson et al. Citation2007). Gas-phase oxidation of I/SVOCs proposed by Robinson et al. (Citation2007) and updated by Grieshop et al. (Citation2009) lumps all species with the same effective saturation concentration into a single volatility bin and assumes a constant mass increase for species in a given bin through each oxidation step. However, it is likely that a large number of isomers are present for each carbon number in the vapor pressure range of I/SVOCs (Goldstein and Galbally Citation2007; Isaacman et al. Citation2012), and laboratory experiments have shown that SOA yields from gas-phase OH radical initiated oxidation of alkane isomers differ according to their molecular structure in the order cyclic > linear > branched (Lim and Ziemann Citation2009). In addition to absorptive gas-to-particle partitioning, previous studies have shown that other formation pathways, such as reactive uptake of small carbonyls on acidic particles (Jang et al. Citation2002; Tolocka et al. Citation2004) and reactions between carboxylic acids and ammonia (Na et al. Citation2007), are important for SOA formation. Presto et al. (Citation2012) recently introduced a chromatography-based approach to determine the volatility distribution of POA emissions without speciation analysis based on the relationship between the retention time of known compounds and their saturation concentration. Observational constraints regarding the volatility distribution of I/SVOCs can be provided using this approach, but I/SVOCs first need to be quantitatively collected and analyzed. To better predict SOA production from the oxidation of I/SVOCs, it is critical to apply observational constraints to emissions, speciation, and gas-phase oxidation mechanisms and pathways of gas-to-particle partitioning of I/SVOCs.
The thermal desorption aerosol gas chromatograph (TAG, Williams et al. Citation2006) has shown the capability to capture hourly trends in the gas-to-particle partitioning of speciated organics and the variability of low-volatility compound vapors (Williams et al. Citation2010). However, the original TAG, with its impactor-based collector, was not designed for capture of vapor phase organics. As a result, this instrument could not quantitatively measure speciated I/SVOCs. Multiple SOA formation pathways are present in the atmosphere, and oxygenated compounds in the vapor pressure range of IVOCs are known to partition into particles through nonabsorptive gas-to-particle partitioning (Jang et al. Citation2002). As a result, there is no clear split between IVOCs and SVOCs in the atmosphere according to gas-to-particle partitioning. We therefore define “SVOCs” to include both IVOCs and SVOCs for our new instrument. There are other operational definitions of SVOCs based on the vapor pressure and air-sampling strategy (Turpin et al. Citation2000). However, SVOCs are generally in the vapor pressure range approximately equivalent to C12-C32 n-alkanes (Turpin et al. Citation2000; Sihabut et al. Citation2005; Robinson et al. Citation2007; de Gouw et al. Citation2011). In this article, we present the development and evaluation of a new configuration for TAG, called semi-volatile TAG (SV-TAG), that enables the quantitative measurement of speciated SVOCs with hourly time resolution.
In selecting the sampling methodology for SV-TAG, consideration was given to denuder-based methods, wherein a sorbent tubular system is used to remove vapor-phase organics prior to particle collection, and to filter-based methods that consist of a filter and sorbent trap in series. Filter-sorbent trap methods have been used extensively (Fraser et al. Citation1997; Simcik et al. Citation1998; Schauer et al. Citation1999, Citation2002; Subramanian et al. Citation2004) but are subject to positive artifacts caused by adsorption of gas-phase organics on a filter as well as negative artifacts due to evaporation of particulate organics. Although sorption onto the filter media may be assessed experimentally (Turpin et al. Citation2000), it is not possible to determine the magnitude of volatilization from the filter, nor the absorption onto already-deposited organic material. The latter could be an even more important pathway contributing to underestimation of gas-phase organics (Storey et al. Citation1995). Denuder-based methods, with laboratory extraction and analysis of the substrates, have been used to measure SVOCs in the atmosphere (Gundel et al. Citation1995; Cui et al. Citation1998; Possanzini et al. Citation2004) and from primary emission sources (Schauer et al. Citation1999, Citation2002). In addition, thermally extractable, multicapillary denuders have been developed using short sections of gas chromatography (GC) capillary columns (Krieger and Hites Citation1992; Tobias et al. Citation2007), and stainless steel (SS) honeycombs coated by cross-linked and bonded polydimethylsiloxane (Rowe and Perlinger Citation2010). If a denuder efficiently removes and retains vapor compounds of interest, the artifacts are smaller because gas-phase organics are collected by the denuder surface while particles pass through and are subsequently captured by the filter or the adsorbent bed downstream (Eatough et al. Citation1993; Gundel et al. Citation1995; Cui et al. Citation1998; Turpin et al. Citation2000). Concerns have been raised concerning the disequilibrium of gas-to-particle partitioning after the removal of gas-phase organics, but this is minimized if the residence time inside the denuder is short (Kamens and Coe Citation1997). Moreover, vapor collection efficiencies and particle losses within the denuder can be experimentally determined (Eatough et al. Citation1993; Gundel et al. Citation1995). For these reasons, we selected a denuder-based method for SV-TAG.
Our SV-TAG instrument combines denuder-difference sampling with in situ thermal desorption and GC/mass spectrometry (MS) analysis. The collection cell is designed to quantitatively collect both organic vapors and particles. The gas-to-particle partitioning is determined by sampling alternately with and without the denuder in-line upstream. Although thermally extractable denuders have been discussed in the literature (Tobias et al. Citation2007; Rowe and Perlinger Citation2010), we opted for the difference approach because it is a less complex system that allows us to combine the gas and particle collection and thermal desorption (CTD) parts into a single component. This article describes the evaluation of the SV-TAG denuder and collector components and procedures and presents example ambient data.
EXPERIMENTAL METHODS
The SV-TAG System
A schematic of the SV-TAG is shown in . Components of the SV-TAG that differ from the original TAG are (1) an activated carbon denuder (2) a metal-fiber filter CTD (F-CTD) cell, and (3) a valve-less injection interface/valve-less injector (VLI) with a secondary focusing trap (FT). The denuder is switched in and out of the sample line, upstream of the F-CTD, and removes organic vapors while allowing particles to penetrate. The F-CTD enables efficient collection of SVOCs from the gas phase while simultaneously enabling efficient collection of all particles, all on a filter surface that can be directly thermally desorbed and then reused for the next sample. The FT facilitates the use of a much higher desorption flow rate (up to 200 ml/min) than is possible with the direct GC column transfer where injection flow rates are typically limited to ∼1–2 ml/min.
FIG. 1 (a) Schematic of the SV-TAG system with the major components labeled. (b) Schematic of the F-CTD, FT, and valveless injector showing the flow direction, valve states, and temperature states in each mode of operation.
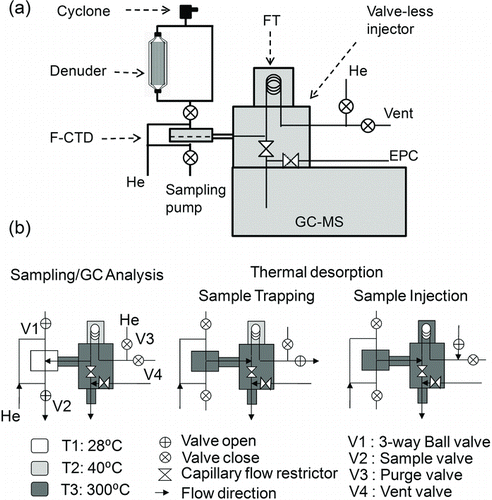
The SV-TAG sample inlet uses a sharp cut PM2.5 cyclone (BGI Inc., Waltham, MA, USA), followed by two parallel sampling lines (a denuder line and a bypass line) and a three-way ball valve (V1, ). The denuder is a multichannel, activated carbon cylindrical monolith made from pyrolized phenolic extrusions (3-cm OD, 20-cm length, ∼490 channels, Mast carbon, U.K.). It is housed inside an aluminum tube and sealed by Viton o-rings with tapered caps at each end connecting to ∼1.0-cm (3/8 inch) OD sampling lines upstream and downstream. The bypass sampling line is made of ∼1.0-cm OD SS tubing.
The F-CTD uses a 37 mm in diameter Bekipor® SS fiber filter (3AL3, Bekaert Fiber Technology, Belgium). This filter has a total fiber surface area of ∼160 cm2 per cm2 of filter surface based on manufacturer specifications. This is higher than the total fiber surface area of ∼130 cm2 per cm2 of filter surface for quartz fiber filters (Mader and Pankow Citation2001). The goal of this study is to develop an in situ instrument capable of collecting SVOC samples. We hypothesized that a passivated SS fiber filter should be quantitatively capable of collecting SVOCs if it had large enough surface area, and it would be suitable for repeated thermal desorption of the sample for analysis. This hypothesis was based on previous observations that breakthrough of SVOCs on quartz filters is small in a 4- or 6-h sampling duration (Subramanian et al. Citation2004), and the demonstration in our original TAG that passivated SS is a suitable material for thermal desorption of organics (Williams et al. Citation2006; Kreisberg et al. Citation2009).
The F-CTD is fabricated by permanently sealing this filter inside a 316 SS, brazed capsule with integrated sampling inlet and outlet tubes and a small bore tube for transferring desorbed organics to the GC (see the online supplemental information; Figure S1). Ridges located inside the capsule clamp the filter in place and ensure rapid heat transfer to the entire filter surface during thermal desorption. After fabrication, the F-CTD is passivated with an Inertium® coating (Advanced Materials Components Express, Lemont, PA, USA). The F-CTD is housed between two aluminum disks outfitted with four cartridge heaters and two temperature sensing thermocouples. Cooling fins are machined into the edges of the aluminum disks. A time programmed blower delivers room air to cool the F-CTD to room temperature after completion of thermal desorption.
The VLI and FT allow the collected sample to be transferred from the F-CTD, focused, and injected into the GC for analysis () without having to pass through a valve. As described by Kreisberg et al. (Citation2012), the VLI provides more reproducible sample transfer than did the six-port valve of the original TAG.
The VLI used in this study consists of two 50-cm-long SS capillaries with inner diameters of 125 μm connected by a pair of VICI® tees (Valco Instruments Co. Inc., Houston, TX, USA) housed in a square aluminum heating block () held at 300°C. Similar to the F-CTD, the capillaries and VICI® tees are passivated with an Inertium® treatment.
The FT for the final configuration is made of a 50-cm section of an SS GC capillary column (MXT-5, 0.5 m × 0.53 mm ID, 5-μm film thickness, Restek, Bellefonte, PA, USA) and integrated into the vent line of the VLI as shown in . The FT has its own aluminum heating and cooling block with heat sink fins on both vertical edges (see the online supplemental information; Figure S2). A thin layer of insulation between the VLI and the FT housing minimizes heat transfer. A time programmed box fan is used to maintain the FT at a low enough temperature to refocus organics desorbed from the F-CTD. This FT does not require liquid nitrogen or other consumables.
SV-TAG Operation
The SV-TAG alternates between two modes of operation: (1) GC sample analysis with simultaneous collection of the next sample and (2) thermal desorption of a sample into the GC. After collection, the deposited organics are thermally desorbed from the F-CTD and refocused onto the FT (“sample trapping”) followed by thermal transfer from the FT to the GC (“sample injection”). After sample injection, the F-CTD is cooled in preparation for collection of the next sample.
Sample collection alternates between the bypass (undenuded) and denuder inlet lines. Gas-to-particle partitioning is determined by difference, with the undenuded sample providing “total organics” and the denuded sample providing “particle-phase organics.”
For sampling, ambient air at 10 L/min is pulled from the center of a larger transport air stream, through the cyclone, through either the denuder or the bypass line, and then delivered to the F-CTD, which is maintained at 28°C. After completion of ambient sampling, the three-way valve (V1) and sample valve (V2) are closed, and the helium flow is set to 200 ml/min for 1 min with the cell still at 28°C to flush any residual air from the F-CTD. The helium flow is then reduced to 10 ml/min, and organics are thermally desorbed by heating the F-CTD from 28°C to 305°C at a rate of ∼35°C/min, then to 315°C at a rate of 5°C/min, and holding at 315°C for 4 minutes. Midway through the desorption, the helium flow is increased from 10 to around 150 ml/min to facilitate the transfer of the less volatile material. The majority (∼80%) of the helium desorption flow enters the F-CTD through its outlet and the remainder through the inlet such that the filter is predominantly back flushed. Desorbed organics are refocused onto the FT, which is held at 40°C. After desorption, the sample is transferred from the FT to the GC, which is initiated while the VLI switches to the sample injection mode by closing the vent valve (V4) and opening the purge valve (V3) (description of VLI operation is provided in the online supplemental information). The FT is heated from 40°C to 315°C in 3 min and held at 315°C for 2 min. The total injection flow is about 3 ml/min consisting of ∼2 ml/min from the FT, ∼0.5 ml/min from the F-CTD, and ∼0.5 ml/min from the electronic pressure controller. During sample injection, the GC oven is held at 45°C and the F-CTD at 315°C.
After sample injection, the system is switched back to the GC analysis mode for organic chemical analysis (see the online supplemental information). The F-CTD and FT heaters are turned off, and fans are turned on to cool the F-CTD and FT. Typically, sample collection time is 30 min (or 90 min for 2-h cycle time), desorption and transfer step is about 20 min, followed by an 8-min cooling step. The GC analysis is typically 35 min and is initiated at the conclusion of the desorption and continues during the collection of the next sample.
System Evaluation
The combined thermal desorption and transfer efficiency of organics from the F-CTD to the head of the GC column was evaluated using a C8-C40 n-alkane liquid standard, which covered the SVOC vapor pressure range. The liquid standard was introduced into the F-CTD via a microliter syringe through a customized injection port and thermally desorbed and transferred to the GC/MS for analysis. The thermal desorption flow was varied for these tests. Initially, transfer efficiencies were evaluated without an FT, which restricted the thermal desorption flow to the GC column flow of 1–2 ml/min. Subsequent experiments, reported here, employed an FT, which decoupled the thermal desorption flow from the chromatographic flow and facilitated evaluation of the transfer efficiencies at higher transfer gas flows, as much as 200 ml/min.
After the addition of an FT into the system, the combined thermal desorption and transfer efficiency from the F-CTD to the head of the column, defined as the recovery, depended on the thermal desorption efficiency of particle-phase organics from the F-CTD, the efficiency of the FT to trap the most volatile compounds in the vapor pressure range of SVOCs, and the transfer efficiency of organics from the FT to the column. Initially, the thermal desorption from the F-CTD was optimized by examining the recovery of the particle-phase n-alkanes, followed by optimizing the recovery of SVOCs from the FT. Our evaluation compared the relative recovery of individual alkanes from a standard comprised of equal alkane masses at each carbon number. The relative recovery of each n-alkane was expressed as the ratio of the abundance of this n-alkane to the maximum abundance of n-alkanes in the C14-C40 range for each run.
The selection of the FT materials was based on minimizing any extra losses caused by the addition of an FT. Since all of the components exposed to organics after thermal desorption were made of SS and the GC column, three different media were evaluated: unpassivated SS tubing and two SS MXT-5 columns with 3- and 5-μm film thickness, respectively. FTs made of each material were evaluated separately to determine which one would be used to optimize for the recovery of SVOCs by increasing the length of the FT and reducing the thermal desorption flow rate. For all experiments, the temperature of the FT was held at 40°C during sample trapping because of its proximity to the heated VLI.
The collection efficiency (CE) and volatilization losses of the F-CTD were evaluated using authentic standards (see the online supplemental information; Table S1) under simulated ambient sampling conditions. The authentic standards covered the vapor pressure range of SVOCs and spanned several different functional groups, typically observed in the atmosphere. These standards were introduced using an impactor CTD (I-CTD) cell placed immediately upstream of the F-CTD to act as a vaporizing source. The I-CTD was used in the original TAG and was a compact collection cell equipped with an injection port for liquid standards (Kreisberg et al. Citation2009).
To evaluate volatilization losses, we utilized a “thermal desorption transfer” method whereby liquid standards introduced into the I-CTD with a microsyringe were transferred to the F-CTD by vaporizing them at 300°C in a 150-ml/min helium flow. This low flow rate ensured efficient capture by the F-CTD. Then, particle and hydrocarbon free air from a zero air generator (model 737, AADCO, Cleves, OH, USA) was drawn at 10 L/min through the F-CTD for either 30 or 90 min, to simulate normal ambient sampling (either 30 or 90 min). Profiles of the flows, F-CTD, and FT temperatures are shown in the online supplemental information (Figure S3).
To evaluate the overall CE of the F-CTD, including both CE and volatilization losses, we utilized an “evaporation transfer” method whereby organic standards deposited in the I-CTD were transferred to the F-CTD over either 30 or 90 min period in a 10 L/min zero air flow at room temperature. This simulated organic vapor collection during real ambient sampling. The material on the F-CTD was analyzed at the end of the room temperature transfer. Then the I-CTD was heated in a 150-ml/min He flow to transfer the remaining standard, and the F-CTD was analyzed a second time. These data provide the total retention by the F-CTD by compounds. This retention is the sum of collected material. Details are shown in the online supplemental information (Figure S4). The detailed description of the thermal desorption method and evaporation transfer method are also provided in the online supplemental information.
The gas CE of the denuder was determined by examining the difference between the undenuded and denuded samples in Berkeley, CA, USA, when a Teflon filter was placed upstream of the cyclone to remove particles. The size-dependent penetration of particles through the denuder and the particle CE by the F-CTD were evaluated through measurements of upstream and downstream particle size distributions as presented in the online supplemental information.
Ambient Sampling
The SV-TAG was first tested in Berkeley, CA, USA, in an hourly automated in situ mode of operation for two periods. In the first period of 13–20 June 2011, both denuded and undenuded samples were collected to investigate gas-to-particle partitioning and overall denuder performance. In the second period of 1–4 October 2011, only undenuded samples were collected to investigate the variability of speciated SVOCs in the atmosphere. In addition, in the second period deuterated internal standards (ISs) were coinjected into the F-CTD with every ambient sample to evaluate the reproducibility of the thermal transfer efficiency. For all ambient runs, the SV-TAG was operated with the 50-cm, 5-μm film FT with the two step purge. The exact temperature and flow settings are presented in the online supplemental information (Figure S5). The denuder performance was also evaluated as part of the California Research at the Nexus of Air Quality and Climate Change (CalNex) campaign in Bakersfield during June–July 2010.
EVALUATION RESULTS
Thermal Desorption and Transfer Efficiency
Tests of the thermal desorption from the F-CTD were first conducted using a 15-cm-long section of unpassivated SS tube as the FT. shows the recovery of n-pentadecane (C15H32, a gas-phase alkane) and n-tritriacontane (C33H68, a particle-phase alkane) as a function of the thermal desorption flow rate (recoveries of the full range of n-alkanes are shown in the online supplemental information [Figure S6]). The recovery of n-tritriacontane was significantly enhanced by increasing the thermal desorption flow rate up to 150 ml/min. The recovery of n-pentadecane decreased with increasing the desorption flow rate, which was attributed to its breakthrough from the unpassivated SS FT.
FIG. 2 Recoveries of representative compounds as a function of flow rate through the F-CTD and the unpassivated SS FT.
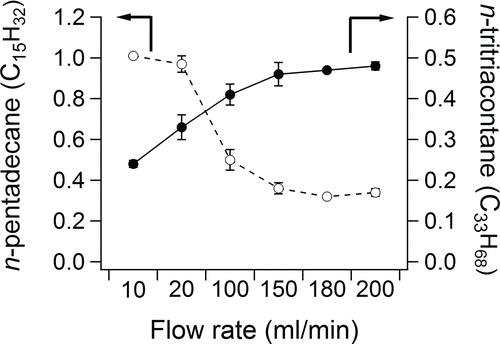
FIG. 3 Recoveries of n-alkanes as a function of carbon number (C13-C26) measured with different FTs: (a) selection of FT materials, (b) optimization of FT length and purge method. For the one-step purge, the thermal desorption flow was set to 150 ml/min for 18 min. For the two-step purge, the thermal desorption flow rate was set to 10 ml/min for 6 min, and then 150 ml/min for 8 min.
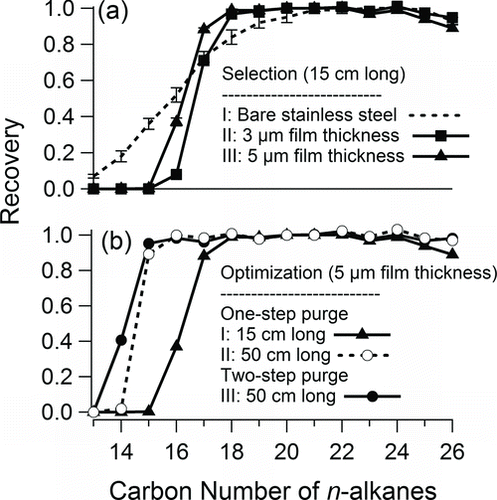
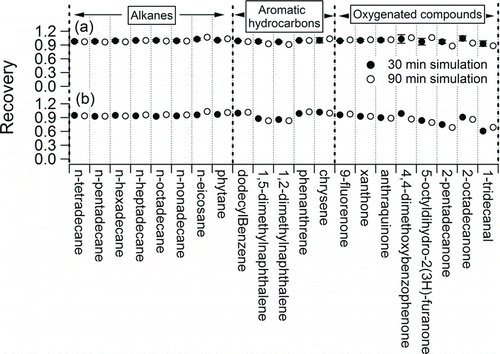
FIG. 4 Overall gas collection efficiency of the F-CTD measured by (a) thermal desorption transfer and (b) evaporation transfer. The compounds are generally grouped by alkanes, aromatic hydrocarbons, and oxygenated compounds, and their chemical formulae are listed in Table S1.
The dependence of the recovery on the desorption flow rate was also evaluated with two other types of FTs made of 15-cm-long GC columns (MXT-5 0.53-mm OD, 3- and 5-μm film thickness). Although the recovery of >C20 n-alkanes was observed to be similar for all three types of FTs under the same thermal desorption method, the recovery of n-alkanes with carbon number ≤20 from the FTs made of the SS GC columns was significantly different from the FT made of the unpassivated SS tube (). The relationship between the recovery and carbon number from the FTs made of GC columns exhibited a steeper drop, relative to the unpassivated SS tube in the lower end of carbon numbers. The steeper drop reduces the number of compounds whose recoveries would be estimated by interpolating the recovery curve of n-alkanes. In addition, the recovery of untested compounds from the FT made of a section of GC column can be derived using their retention time determined through GC analysis and the recovery curve constructed using the recovery and retention time of n-alkanes. The thicker film (5 μm) FT improved the recovery of more volatile n-alkanes, relative to the FT made of the GC column with 3-μm film thickness, due to the increased capacity of the film to trap organics.
The recovery of <C18 n-alkanes from the FT was optimized using the FT made of a section of GC column with 5 μm film thickness. Optimal recovery for SVOCs was achieved when the length of the FT was increased to 50 cm and the total volume of desorption flow was reduced by shortening the total desorption time from 18 to 14 min and by using a two step purge with the flow rate of 10 ml/min for 6 min followed by the higher flow rate of 150 ml/min for 8 min. With the two step purge and 50 cm long FT, the recovery of n-tetradecane (C14H30) from the FT was about 40%, while the recovery of C15-C26 n-alkanes was above 95% () and C27-C32 n-alkanes was 50–90% (Figure S6). Assuming a Gaussian elution profile, we estimate that recovering close to 100% of n-tetradecane would require doubling the length of the FT, improvements for more volatile components would best be achieved through redesign of the FT housing to facilitate active cooling.
SVOC Collection by the F-CTD
The upper limit for losses of collected organics due to volatilization was evaluated using the thermal desorption transfer method. The average recovery (n = 5) of most compounds was around 100% after a 30-min zero air purge (). The recovery of each compound with a 90-min purge was approximately the same as with a 30-min purge, and the average recovery of all compounds after 30- and 90-min purges was 0.99 ± 0.02 and 0.99 ± 0.05, respectively. Except for the concentrations listed in Table S1, volatilization of collected n-alkanes from the F-CTD was evaluated at concentrations of both 16.7 and 66.7 ng/m3 for 30-min ambient sampling. This range of concentrations was chosen to mimic the ambient concentration range of n-pentadecane (0–55 ng/m3) reported by Fraser et al. (Citation1997). The fraction of each compound observed to be retained on the F-CTD was in the range of 0.9 to 1.05. Since organic compounds with lower vapor pressures show stronger adsorption constants on the surface of sampling substrates (Goss and Schwarzenbach Citation1998), we conclude that losses due to volatilization from the surface of the F-CTD are negligible for organics with vapor pressures lower than n-tetradecane under these sampling conditions.
The overall CE of the F-CTD for gas-phase organics was directly evaluated using the evaporation transfer method. All compounds had CE in the range of 0.8 to 1.0 for 30-min simulated ambient sampling, with the exceptions of 1-tridecanal and 2-pentadecanone (). A similar overall CE was also observed for 90-min simulated ambient sampling, and the average ratio of overall CE for all compounds between 30 and 90-min simulation was 1.00 ± 0.05 (). The similar overall CE for all other compounds observed in both 30 and 90-min simulation demonstrates that the F-CTD is capable of quantitative measurements of SVOCs with vapor pressures lower than n-tetradecane for 90-minute sampling at 10 L/min.
It is unclear what caused the lower measured overall CE for 1-tridecanal and 2-pentadecanone. The diffusivity of 1-tridecanal and 2-pentadecanone is larger than most other compounds, such as n-heptadecane, yet the recovery of n-heptadecane is close to 100% in the same evaluation. In addition, we have demonstrated that the losses of collected organics due to volatilization are negligible. However, the measured overall CE was reproducible, allowing for an empirical correction.
F-CTD Particle Collection Efficiency
Particle collection efficiencies for the F-CTD are presented in the online supplemental information (Figures S7 and S8). Mobility-based measurements with test aerosols showed collection of 94% to 100%, with a minimum at 100-nm diameter for sampling at 10 L/min (Figure S7). Optical counter data with ambient particles showed a similar profile but with a minimum CE of 90% at 100 nm (Figure S8). On a mass basis, the average mass CE for PM1.0 ambient particles was 98% as estimated from the upstream and downstream particle volume distribution. In contrast to the particle CE of the original I-CTD, which had a 50% cut-point at 0.1 μm, the F-CTD improved the CE of ultrafine particles to over 90%.
Denuder Vapor Collection and Particle Penetration
For atmospheric measurements, the concentration of gas-phase organics measured downstream of a Teflon filter followed by the denuder was less than 1% of that measured downstream of the Teflon filter alone, indicating a vapor CE of over 99%. Moreover, these measurements were observed to be stable for more than a month of continuous ambient sampling during the CalNex campaign in Bakersfield during June–July 2010 (see the online supplemental information [Figure S9]). Average losses of particles inside the denuder were less than 10% by number for particle sizes spanning the range of 0.05 to 1.00 μm (see the online supplemental information [Figure S10]).
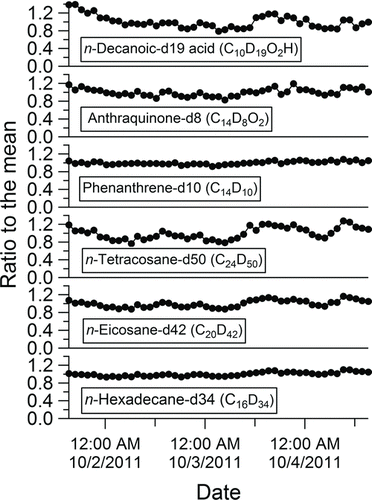
FIG. 5 Stability of the SV-TAG collection and detection system over a 3-day period of continuous ambient sampling as indicated by the responses of 6 perdeuterated internal standards that were coinjected with each ambient sample.
Measurements of Ambient Air
The stability of SV-TAG for ambient run of 3 days was evaluated by examining the consistency of ISs injected with each sample. As shown in , the variability of an IS around its mean value was within 10% for the more volatile ISs (hexadecane-d34, eicosane-d42, and phenanthrene-d8). The range of variability for later eluting ISs, such as tetracosane-d50, was greater and was found to be strongly related to changes in the total organics collected by the F-CTD (organic loading) (see the online supplemental information [Figure S11]). The effect of the organic loading on the recovery of individual compounds has been reported in measurements of ambient organic aerosol with a previous version of the TAG (Lambe et al. Citation2010) and an independent study of thermal desorption of organics from charcoal (Senf and Frank Citation1990). The observed relationship between the variability of each IS and the organic loading establishes the need to use ISs introduced on the top of ambient samples to correct for the effect of varying organic loadings. It should be noted that the effect of the organic loading could be overestimated in the current system because ISs were introduced into the F-CTD from its downstream side and thus increasing the surface area to which ISs were exposed, relative to the ambient samples collected on the upstream surface. The injection of ISs will be changed in future designs to better mimic ambient sampling.
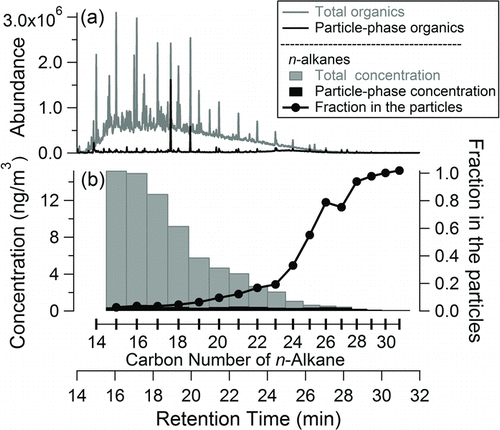
A pair of undenuded and denuded samples are shown in . More than one hundred compounds were identified in the undenuded sample, using authentic standards and according to the best matches available with the National Institute of Standards and Technology (NIST) 2008 mass spectral database. The identified compounds span a broad vapor pressure range and many different functional groups, such as alkanes, acids, and aromatics. For example, observed n-alkanes demonstrate the wide vapor pressure range from SVOCs to particle-phase organics that are possible to detect and quantify with this SV-TAG instrument, and they also highlight the capability to quantify gas-to-particle partitioning of organic compounds (). Although the undenuded and denuded samples were not collected simultaneously, the previous version of the TAG has demonstrated that this sampling strategy is able to capture the variability of gas-to-particle partitioning in a long timeline of observations (Zhao et al. Citation2012).
FIG. 6 (a) Total ion chromatograms of undenuded and denuded ambient samples collected in Berkeley, CA, USA. The sampling volumes were 0.5 m3 each, collected at 10 L/min for 50 min, and the denuded sample was collected 40 min after the undenuded sample; two large peaks in the chromatogram of the denuder sample were peaks of column bleed. (b) Measured concentrations of n-alkanes (left axis) and the particle phase fraction (right axis) of each n-alkane calculated by dividing the particle-phase concentration (denuded sample) by the total concentration (undenuded sample).
Some of the identified compounds, which are predominantly present in the gas phase, can be used as organic markers to provide observational constraints on the abundance of primary emissions of gas-phase SVOCs. Pristane and phytane are potential tracers for the emissions of SVOCs from diesel and gasoline powered vehicles (Fraser et al. Citation1997; Schauer et al. Citation1999, Citation2002). Other identified organic compounds, such as phthalic acid, 6, 10, 14-trimethyl-2-pentadecanone, alkyl cyclohexanes, and dibenzothiophene, are frequently used as source markers for organic aerosol in source apportionment models and are traditionally considered to be only present in particles (Shrivastava et al. Citation2007). However, our observations of gas-to-particle partitioning of these compounds clearly show that they are present in both gas and particle phases as SVOCs. With the SV-TAG, we can clearly separate and quantify both the gas phase and particle phase abundance of these compounds and thus investigate their partitioning and use them with less uncertainty for source apportionment with hourly resolution.
The measured abundance of gas-phase organics was about an order of magnitude larger than the particle-phase organics in Berkeley, CA, USA, over the SVOC range observed by the SV-TAG. The gas-phase organics, determined by the difference between the total (undenuded) and particle-phase (denuded) organics, were dominant early in the chromatogram and mostly formed an unresolved complex mixture (). The sum of measured n-alkanes (C14-C20), which were dominantly present in the gas phase, accounted for ∼7% of total measured organics in the same retention time range as n-alkanes (C14-C20), defined as high-volatility SVOCs, which were dominantly present in the gas phase and calculated by integrating the total ion signal in the retention time range of n-alkanes (C14-C20). The sum of measured of n-alkylcyclohexanes (C14-C20) accounted for less than 1% of high-volatility SVOCs. The upper limit of the contribution from straight-chain and branched alkanes to high-volatility SVOCs was estimated to be ∼32% by assuming that m/z 57 (C4H9 +) was just contributed by straight-chain and branched alkanes. This restricts the upper limit for branched alkanes to be ∼25% of measured high-volatility SVOCs and indicates that branched hydrocarbons likely made up a substantial fraction of organics in the ambient air in Berkeley, CA, USA.
SUMMARY AND ATMOSPHERIC IMPLICATIONS
In this article, we have demonstrated that the SV-TAG is capable of hourly automated quantitation of speciated gas- and particle-phase organics with vapor pressures lower than tetradecane (C14). The recovery of n-alkanes relative to eicosane (C20) is 40% for C14, 95% to 100% for C15-C26, and 50% to 90% for C27-C32. The recovery of individual compounds during the ambient measurements can be tracked and corrected using ISs introduced into each sample. The F-CTD cell efficiently collects the entire particle size range of PM2.5 with CE near 100%, except for a slight dip to ∼90% in a narrow range around 0.1 μm. The SV-TAG facilities quantitative measurements of the gas-to-particle partitioning of SVOC compounds through a denuder difference method. We also showed that traditional molecular markers for organic aerosol can be routinely measured by the SV-TAG with minimal sampling artifacts. We observed that many traditional organic aerosol markers were actually SVOCs with a significant fraction in the gas phase, which has important implications for the use of these organic tracers in source apportionment models. The SV-TAG is now ready for field deployments and will provide unique datasets for the study of emissions, transformation, and partitioning of SVOCs and for studies of SOA formation and transformation in the atmosphere over hourly to seasonal timescales.
uast_a_747673_sm0035.zip
Download Zip (1.8 MB)Acknowledgments
The authors would like to thank Gabriel Isaacman and Arthur W. H. Chan (UC Berkeley) for their assistance in integrating the automated liquid calibration system into the SV-TAG system. This work was supported by the Department of Energy STTR program award number DE-FG02-08ER86335.
[Supplementary materials are available for this article. Go to the publisher's online edition of Aerosol Science and Technology to view the free supplementary files.]
REFERENCES
- Chan , A. W. H. , Kautzman , K. E. , Chhabra , P. S. , Surratt , J. D. , Chan , M. N. , Crounse , J. D. , Kurten , A. , Wennberg , P. O. , Flagan , R. C. and Seinfeld , J. H. 2009 . Secondary Organic Aerosol Formation from Photooxidation of Napthalene and Alkylnaphthalenes: Implications for Oxidation of Intermediate Volatility Organic Compounds (IVOCs) . Atmos. Chem. Phys. , 9 : 3049 – 3060 .
- Cui , W. , Eatough , D. J. and Eatough , N. L. 1998 . Fine Particulate Organic Material in the Los Angeles Basin - I: Assessment of the High-Volume Brigham Yong University Organic Sampling System, BIG BOSS . J. Air Waste Manage. Assoc. , 48 : 1024 – 1037 .
- de Gouw , J. A. , Middlebrook , A. M. , Warneke , C. , Ahmadov , R. , Atlas , E. L. , Bahreini , R. , Blake , D. R. , Brock , C. A. , Brioude , J. , Fahey , D. W. , Fehsenfeld , F. C. , Holloway , J. S. , Henaff , M. Le , Lueb , R. A. , Mckeen , S. A. , Meagher , J. F. , Murphy , D. M. , Paris , C. , Parrish , D. D. , Perring , A. E. , Pollack , I. B. , Ravishankara , A. R. , Robinson , A. L. , Ryerson , T. B. , Schwarz , J. P. , Spackman , J. R. , Srinivasan , A. and Watts , L. A. 2011 . Organic Aerosol Formation Downwind from the Deepwater Horizon Oil Spill . Science , 331 : 1295 – 1299 .
- Dzepina , K. , Volkamer , R. M. , Madronich , S. , Tulet , P. , Ulbrich , I. M. , Zhang , Q. , Cappa , C. D. , Ziemann , P. J. and Jimenez , J. L. 2009 . Evaluation of Recently-Proposed Secondary Organic Aerosol Models for a Case Study in Mexico City . Atmos. Chem. Phys. , 9 : 5681 – 5709 .
- Eatough , D. J. , Wadsworth , A. , Eatough , D. A. , Crawford , J. W. , Hansen , L. D. and Lewis , E. A. 1993 . A Multiple-System, Multichannel Diffusion Denuder Sampler for the Determination of Fine-Particulate Organic Material in the Atmosphere . Atmos. Environ. , 27 : 1213 – 1219 .
- Fraser , M. P. , Cass , G. R. , Simoneit , B. R. T. and Rasmussen , R. A. 1997 . Air Quality Model Evaluation Data for Organics. 4. C2-C36 Non-Aromatic Hydrocarbons . Environ. Sci. Technol. , 31 : 2356 – 2367 .
- Goldstein , A. H. and Galbally , L. E. 2007 . Known and Unexplored Organic Constituents in the Earth's Atmosphere . Environ. Sci. Technol. , 41 : 1514 – 1512 .
- Goss , K. U. and Schwarzenbach , R. P. 1998 . Gas/Solid and Gas/Liquid Partitioning of Organic Compounds: Critical Evaluation of the Interpretation of Equilibrium Constants . Environ. Sci. Technol. , 32 : 2025 – 2032 .
- Grieshop , A. P. , Logue , J. M. , Donahue , N. M. and Robinson , A. L. 2009 . Laboratory Investigation of Photochemical Oxidation of Organic Aerosol from Wood Fires 1: Measurement and Simulation of Organic Aerosol Evolution . Atmos. Chem. Phys. , 9 : 1263 – 1277 .
- Gundel , L. A. , Lee , V. C. , Mahanama , K. R. R. , Stevens , R. K. and Daisey , J. M. 1995 . Direct Determination of the Phase Distributions of Semi-Volatile Polycyclic Aromatic Hydrocarbons Using Annular Denuders . Atmos. Environ. , 29 ( 14 ) : 1719 – 1733 .
- Hodzic , A. , Jimenez , J. L. , Madronich , S. , Canagaratna , M. R. , DeCarlo , P. F. , Kleinman , L. and Fast , J. 2010 . Modeling Organic Aerosols in a Megacity: Potential Contribution of Semi-Volatile and Intermediate Volatility Primary Organic Compounds to Secondary Organic Aerosol Formation . Atmos. Chem. Phys. , 10 : 5491 – 5514 .
- Isaacman , G. , Wilson , K. R. , Chan , A. W. H. , Worton , D. R. , Kimmel , J. R. , Nah , T. , Hohaus , T. , Gonin , M. , Kroll , J. H. , Worsnop , D. R. and Goldstein , A. H. 2012 . Improved Resolution of Hydrocarbon Structures and Constitutional Isomers in Complex Mixtures Using Gas Chromatography-Vacuum Ultraviolet-Mass Spectrometry . Anal. Chem. , 84 : 2335 – 2342 .
- Jang , M. S. , Czoschke , N. M. , Lee , S. and Kamens , R. M. 2002 . Heterogeneous Atmospheric Aerosol Production by Acid-catalyzed Particle-Phase Reactions . Science , 298 : 814 – 817 .
- Kamens , R. M. and Coe , D. L. 1997 . A Large Gas-Phase Stripping Device to Investigate Rates of PAH Evaporation from Airborne Diesel Soot Particles . Environ. Sci. Technol. , 31 : 1830 – 1833 .
- Kreisberg , N. M. , Hering , S. V. , Williams , B. J. , Worton , D. R. and Goldstein , A. H. 2009 . Quantification of Hourly Speciated Organic Compounds in Atmospheric Aerosols, Measured by an In Situ Thermal Desorption Aerosol Gas Chromatograph (TAG) . Aerosol Sci. Technol. , 43 : 38 – 52 .
- Kreisberg , N. M. , Worton , D. R. , Zhao , Y. , Ruehl , C. R. , Goldstein , A. H. and Hering , S. V. 2012 . Development of an Automated High Temperature Valve-Less Injection System for In Situ Gas Chromatography . Prepared for submission to Journal of Chromatography A ,
- Krieger , M. S. and Hites , R. A. 1992 . Diffusion Denuder for the Collection of Semivolatile Organic-Compounds . Environ. Sci. Technol. , 26 : 1551 – 1555 .
- Lambe , A. T. , Chacon-Madrid , H. J. , Nguyen , N. T. , Weitkamp , E. A. , Kreisberg , N. M. , Hering , S. V. , Goldstein , A. H. , Donahue , N. M. and Robinson , A. L. 2010 . Organic Aerosol Speciation: Intercomparison of Thermal Desorption Aerosol GC/MS (TAG) and Filter-Based Techniques . Aerosol Sci. Technol. , 44 : 141 – 151 .
- Lim , Y. B. and Ziemann , P. J. 2009 . Effects of Molecular Structure on Aerosol Yield from OH Radical-Initiated Reactions of Linear, Branched, and Cyclic Alkanes in the Presence of NOx . Environ. Sci. Technol. , 43 : 2328 – 2334 .
- Mader , B. T. and Pankow , J. F. 2001 . Gas/Solid Partitioning of Semivolatile Organic Compounds (SOCs) to Air Filters. 3. An Analysis of Gas Adsorption Artifacts in Measurements of Atmospheric SOCs and Organic Carbon (OC) When Using Teflon Membrane Filters and Quartz Fiber Filters . Environ. Sci. Technol. , 35 : 3422 – 3432 .
- Na , K. , Song , C. , Switzer , C. and Cocker , D. R. 2007 . Effect of Ammonia on Secondary Organic Aerosol Formation from Alpha-Pinene Ozonolysis in Dry and Humid Conditions . Environ. Sci. Technol. , 41 : 6096 – 6102 .
- Possanzini , M. , Di Palo , V. , Gigliucci , P. , Concetta , M. , Sciano , T. and Cecinato , A. 2004 . Determination of Phase-Distributed PAH in Rome Ambient Air by Denuder/GC-MS Method . Atmos. Environ. , 38 : 1727 – 1734 .
- Presto , A. A. , Hennigan , C. J. , Nguyen , N. T. and Robinson , A. L. 2012 . Determination of Volatility Distributions of Primary Organic Aerosol Emissions from Internal Combustion Engines Using Thermal Desorption Gas Chromatography Mass Spectrometry . Aerosol Sci. Technol. , 46 : 1129 – 1139 .
- Presto , A. A. , Miracolo , M. A. , Donahue , N. M. and Robinson , A. L. 2010 . Secondary Organic Aerosol Formation from High-NOx Photo-Oxidation of Low Volatility Precursors: n-Alkanes . Environ. Sci. Technol. , 44 : 2029 – 2034 .
- Robinson , A. L. , Donahue , N. M. , Shrivastava , M. K. , Weitkamp , E. A. , Sage , A. M. , Grieshop , A. P. , Lane , T. E. , Pierce , J. R. and Pandis , S. N. 2007 . Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Ageing . Science , 315 : 1259 – 1262 .
- Rowe , M. D. and Perlinger , J. A. 2010 . Performance of a High Flow Rate Thermally Extractable Multicapillary Denuder for Atmospheric Semivolatile Organic Compound Concentration Measurement . Environ. Sci. Technol. , 44 : 2098 – 2104 .
- Schauer , J. J. , Kleeman , M. J. , Cass , G. R. and Simoneit , B. R. T. 1999 . Measurement of Emissions from Air Pollution Sources. 2. C1 through C30 Organic Compounds from Medium Duty Diesel Trucks . Environ. Sci. Technol. , 33 : 1578 – 1587 .
- Schauer , J. J. , Kleeman , M. J. , Cass , G. R. and Simoneit , B. R. T. 2002 . Measurement of Emissions from Air Pollution Sources. 5. C1-C32 Organic Compounds from Gasoline-Powered Motor Vehicles . Environ. Sci. Technol. , 36 : 1169 – 1180 .
- Senf , L. and Frank , H. 1990 . Thermal-Desorption of Organic Pollutants Enriched on Activated Carbon.5. Desorption Behavior and Temperature Profile . J. Chromatogr. , 520 : 131 – 135 .
- Shrivastava , M. K. , Lane , T. E. , Donahue , N. M. , Pandis , S. N. and Robinson , A. L. 2008 . Effects of Gas Particle Partitioning and Aging of Primary Emissions on Urban and Regional Organic Aerosol Concentrations . J. Geophys. Res. , 113 : D18301 doi: 10.1029/2007JD009735
- Shrivastava , M. K. , Subramanian , R. , Rogge , W. F. and Robinson , A. L. 2007 . Sources of Organic Aerosol: Positive Matrix Factorization of Molecular Marker Data and Comparison of Results from Different Source Apportionment Models . Atmos. Environ. , 41 : 9353 – 9369 .
- Sihabut , T. , Ray , J. , Northcross , A. and McDow , S. R. 2005 . Sampling Artifact Estimates for Alkanes, Hopanes, and Aliphatic Carboxylic Acids . Atmos. Environ. , 39 : 6945 – 6956 .
- Simcik , M. F. , Franz , T. P. , Zhang , H. X. and Eisenreich , S. J. 1998 . Gas-Particle Partitioning of PCBs and PAHs in the Chicago Urban and Adjacent Coastal Atmosphere: States of Equilibrium . Environ. Sci. Technol. , 32 : 251 – 257 .
- Storey , J. M. , Luo , W. , Isabelle , L. M. and Pankow , J. F. 1995 . Gas/Solid Partitioning of Semivolatile Organic Compounds to Model Atmospheric Solid Surfaces As a Function of Relative Humidity. 1. Clean Quartz . Environ. Sci. Technol. , 29 : 2420 – 2428 .
- Subramanian , R. , Khlystov , A. Y. , Cabada , J. C. and Robinson , A. L. 2004 . Positive and Negative Artifacts in Particulate Organic Carbon Measurements with Denuded and Undenuded Sampler Configurations . Aerosol Sci. Technol. , 38 : 27 – 48 .
- Tobias , D. E. , Perlinger , J. A. , Morrow , P. S. , Doskey , P. V. and Perram , D. L. 2007 . Direct Thermal Desorption of Semivolatile Organic Compounds from Diffusion Denuders and Gas Chromatographic Analysis for Trace Concentration Measurement . J. Chromatogr. A , 1140 : 1 – 12 .
- Tolocka , M. P. , Jang , M. , Ginter , J. M. , Cox , F. J. , Kamens , R. M. and Johnston , M. V. 2004 . Formation of Oligomers in Secondary Organic Aerosol . Environ. Sci. Technol. , 38 : 1428 – 1434 .
- Tsimpidi , A. P. , Karydis , V. A. , Zavala , M. , Lei , W. , Molina , L. , Ulbrich , I. M. , Jimenez , J. L. and Pandis , S. N. 2010 . Evaluation of the Volatility Basis-Set Approach for the Simulation of Organic Aerosol Formation in the Mexico City Metropolitan Area . Atmos. Chem. Phys. , 10 : 525 – 546 .
- Turpin , B. J. , Saxena , P. and Andrews , E. 2000 . Measuring and Simulating Particulate Organics in the Atmosphere: Problems and Prospects . Atmos. Environ. , 34 : 2983 – 3013 .
- Volkamer , R. , Jimenez , J. L. , San Martini , F. , Dzepina , K. , Zhang , Q. , Salcedo , D. , Molina , L. T. , Worsnop , D. R. and Molina , M. J. 2006 . Secondary Organic Aerosol Formation from Anthropogenic Air Pollution: Rapid and Higher Than Expected . Geophys. Res. Lett. , 33 : L17811
- Weitkamp , E. A. , Sage , A. M. , Pierce , J. R. , Donahue , N. M. and Robinson , A. L. 2007 . Organic Aerosol Formation from Photochemical Oxidation of Diesel Exhaust in a Smog Chamber . Environ. Sci. Technol. , 41 : 6969 – 6975 .
- Williams , B. J. , Goldstein , A. H. , Kreisberg , N. M. and Hering , S. V. 2006 . An In-Situ Instrument for Speciated Organic Composition of Atmospheric Aerosols: Thermal Desorption Aerosol GC/MS-FID (TAG) . Aerosol Sci. Technol. , 40 : 627 – 638 .
- Williams , B. J. , Goldstein , A. H. , Kreisberg , N. M. and Hering , S. V. 2010 . In Situ Measurements of Gas/Particle-Phase Transitions for Atmospheric Semivolatile Organic Compounds . P. Natl. Acad. Sci. , 107 : 6676 – 6681 .
- Zhao , Y. , Kreisberg , N. M. , Worton , D. R. , Isaacman , G. , Weber , R. J. , Liu , S. , Day , D. A. , Russell , L. M. , Markovic , M. Z. , Vandenboer , T. C. , Murphy , J. G. , Hering , S. V. and Goldstein , A. H. 2012 . Insights into SOA Formation Mechanisms from Measured Gas/Particle Partitioning of Specific Organic Tracer Compounds . Environmetal Science and Technology , in review