Abstract
Spectrophotometric methods were developed to quantify carbonyl, hydroxyl, carboxyl, and ester groups in samples with composition typical of oxidized atmospheric organic aerosol. The methods employ derivatizing agents to convert functional groups to characteristic colored derivatives that are quantified by spectrophotometry. Effects of molecular structure on quantification have been evaluated by measuring calibration curves for a variety of monofunctional and multifunctional compounds. In addition, potential interferences from compounds containing nontarget functional groups have been determined and methods developed to eliminate these interferences. Detection limits are approximately 0.01, 0.02, 0.03, and 0.1 μmoles for carbonyl, hydroxyl, carboxyl, and ester groups, respectively. The use of these methods for analysis of secondary organic aerosol (SOA) composed of a complex mixture of oxidized compounds is also demonstrated.
© 2013 American Association for Aerosol Research
INTRODUCTION
Oxidized organic particulate matter is an important component of the atmosphere that can consist of up to thousands of compounds that are emitted directly from anthropogenic and biogenic sources or are formed in the atmosphere from the condensation of products of the oxidation of volatile organic compounds (VOCs) (Jacobson et al. Citation2000; Kroll and Seinfeld Citation2008; Hallquist et al. Citation2009). The chemical composition of organic aerosol (OA) is most commonly characterized using online or offline mass spectrometry (MS), with offline analyses sometimes including gas or liquid chromatography (Hallquist et al. Citation2009; Hoffmann et al. Citation2011). These methods span a range in chemical specificity from individual molecules to classification according to the degree of oxidation or as hydrocarbon-like or oxidized OA. Each method has its strengths and weaknesses, in terms of chemical specificity, the fraction of OA mass that can be analyzed, real-time or offline analysis, sample size, measurement time resolution, and the cost and difficulty of analysis (Hallquist et al. Citation2009). Alternately, a few groups have employed Fourier transform infrared (FTIR) spectroscopy (Blando et al. Citation2001; Coury and Dillner Citation2008; Russell et al. Citation2011) and nuclear magnetic resonance (NMR) spectroscopy (Decesari et al. Citation2000) to quantify specific oxygen-, nitrogen-, and sulfur-containing functional groups in OA, including carbonyl, hydroxyl, carboxyl, nitrate, amine, and sulfate, as well as saturated, unsaturated, and aromatic hydrocarbon groups. This approach is generally intermediate to those mentioned above in terms of chemical specificity and also with regard to other analytical strengths and weaknesses (Hallquist et al. Citation2009). In spite of the value of the functional group analysis for characterizing OA in cases where molecular identification is not necessary, but more chemical detail is sought than can be obtained by aerosol mass spectrometry (AMS) methods, FTIR and NMR spectroscopy have seen relatively little use, especially in laboratory studies. The reasons for this are not entirely clear, but may be in part due to the considerable skill and experience needed to interpret FTIR and NMR spectra. Identification and quantification can be difficult because of overlapping peaks, the potential for matrix effects, and the requirement for functional group standards appropriate for the mix of organic compounds being analyzed (Blando et al. Citation2001).
Prior to the development of sophisticated FTIR, NMR, and MS instrumentation, organic mixtures of interest in many research fields and in industrial applications were often routinely characterized using spectrophotometric methods that involved reactions with derivatizing reagents to yield products that were characteristic of specific functional groups and were also colored, so that they could be quantified by measuring their absorbance. Although derivatization is still commonly used in atmospheric chemistry research to improve the properties of organic compounds for gas chromatography–mass spectrometry (GC-MS) analysis (Yu et al. Citation1999; Jaoui et al. Citation2005), because of the time-consuming chemical processing and the generally poorer sensitivity of spectrophotometry compared with other methods, derivatization–spectrophotometric methods have mostly fallen out of favor. Nevertheless, over the past few years, we have worked to revive this approach for analysis of mixtures of atmospheric organic compounds, believing that if a suite of simple, reliable methods were available for routine analysis of the major functional groups, then they would be used. The approach we used was to identify the most promising methods for quantifying carbonyl [C(O)], hydroxyl [CHOH], carboxyl [C(O)OH], and ester [C(O)OR] groups from among the many available in the literature on organic analysis and then evaluate these for specificity, interferences, and detection limits, modifying the methods when necessary or identifying better ones. The recommended methods and the results of the evaluations made using a variety of monofunctional and multifunctional compounds are described in detail below. An example of their application, together with methods we have used previously for peroxides [CHOOR] (Docherty et al. Citation2005) and nitrates [CHONO2] (Matsunaga and Ziemann Citation2009), to the analysis of secondary organic aerosol (SOA) formed in an environmental chamber reaction is also presented.
EXPERIMENTAL SECTION
General Information
The following chemical abbreviations are used below: 2-nitrophenylhydrazine (2-NPH), 2-nitrophenylhydrazine hydrochloride (2-NPH.HCl), 2-nitrophenyl hydrazide (2-NP hydrazide), N,N′-dicyclohexylcarbodiimide (DCC), 2,4-dinitrophenylhydrazine (2,4-DNPH), 2,4-dinitrophenyl hydrazone (2,4-DNP hydrazone), triphenylphosphine (TPP), 4-nitrobenzoyl chloride (4-NBC), 4-dimethylaminopyridine (4-DMAP), and trimethylsilyldiazomethane (TMSDM). Most of the reagent solutions described below were freshly prepared for each set of analyses, except for the 2-NPH.HCl, DCC, pyridine, and 2,4-DNPH solutions since contamination of these would be apparent from reagent blank analyses. It is possible that many of the other solutions do not need to be freshly prepared, but we did not evaluate this. The amounts of each of the freshly prepared reagent solutions listed below are those required for analysis of the following solutions: (1) two reagent blanks (analysis conducted with all reagents but without sample), (2) one sample (two analyses, with the second diluted by a factor of 2 relative to the first), and (3) three standard solutions. The sample is analyzed at two different concentrations in order to verify that excess derivatizing agent is present. This will be the case if the two analyses give the same result, within uncertainties, and are within the linear range of the calibration curve. Absorbance was measured using an USB4000-UV-Vis Miniature Fiber Optic spectrophotometer from Ocean Optics with a 1-cm quartz cell.
Carboxyl Groups
The following chemicals with specific purities or grades (with their supplier name) were used: 2-NPH.HCl (98%) (TCI); DCC (99%), TPP (99%), cumene hydroperoxide (80%), benzoyl peroxide (97%), tridecanoic acid (98%), decanedioic acid (99%), 2-ketopentanedioic acid (98%), 2-ketohexanedioic acid (98%), (9Z)-octadec-9-enoic acid (99%), and 11-hydroxyundecanoic acid (96%) (Sigma-Aldrich); thiourea (99%), extra dry pyridine (99.5%), and KOH (“for analysis;” ≥85% with remainder water and <1% K2CO3 and trace metals) (Acros Organics); and absolute ethanol (molecular biology grade) and water (optima grade) (Fisher Scientific).
The following reagent solutions were used: (1) 2-NPH.HCl solution: 25 mL of 0.02 M 2-NPH.HCl in ethanol (requires warming at ∼40°C for complete dissolution), (2) DCC solution: 25 mL of 0.25 M DCC in ethanol, (3) Pyridine solution: 25 mL of 6% v/v pyridine in ethanol, (4) TPP solution: 5 mL of 0.01 M TPP in ethanol, (5) Thiourea solution: 10 mL of 10 g L−1 thiourea in ethanol, and (6) KOH solution: 5 mL of 100 g L−1 KOH in 10% v/v water in ethanol.
The procedure is as follows: a sample or carboxylic acid standard containing ≥0.03 μmoles of carboxyl groups dissolved in 0.2 mL of ethanol is added to a 5 mL volumetric flask with 0.2 mL of TPP solution. This solution is kept at room temperature for 30 min to decompose peroxide compounds. In order, 0.5 mL each of the 2-NPH.HCl, pyridine, and DCC solutions are added to the flask and the solution is kept at room temperature for 24 h. Then, 1 mL of thiourea solution and 0.25 mL of KOH solution are added to the flask, whereupon a turbid brown-violet solution is formed. The solution is warmed in a water bath at 60°C for 15 min to reduce the brown color, which masks the violet color and interferes with the absorbance measurement. After allowing the solution to cool to room temperature, the volume is made up to the 5 mL mark with water. The absorbance of the solution is measured at 550 nm.
Carbonyl Groups
The following chemicals with specific purities or grades (with their supplier name) were used: 2,4-DNPH (97%), TPP (99%), 3,4-hexanedione (95%), 2-ketopentanedioic acid (98%), 2-ketohexanedioic acid (98%), and pentane-1,5-dial in water (50%) (Sigma-Aldrich); KOH (“for analysis”; ≥85% with remainder water and <1% K2CO3 and trace metals) (Acros Organics); concentrated HCl (certified ACS plus grade), absolute ethanol (molecular biology grade), hexane (optima grade), and water (optima grade) (Fisher Scientific); nonanal (95%), undec-10-enal (95%), and 3-hexadecanone (99%) (Alfa Aesar); and 6-hydroxy-5-decanone (97%) and 5-hydroxy-2-pentanone (96%) (TCI). A mixture of 9,10-ketonitrate octadecanoic acid isomers were synthesized by reacting (9Z)-octadec-9-enoic acid (oleic acid) particles with NO3 radicals in the environmental chamber (Docherty and Ziemann Citation2006) and then collecting the aerosol on a filter and purifying by high-performance liquid chromatography (HPLC) (Matsunaga and Ziemann Citation2009).
The following reagent solutions were used: (1) Hexane/ethanol solution: 30 mL of 30% v/v hexane in ethanol, (2) 2,4-DNPH solution: 25 mL of 0.005 M 2,4-DNPH in 4% v/v concentrated HCl in ethanol, (3) TPP solution: 5 mL of 0.01 moles TPP in ethanol, and (4) KOH solution: 50 mL of 100 g L−1 KOH in 20% v/v water in ethanol.
The procedure is as follows: A sample or carbonyl standard containing ≥0.01 μmoles of carbonyl groups dissolved in 1 mL of hexane/ethanol solution is added to a 5 mL volumetric flask with 0.2 mL of TPP solution. This solution is kept at room temperature for 30 min to decompose peroxide compounds. Then, 0.4 mL of 2,4-DNPH solution is added and the solution is kept at room temperature for 24 h. Upon adding 3 mL of KOH solution, a KCl precipitate forms, which is dissolved by adding water to the 5 mL mark. The absorbance is immediately measured at 480 nm.
Two points regarding this analysis need to be emphasized. First, sufficient water must be added to completely dissolve the KCl precipitate and, because the absorbing quinoidal ion that forms upon making the solution alkaline is unstable, the absorbance needs to be measured within 10 min of adding the KOH solution to achieve accurate results (Lappin Citation1951; Jordan and Veatch Citation1964; Yukawa et al. Citation1993; Meyer and Rebrovic Citation1995).
Ester Groups
The following chemicals with specific purities or grades (with their supplier name) were used: hydroxylamine hydrochloride (99%), KOH (“for analysis;” ≥85% with remainder water and <1% K2CO3 and trace metals) and FeCl3·6H2O (99%) (Acros Organics); concentrated HCl (certified ACS plus grade), absolute ethanol (molecular biology grade), bis(2-ethylhexyl)-decanedioate (97%), and water (optima grade) (Fisher Scientific); and methyl tridecanoate (98%), bis(2-ethylhexyl) phthalate (99%), methyl (Z)-octadec-9-enoate (99%), and dimethyl-3-oxopentanedioate (96%) (Sigma-Aldrich).
The following reagent solutions were used: (1) Ethanol/water solution: 100 mL of 50% v/v ethanol in water, (2) Hydroxylamine hydrochloride solution: 10 mL of 2.0 M hydroxylamine hydrochloride in 50% v/v ethanol in water, (3) KOH solution: 10 mL of 2.4 M KOH in 50% ethanol in water, (4) HCl solution: 5 mL of 50% v/v concentrated HCl in 50% v/v ethanol in water, and (5) FeCl3 solution: 5 mL of 0.44 M FeCl3⋅6H2O in 6% v/v HCl solution in ethanol.
The procedure is as follows: A sample or ester standard containing ≥0.1 μmoles of ester groups dissolved in 1 mL of ethanol is added to a 5 mL volumetric flask. The hydroxylamine hydrochloride solution and KOH solution are then mixed, and 2 mL of that solution are added to the flask, followed by 0.6 mL of water to dissolve the precipitate of hydroxylamine hydrochloride that forms. This solution is kept at room temperature for 1 h. Successively, 0.5 mL each of HCl solution and FeCl3 solution are added, followed by water up to the 5 mL mark. The absorbance of the solution is immediately measured at 540 nm.
It is worth noting that this method consists of two steps: the formation of hydroxamic acids by reaction of esters with hydroxylamine in alkaline medium, and a color-developing step involving the reaction of ferric ion with the hydroxamic acids in acidic medium (see the discussion in the online supplementary information). In the hydroxamic acid formation step, the rate of reaction depends on the concentration of hydroxylamine, pH, temperature, and reaction time. Thus, the stated reaction temperature and time are important. Furthermore, the color intensity produced from the reaction of ferric ion with hydroxamic acids depends on the concentration of ferric iron and pH (Hestrin Citation1949; Goddu et al. Citation1955; Verheyden and Nys Citation1962). Loss of color upon standing appears to be inevitable, however (Thompson Citation1950; Stern and Shapiro Citation1953; Goddu et al. Citation1955; Goldenberg Citation1958; Morgan and Kingsbury Citation1959; Verheyden and Nys Citation1962; Feldmann and Robb Citation1970; Feldmann et al. Citation1971), including for our method, so absorbance should be measured immediately after adding HCl and FeCl3.
Hydroxyl Groups
The following chemicals with specific purities or grades (with their supplier names) were used: 4-NBC (97%), 2 M TMSDM in diethyl ether, 2-hexadecanone (99%), 1,10-decanediol (98%), 1,2-tetradecanediol (90%), 12-hydroxyoctadecanoic acid (99%), and 16-hydroxyhexadecanoic acid (98%) (Sigma-Aldrich); 5-hydroxy-2-pentanone (96%) and 6-hydroxy-5-decanone (97%) (TCI); 4-DMAP (99%) and extra dry pyridine (99.5%) (Acros Organics); Na2CO3 (99%) (Baker & Adamson); NaHCO3 (99.7%) (Mallinickrodt); NaCl (UPC/FCC); concentrated HCl (certified ACS plus grade), hexane (optima grade), dichloromethane (optima grade), methanol (optima grade), toluene (certified ACS grade), and water (optima grade) (Fisher Scientific). A mixture of 9,10-hydroxynitrate octadecanoic acid isomers were synthesized by reacting (9Z)-octadec-9-enoic acid (oleic acid) particles with nitrate radicals in the environmental chamber (Docherty and Ziemann Citation2006) and then collecting the aerosol on a filter and purifying by HPLC (Matsunaga and Ziemann Citation2009).
The following reagent solutions were used: (1) Methanol/toluene solution: 30 mL of 40% v/v methanol in toluene, (2) TMSDM solution: 5 mL of 0.04 M TMSDM (in diethyl ether) in methanol/toluene solution, (3) 4-NBC solution: 2 mL of 100 g L−1 4-NBC in pyridine, (4) 4-DMAP solution: 25 mL of 0.02 M 4-DMAP and 50 g L−1 Na2CO3in water, (5) NaHCO3 solution: 25 mL of 50 g L−1 NaHCO3 in water, (6) HCl solution: 200 mL of 0.24 N HCl and 25 g L−1 NaCl in water, and (7) dichloromethane/hexane solution: 50 mL of 9% v/v hexane in dichloromethane.
The procedure is as follows: A sample or alcohol standard containing ≥0.02 μmoles of hydroxyl groups dissolved in 100 μL of pyridine is added to a 5 mL reaction vial with 150 μL of 4-NBC solution. The solution is kept at room temperature for 1 h. The pyridine is then evaporated in ∼10 min using an N2 stream and 2 mL of 4-DMAP solution are added to hydrolyze the excess 4-NBC. The vial is treated in an ultrasonic bath for 2 min and then left for 30 min to completely dissolve the reagent. Because the derivatives are poorly soluble in water and give a turbid solution, they are extracted into 5 mL of dichloromethane/hexane solution in a separatory funnel and the organic phase is washed once with 2 mL of NaHCO3 solution and three times with 5 mL of HCl solution. The absorbance of the dichloromethane/hexane layer, which contains the derivatives, is measured at 253 nm.
Carboxyl and hydroperoxy groups can interfere with this analysis. Carboxylic acids are removed prior to the hydroxyl group analysis by dissolving 100 μL of sample in the methanol/toluene solution and adding this to a 5 mL reaction vial with 200 μL of TMSDM solution. The mixture is kept at room temperature for 1 h. The methanol and toluene are then evaporated in ∼10 min using an N2 stream and the residue is dissolved in 100 μL pyridine. Hydroperoxides cannot be removed by reaction with TPP, as is done in some of our other methods, because alcohols are formed in the process. Instead, since hydroperoxides only interfere at concentrations ≥0.02 mM, if peroxide interference is a concern, then the sample is diluted to achieve a concentration of peroxides ≤0.02 mM prior to the hydroxyl group analysis. The method of Docherty et al. (Citation2005) is used to quantify total peroxides prior to dilution and thus obtain an upper limit to the hydroperoxide concentration.
Analysis of SOA Formed from the Reaction of α-Pinene with O3
Replicate experiments were performed of reactions of α-pinene with O3 in an 8.3 m3 FEP Teflon environmental chamber filled with clean, dry air at room temperature and atmospheric pressure. Cyclohexane and O3 were added to achieve chamber concentrations of 1200 and 2 ppmv, respectively, and then 1 ppmv of α-pinene was added to initiate the reaction, which consumed all the α-pinene. The cyclohexane scavenged >99% of the OH radicals formed in the reaction (Docherty et al. Citation2005). (1S)-(-)-α-pinene (99%) and cyclohexane (HPLC grade) were obtained from Sigma-Aldrich and O3 was generated using a Welsbach T-408 O3 generator.
Replicate particle samples were collected after each experiment on preweighed Millipore filters (0.45-μm pore size, Fluoropore FHLP, 47 mm). Sample air volumes were ∼1 m3 collected over ∼1 h through stainless steel tubing. Immediately after sampling, each filter was reweighed to calculate the mass of sample collected and then extracted twice in 4 mL of ethyl acetate at room temperature for >10 min. For each filter, extracts were combined in a preweighed brown vial and then dried using an N2 stream. The vial was reweighed to determine the mass of sample extracted. The mass increase of the filter, the mass of dried extract, and the calculated mass collected based on scanning mobility particle sizer measurements made during sampling were the same within uncertainties, indicating that the extraction method was quantitative (>90% efficient) and no sample was lost by volatilization, so no corrections were made for extraction efficiencies. This appears to be the case for many other types of SOA we have studied (Docherty et al. Citation2005; Matsunaga and Ziemann Citation2009). The dried sample was dissolved in an appropriate volume of ethyl acetate to achieve a sample concentration of ∼1 mg mL−1, and separate aliquots were taken for analysis of each functional group, dried using an N2 stream to remove ethyl acetate, and then dissolved in an appropriate solvent for each analysis, as noted above. A clean blank filter was also prepared and analyzed similarly. The methods used for nitrate and peroxide analysis were described previously (Matsunaga and Ziemann Citation2009; Docherty and Ziemann 2006). Dried filter extracts were also analyzed for C and N using a Costech ECS 4010 CHNSO high-temperature combustion elemental analyzer and for O and H using a Thermo TC/EA temperature conversion elemental analyzer interfaced to a Thermo Delta V isotope ratio mass spectrometer.
RESULTS AND DISCUSSION
The chemical abbreviations used below are defined in the General Information section. A more detailed discussion of the considerations that led to the development of the methods presented here is given in the online supplementary information. Readers are encouraged to consult that discussion to better appreciate the basis for the methods and their sensitivities to variations in procedures and conditions.
Analysis of Carboxyl Groups
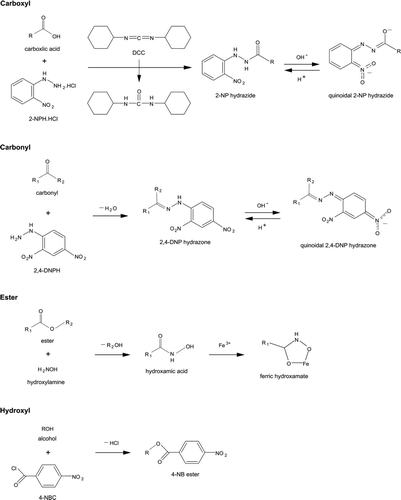
The method presented here for the analysis of carboxyl groups is a modified version of the method of Miwa et al. (Citation1980). The basis of the method is that carboxylic acids are reacted with 2-NPH.HCl by using DCC as a coupling agent in the presence of pyridine, forming 2-NP hydrazide derivatives that give an intense violet color in an alkaline medium (pH > 12) due to the quinoidal resonance form of 2-NP hydrazides (Miwa et al. Citation1980, Citation1985; Korany et al. Citation1989). The chemistry of the carboxyl group analysis is shown in .
FIG. 1 Chemistry employed in the derivatization and analysis of carboxyl, carbonyl, ester, and hydroxyl groups.
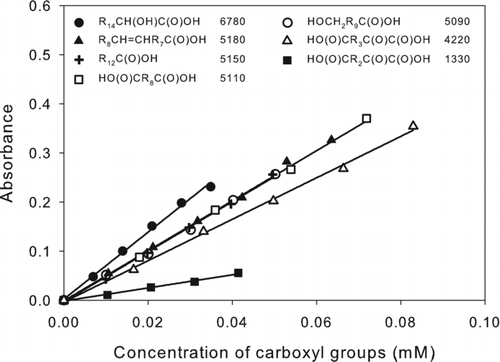
The effect of molecular structure on the method results were evaluated by preparing calibration curves for a variety of monocarboxylic and dicarboxylic acids, some of which also contained carbonyl or hydroxyl groups or C˭C double bonds. The results are shown in . Linearity was excellent over the range of concentrations measured. Values of ϵ (M−1 cm−1) for the compounds tested were in the range 4200–6800, with the exception of 2-ketopentanedioic acid, for which ϵ ∼ 1300, even though the value for 2-ketohexanedioic acid was ∼4200. Most had values of ϵ ∼ 5100, similar to those reported previously (Horikawa and Tanimura Citation1982). Functional groups adjacent to or conjugated with the chromophore are known to affect UV-Vis absorbance (Shriner et al. Citation1964), as is observed here. In the case of 2-hydroxyhexadecanoic acid, the hydroxyl group increases ϵ by ∼30%, whereas for 2-ketohexanedioic and 2-ketopentanedioc acid, the carbonyl group decreases ϵ by ∼20% and ∼75%, respectively. Functional groups that are not adjacent to or conjugated with the chromophore are expected to have much less effect on ϵ, as observed here where the hydroxyl group in 11-hydroxyundecanoic acid and the C˭C double bond in (9Z)-octadec-9-enoic acid (oleic acid) have negligible effects. For analysis of samples containing carboxylic acids with known structures, appropriate calibration curves can be prepared, whereas for samples containing unidentified compounds, the use of a simple monocarboxylic acid should give reasonably accurate results in most cases. Except for 2-ketopentanedioic acid, values of ϵ for all the carboxylic acids analyzed here were within ∼30% of the value measured for tridecanoic acid.
FIG. 2 Calibration curves of carboxylic acids: 2-hydroxyhexadecanoic acid (•), (9Z)-octadec-9-enoic acid (▴), tridecanoic acid (+), decanedioic acid (□), 11-hydroxyundecanoic acid (○), 2-ketohexanedioic acid (▵), and 2-ketopentanedioic acid (▪). The corresponding structures and values of ϵ (M−1 cm−1) are shown in the legend. R n designates (CH2) n or H(CH2) n depending on whether the alkyl group is in the interior or at the end of the molecule. The values of ϵ were calculated as ϵ = 103 × m, where m is the slope of the least-squares line fit to the calibration curve A = m(C) + b, where A = absorbance = absorbance (sample) – absorbance (blank), and (C) is the concentration of carboxyl groups (mM). For all lines, |b| ≤ 0.005 and R 2 > 0.99.
The method was tested for potential interferences from compounds containing other functional groups that are likely to be present in an SOA mixture. It was determined that nitrate, carbonyl, hydroxyl, ester, and peroxy groups other than hydroperoxy give no or little interference, but that hydroperoxides interfered by giving the same response as carboxylic acids (Figure S1 in the online supplementary information). Therefore, in our method, TPP solution is added to the sample prior to analysis to decompose peroxides, similar to the approach used by Chiba et al. (Citation1989) to remove peroxides prior to the carbonyl group analysis. TPP is a common reducing agent for organic hydroperoxides and is oxidized to TPP oxide while the hydroperoxides are reduced to alcohols that do not interfere with the carboxyl group analysis. The reaction is
The detection limit calculated as 3 × the standard deviation of blank measurements was 0.03 μmoles of carboxyl groups for carboxylic acids with ϵ = 5100. The reproducibility determined from multiple analyses of tridecanoic acid was 6% relative standard deviation.
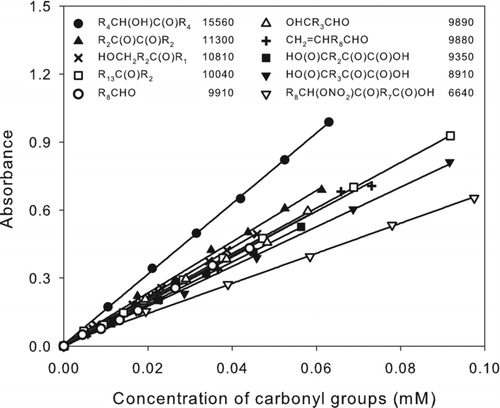
FIG. 3 Calibration curves of carbonyl compounds: 6-hydroxy-5-decanone (•), 3,4-hexadecadione (▴), 5-hydroxy-2-pentanone (×), 3-hexadecanone (□), nonanal (○), pentane-1,5-dial (▵), undec-10-enal (+), 2-ketopentanedioic acid (▪), 2-ketohexanedioic acid (▾), and 9,10-ketonitrate octadecanoic acid isomers (▿). The corresponding structures and values of ϵ (M−1 cm−1) are shown in the legend. R n designates (CH2) n or H(CH2) n depending on whether the alkyl group is in the interior or at the end of the molecule. The values of ϵ were calculated as ϵ = 103 × m, where m is the slope of the least-squares line fit to the calibration curve A = m(C) + b, where A = absorbance = absorbance (sample) – absorbance (blank), and (C) is the concentration of carbonyl groups (mM). For all lines, |b| ≤ 0.011 and R 2 > 0.99.
Analysis of Carbonyl Groups
The method presented here for analysis of carbonyl groups was developed from the American Society of Testing and Materials (ASTM) standard method ASTM International (Citation2005) and the methods of Lappin (Citation1951) and Jordan and Veatch (Citation1964). The method is based on the reaction of aldehydes and ketones with 2,4-DNPH in acidic solution to form 2,4-DNP hydrazone derivatives. These derivatives are then reacted with a base to produce an intense wine-red color due to formation of the quinoidal ion and then the absorbance is measured. The chemistry of the carbonyl group analysis is shown in .
The effect of molecular structure on the method results was evaluated by preparing calibration curves for a selection of ketones, diketones, aldehydes, and dialdehydes, some of which also contained hydroxyl, carboxyl, or nitrate groups or C˭C double bonds. The results are shown in . Linearity was excellent over the range of concentrations measured. Values of ϵ (M−1 cm−1) were the same for ketones and aldehydes, and for all the compounds tested, were in the range 6600–15600. Except for 6-hydroxy-5-decanone and 9,10-ketonitrate octadecanoic acid isomers, which have hydroxyl and nitrate groups adjacent to the carbonyl groups, the range was 8900–11300. The hydroxyl group increased ϵ, as was the case in the carboxyl group analysis, whereas the nitrate group decreased ϵ. Vicinal carboxyl or carbonyl groups or nonvicinal functional groups had little effect on the values of ϵ. For analysis of samples containing carbonyls with known structures, appropriate calibration curves can be prepared, whereas for samples containing unidentified compounds, the use of a simple ketone or aldehyde should give reasonably accurate results in most cases. Except for 6-hydroxy-5-decanone and the 9,10-ketonitrate octadecanoic acid isomers, values of ϵ for all the carbonyls analyzed here were within ∼10% of the values measured for 3-hexadecanone and nonanal, whereas for the other two compounds, the values were within ∼50%.
The method was tested for potential interferences from other compounds containing nitrate, carboxyl, hydroxyl, ester, peroxy, and hydroperoxy groups. Only hydroperoxides interfered with the analysis, by giving the same response as carbonyls. Hydroperoxides are thus decomposed prior to analysis by adding TPP, as suggested by Chiba et al. (Citation1989) and is done in the carboxyl group analysis. This had no or little other effect on the carbonyl group analysis. It is worth noting that ketones and aldehydes present as hemiacetals and peroxyhemiacetals are measured by this method, since peroxyhemiacetals are converted to hemiacetals by reaction with TPP and hemiacetals reversibly dissociate to carbonyls and alcohols under the conditions of the analysis.
The detection limit calculated as 3 × the standard deviation of blank measurements was 0.01 μmoles of carbonyl groups for carbonyl compounds with ϵ = 10,000, the average value for the compounds tested. The reproducibility determined by multiple analyses of 3-hexadecanone was 4% relative standard deviation.
Analysis of Ester Groups
The method presented here for analysis of esters is a modified version of the method of Goldenberg (Citation1958), which was developed for spectrophotometric analysis of carboxylic acids and their corresponding esters. The method is based on the conversion of esters to their hydroxamic acids using hydroxylamine hydrochloride in alkaline medium. After adding an acidified solution of alcoholic FeCl3, a red-colored complex of ferric hydroxamate forms and the absorbance is measured. The chemistry of the ester group analysis is shown in .
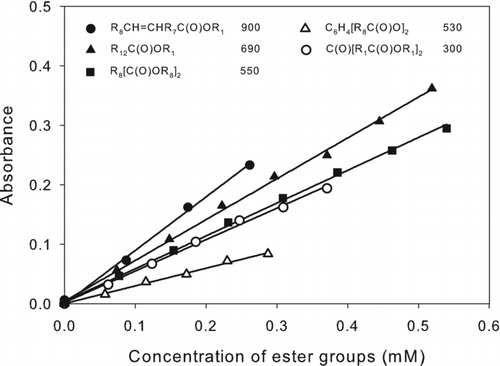
Calibration curves were prepared for a selection of esters with different structures, although no compounds were available with functional groups adjacent to an ester group. Nonetheless, there were considerable differences in measured values of ϵ. The results are shown in . Linearity was excellent over the range of concentrations measured, and the values of ϵ (M−1 cm−1) were in the range 300–900. The range for esters without an additional functional group was 530–690, with the values for the unsaturated ester and ketoester being 300 and 900, respectively. The value of ϵ measured for methyl (Z)-octadec-9-enoate (methyl oleate) was similar to that reported by Goddu et al. (1955). In addition to possible effects of the ketone group and C=C double bond on ϵ, the differences observed among esters might be due to differences in the rates of hydroxamic acid formation, hydrolysis of esters in alkaline medium, and decomposition of ferric hydroxamates. For analysis of samples containing esters with known structures, appropriate calibration curves can be prepared, whereas for samples containing unidentified compounds, the use of a simple ester such as methyl tridecanoate or bis(2-ethylhexyl)decanedioate (dioctyl sebacate) should give reasonably accurate results in most cases. Values of ϵ for all the esters analyzed here were within ∼50% of the values measured for these compounds.
FIG. 4 Calibration curves of esters: methyl (Z)-octadec-9-enoate (•), methyl tridecanoate (▴), bis(2-ethylhexyl)decanedioate (▪), bis(2-ethylhexyl)phthalate (▵), and dimethyl-3-oxopentanedioate (○). The corresponding structures and values of ϵ (M−1 cm−1) are shown in the legend. R n designates (CH2) n or H(CH2) n depending on whether the alkyl group is in the interior or at the end of the molecule. The values of ϵ were calculated as ϵ = 103 × m, where m is the slope of the least-squares line fit to the calibration curve A = m(C) + b, where A = absorbance = absorbance (sample) – absorbance (blank), and (C) is the concentration of ester groups (mM). For all lines, |b| ≤ 0.004 and R 2 > 0.99.
The method was tested for potential interferences from other compounds containing nitrate, carboxyl, hydroxyl, peroxy, and hydroperoxy groups. None of these groups interfered with the ester group analysis, but because the hydroxamic acid method is used to analyze acid anhydrides (Morgan Citation1958), amides (Bergmann Citation1952), and acyl phosphates (Lipmann and Tuttle Citation1945) under similar conditions, these compounds would interfere with the analysis.
The detection limit calculated as 3 × the standard deviation of blank measurements was 0.1 μmoles of ester groups for ester compounds with ϵ = 550, the average value for the compounds tested. The reproducibility determined by multiple analyses of bis(2-ethylhexyl)decanedioate was 5% relative standard deviation.
Analysis of Hydroxyl Groups
The method presented here for analysis of hydroxyl groups is a modified version of the method of Nachtmann (Citation1976). The method is based on the conversion of hydroxyl groups to esters using 4-NBC, which are subsequently solvent extracted, washed, and the absorbance is measured. The chemistry of the hydroxyl group analysis is shown in .
The effect of molecular structure on the results was evaluated by preparing calibration curves for a selection of monoalcohols and diols, some of which also contained carboxyl, carbonyl, or nitrate groups. The results are shown in . Linearity was excellent over the range of concentrations measured. Values of ϵ (M−1 cm−1) for all the compounds tested were in the range 9100–15,900, although except for 6-hydroxy-5-decanone and 5-hydroxy-2-pentanone, the range was 13,400–15,900. The presence of a carbonyl group decreases ϵ by ∼30%, similar to the effect on the carboxyl group analysis. For analysis of samples containing alcohols with known structures, appropriate calibration curves can be prepared, whereas for samples containing unidentified compounds, the use of a simple alcohol such as 2-hexadecanol should give reasonably accurate results in most cases. Values of ϵ for all the alcohols analyzed here were within ∼30% of the value measured for 2-hexadecanol.
FIG. 5 Calibration curves of alcohols: 16-hydroxyhexadecanoic acid (•), 1,2-tetradecanediol (+), 1,10-decanediol (▪), 2-hexadecanol (○), 12-hydroxyoctadecanoic acid (□), 9,10-hydroxynitrate octadecanoic acid isomers (▵), 6-hydroxy-5-decanone (▴), and 5-hydroxy-2-pentanone (×). The corresponding structures and values of ϵ (M−1 cm−1) are shown in the legend. R n designates (CH2) n or H(CH2) n depending on whether the alkyl group is in the interior or at the end of the molecule. The values of ϵ were calculated as ϵ = 103 × m, where m is the slope of the least-squares line fit to the calibration curve A = m(C) + b, where A = absorbance = absorbance (sample) – absorbance (blank), and (C) is the concentration of hydroxyl groups (mM). For all lines, |b| ≤ 0.004 and R 2 > 0.99.
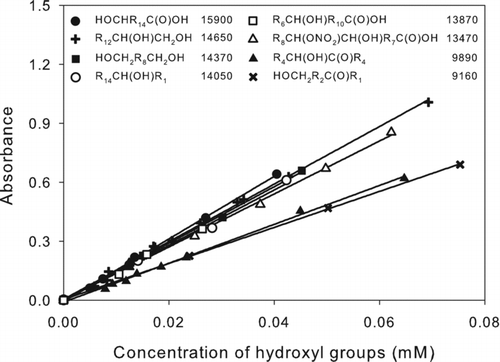
The method was tested for potential interferences from other compounds containing nitrate, carboxyl, hydroxyl, ester, peroxy, and hydroperoxy groups. No interference was observed for nitrates, ketones, or esters, or from peroxides other than hydroperoxides, and only a small interference was observed for aldehydes. Conversely, interferences from carboxyl and hydroperoxy groups were significant (Figure S5). The effect of carboxylic acids is surprising, since the values of ϵ measured for 16-hydroxyhexadecanoic acid and 12-hydroxyoctadecanoic acid were similar to those of monoalcohols and diols. Nonetheless, carboxyl groups are removed prior to derivatization of hydroxyl groups with 4-NBC by converting them to their corresponding methyl esters by reaction with TMSDM. Although TMSDM can also derivatize alcohols, that reaction requires the addition of fluoroboric acid (Presser and Hüfner Citation2004). Hydroperoxides could not be removed by reaction with TPP, as is done in some of our other methods, because alcohols are formed in the process. Instead, since hydroperoxides only interfere at concentrations ≥0.02 mM (Figure S5), the sample is diluted appropriately prior to the hydroxyl group analysis using the initial peroxide concentration measured using the method of Docherty et al. (Citation2005) for guidance.
The detection limit calculated as 3 × the standard deviation of blank measurements was 0.02 μmoles of hydroxyl groups for alcohols with ϵ = 14,400 (on a per hydroxyl group basis), the average value for the compounds tested. The reproducibility determined by multiple analyses of 1,2-tetradecanediol was 7% relative standard deviation.
TABLE 1 Composition of functional groups in SOA formed from reaction of α-pinene with O3 in the presence of cyclohexane
Method for Calculating and Expressing Functional Group Composition
The functional group composition of a sample, including methylene groups (CH2), is obtained by assuming that the only functional groups possibly present in the sample are carboxyl, carbonyl, ester, hydroxyl, peroxide, nitrate, and methylene. The most likely groups omitted by this assumption are ethers and epoxides, but these are expected to be absent or minor in many cases. The approach is to first sum the masses of measured functional groups, calculated as the measured moles × molecular weight using the following molecular weights (g mol−1): carboxyl [C(O)OH] = 45, carbonyl [C(O)] = 28, hydroxyl [CHOH] = 30, nitrate [CHONO2] = 75, ester [C(O)O] = 44, and peroxide [CHOOH] = 46. This value is then subtracted from the measured sample mass to obtain the mass of CH2 groups, which is then divided by 14 to obtain the moles of CH2 groups. This information is then used to calculate the mole fraction of each functional group (moles of functional group/total moles of functional groups). Because the sum of the mole fractions is 1 and each functional group has one C atom, each mole fraction is also equal to the fraction of that functional group per C atom of SOA. In our laboratory studies where the parent volatile organic compound (VOC) is known, we usually find it convenient to express the composition relative to the number of C atoms in the parent VOC by multiplying the mole fractions by the carbon number of the parent VOC. In cases where all SOA products have maintained the original carbon backbone of the parent VOC (i.e., there are no fragmentation products), this approach gives the average number of each functional group added to the parent VOC in the SOA-forming reactions, which makes it easier to visualize the average molecular structure of products. If more detailed information on the molecular weights of products is available, then an average number of C atoms per product can be calculated and used instead of the carbon number of the parent VOC. This approach normally has significant uncertainties, however, since molecular analysis by methods such as GC-MS and LC-MS (liquid chromatography–mass spectrometry) usually give an incomplete quantitative analysis of OA (Yu et al. Citation1999; Iinuma et al. 2004) and the formation of oligomers complicates the assignment of monomer molecular weights. Alternately, the functional group composition of OA can be expressed as mass fractions (Russell et al. Citation2011) or on a per C atom basis, which is preferable when not all functional groups have been quantified but measurements of total OA mass or carbon, respectively, are available. When the functional group analysis is relatively complete, then each functional group can be assigned a molecular weight and the composition can be expressed as mole fractions (Russell et al. Citation2011).
Functional Group Composition of SOA
The methods developed here were used to analyze SOA formed from the reaction of α-pinene with O3 in the presence of cyclohexane added as an OH radical scavenger. α-Pinene is the largest of the monoterpene emissions to the atmosphere (primarily by coniferous trees), which account for ∼10% of nonmethane VOC emissions on a global scale (Guenther et al. Citation1995). Its reaction with O3 efficiently produces low-volatility products that can condense to form SOA. Molecular analysis of SOA formed from this reaction has identified and quantified a variety of multifunctional products containing carboxyl, carbonyl, ester, hydroxyl, and peroxy groups, a significant fraction of which are thought to be present in oligomers (Yu et al. Citation1999; Reinnig et al. Citation2009; Yasmeen et al. Citation2010). The results of our analyses are shown in , along with the O/C and H/C ratios calculated from the functional group analysis and measured by standard combustion methods. Assuming that all products contain 10 C atoms, the same as α-pinene (although some fragmentation products are present [Yu et al. Citation1999; Iinuma et al. 2004], the average numbers of carboxyl, carbonyl, ester, hydroxyl, peroxide, and methylene groups per C10 SOA molecule were 1.63, 0.99, 0.41, 0.20, 0.15, and 6.62, respectively. Average O/C and H/C ratios calculated from the functional group analysis were 0.54 and 1.56, respectively, which compare reasonably well with average values of 0.44 and 1.65 measured by combustion methods. These ratios fall within the major domain of values measured by Heaton et al. (Citation2009) for this reaction using offline high-resolution MS, for which the range was O/C ∼ 0.2–0.6 and H/C ∼ 1.4–1.8, and are at the high end of average values measured for this reaction using online high-resolution aerosol MS, where, e.g., O/C and H/C ratios of ∼0.45 and ∼1.4 have been measured at low mass concentrations and ∼0.30 and ∼1.5 at high mass concentrations, respectively (Shilling et al. Citation2009). Also, for comparison, Russell et al. (Citation2011) have used FTIR spectroscopy to analyze the functional group composition of OA collected in a coniferous forest in Whistler, Canada, where monoterpenes dominate VOC emissions and SOA is expected to derive largely from the oxidation of these compounds. The average number of carboxyl, carbonyl (including esters), hydroxyl, and methylene groups per C10 molecule in the OA in Whistler (calculated by multiplying FTIR-measured mole fractions by 10) were 1.0, 1.6, 2.5, and 4.4, respectively, and the reported average O/C ratio was 0.72 (Russell et al. Citation2011). This composition was somewhat similar to that measured here for SOA formed in the chamber, although the Whistler aerosol had a much higher concentration of hydroxyl groups, fewer carboxyl groups, and overall was more highly oxidized. This may reflect a significant contribution to Whistler aerosol of products formed from reactions of OH radicals with monoterpenes, which should contain more hydroxyl and less carboxyl groups than products of O3 reactions (Capouet et al. Citation2008). In the clean remote atmosphere, the ratio of OH:O3 reaction for α-pinene is ∼0.6:0.4 (Atkinson and Arey Citation2003).
CONCLUSIONS
The spectrophotometric methods developed here can be used to quantify the concentrations of carbonyl, hydroxyl, carboxyl, and ester groups in oxidized OA. The methods employ derivatizing agents to convert functional groups to characteristic colored derivatives that are quantified by measuring the absorbance using spectrophotometry. The effects of molecular structure on quantification have been evaluated by measuring calibration curves for a variety of monofunctional and multifunctional compounds, and potential interferences from other compounds containing nontarget functional groups have been determined, and methods developed to eliminate these interferences. For these methods, there are no artifacts due to ester formation from reactions of carboxylic acids and alcohols or from ester hydrolysis, neither of which occurs under the conditions of these analyses, and hemiacetal and peroxyhemiacetal formation will not affect the quantification of alcohols, carbonyls, and hydroperoxides, which can reversibly form these compounds. For analysis of samples containing compounds with known structures, appropriate calibration curves can be prepared, whereas for samples containing unidentified compounds, the use of a simple monofunctional carboxylic acid, ketone or aldehyde, ester, and alcohol should give results that are within ∼30% (and often much less) of the correct value, and at worst ∼50%. Detection limits are approximately 0.01, 0.02, 0.03, and 0.1 μmoles for carbonyl, hydroxyl, carboxyl, and ester groups, respectively. A complete set of replicate analyses for these groups as well as peroxide (Docherty et al. Citation2005) and nitrate (Matsunaga and Ziemann Citation2009) requires ∼1 mg of sample, and with standard curves, takes ∼3 days, although additional samples can be analyzed in parallel. Unfortunately, it is unlikely that detection limits could be substantially improved by using longer cell path lengths, because the reagent blanks already have sufficiently high absorbance with a 1-cm cell (carbonyl and carboxyl ∼ 0.1, ester ∼ 0.2, and hydroxyl ∼ 0.3) that the use of, e.g., a 50-cm waveguide fully saturates the blank absorbance. We did not attempt to determine the source of this absorbance, but since the chemicals we used were of very high purity, it was probably not due to impurities. Although these methods were demonstrated here by analyzing SOA samples, they should also be applicable to the analysis of organic gas samples, although care must be taken to avoid evaporation of the more volatile compounds during sample processing. To analyze organic compounds in ambient air one would need to correct absorption measurements of derivatized samples for the background absorbance in the sample due to species such as brown carbon and humic-like substances. Also, because of the potential for effects of materials such as black carbon or mineral dust on extraction efficiencies, it would also be necessary to evaluate and optimize this aspect of sample preparation, as is done for other offline methods of organic analysis. The organic extract obtained can then be dried and analyzed using the methods described here. These methods should complement the use of FTIR and NMR spectroscopy for the functional group analysis, since although those methods have lower detection limits, they are less specific for some functional groups. The derivatives formed by these methods should also be suitable for HPLC-MS analysis, providing a better means to identify compounds by MS and also to quantify compounds using UV-Vis absorbance. As is the case with all analytical methods, it is useful to have complimentary means for checking the validity (or at least the “reasonableness”) of the results obtained with these methods, such as functional group composition measured by FTIR, which does not require filter extraction, and elemental analyses obtained using offline combustion techniques and online high-resolution aerosol MS.
Supplemental Information.zip
Download Zip (227.1 KB)Acknowledgments
This research was funded by the Office of Science (BER), U.S. Department of Energy, under DE-FG02-05ER63984 and DE-SC 0006633. Sukon Aimanant would like to thank the Government of Thailand for fellowship support.
[Supplementary materials are available for this article. Go to the publisher's online edition of Aerosol Science and Technology to view the free supplementary files.]
REFERENCES
- ASTM International . 2005 . Standard Test Method for Trace Quantities of Carbonyl Compounds with 2,4-Dinitrophenylhydrazine . Designation , E411-05 : 502 – 505 .
- Atkinson , R. and Arey , J. 2003 . Atmospheric Degradation of Volatile Organic Compounds . Chem. Rev. , 103 : 4605 – 4638 .
- Blando , J. D. , Porcja , R. J. and Turpin , B. J. 2001 . Issues in the Quantification of Functional Groups by FTIR Spectroscopic Analysis of Impactor-Collected Aerosol Samples . Aerosol Sci. Technol. , 35 : 899 – 908 .
- Bergmann , F. 1952 . Colorimetric Determination of Amides as Hydroxamic Acids . Anal. Chem. , 24 : 1367 – 1369 .
- Capouet , M. , Müller , J.-F. , Ceulemans , K. , Compernolle , S. , Vereecken , L. and Peeters , J. 2008 . Modeling Aerosol Formation in Alpha-Pinene Photo-Oxidation Experiments . J. Geophys. Res. , 113 : D02308
- Chiba , T. , Takazawa , M. and Fujimoto , K. 1989 . A Simple Method for Estimating Carbonyl Content in Peroxide-Containing Oils . J. Am. Oil. Chem. , 66 : 1588 – 1592 .
- Coury , C. and Dillner , A. M. 2008 . A Method to Quantify Organic Functional Groups and Inorganic Compounds in Ambient Aerosols Using Attenuated Total Reflectance FTIR Spectroscopy and Multivariate Chemometric Techniques . Atmos. Environ. , 42 : 5923 – 5932 .
- Decesari , S. , Facchini , M. C. , Fuzzi , S. and Tagliavini , E. 2000 . Characterization of Water-Soluble Organic Compounds in Atmospheric Aerosol: A New Approach . J. Geophys. Res. , 105 : 1481 – 1489 .
- Docherty , K. S. , Wu , W. , Lim , Y. B. and Ziemann , P. J. 2005 . Contributions of Organic Peroxides to Secondary Aerosol Formed from Reactions of Monoterpenes with O3 . Environ. Sci. Technol. , 39 : 4049 – 4059 .
- Docherty , K. S. and Ziemann , P. J. 2006 . Reaction of Oleic Acid Particles with NO3 Radicals: Products, Mechanism, and Implications for Radical-Initiated Organic Aerosol Oxidation . J. Phys. Chem. A , 110 : 3567 – 3577 .
- Feldman , J. A. , Frank , S. G. and Holmes , J. H. 1971 . Determination of Di-(2-ethylexyl) Sodium Sulfonsuccinate and Related Dialkyl Esters in Aqueous Solutions by the Ferric Hydroxamate Procedure . J. Pharm. Sci. , 60 : 920 – 921 .
- Feldmann , J. A. and Robb , B. J. 1970 . Colorimetric Determination of Atropine, Homatropine, Scopolamine and Their Derivatives by the Ferric Hydroxamate Method . J. Pharm. Sci. , 59 : 1646 – 1647 .
- Goddu , R. F. , LeBlanc , N. F. and Wright , C. M. 1955 . Spectrophotometric Determination of Esters and Anhydrides by Hydroxamic Acid Reaction . Anal. Chem. , 27 : 1251 – 1255 .
- Goldenberg , V. 1958 . Colorimetric Determination of Carboxylic Acid Derivatives as Hydroxamic Acids . Anal. Chem. , 30 : 1327 – 1330 .
- Greenbaum , M. A. , Denney , D. B. and Hoffmann , A. K. 1956 . A Study of the Mechanism of the Conversion of Benzoyl Peroxide to Benzoic Anhydride by Triphenylphosphine . J. Am. Chem. Soc. , 78 : 2563 – 2565 .
- Guenther , A. , Hewitt , C. N. , Erickson , D. , Fall , R. , Geron , C. Graedel , T. 1995 . A Global Model of Natural Volatile Organic Compound Emissions . J. Geophys. Res. , 100 : 8873 – 8892 .
- Hallquist , M. , Wenger , J. C. , Baltensperger , U. , Rudich , Y. , Simpson , D. Claeys , M. 2009 . The Formation, Properties and Impact of Secondary Organic Aerosol: Current and Emerging Issues . Atmos. Chem. Phys. , 9 : 5155 – 5236 .
- Heaton , K. G. , Sleighter , R. L. , Hatcher , P. G. , Hall , W. A. IV and Johnston , M. V. 2009 . Composition Domains in Monoterpene Secondary Organic Aerosol . Environ. Sci. Technol. , 43 : 7797 – 7802 .
- Hestrin , S. 1949 . The Reaction of Acetylcholine and Other Carboxylic Acid Derivates with Hydroxylamine and Its Analytical Application . J. Biol. Chem. , 180 : 249 – 261 .
- Hoffmann , T. , Huang , R-J. and Kalberer , M. 2011 . Atmospheric Analytical Chemistry . Anal. Chem. , 83 : 4649 – 4664 .
- Horikawa , R. and Tanimura , T. 1982 . Spectrophotometric Determination of Carboxylic Acids with 2-Nitrophenylhydrazine in Aqueous Solution . Anal. Lett. , 15 : 1629 – 1642 .
- Iinuma , Y. , Böge , O. , Gnauk , T. and Herrmann , H. 2004 . Aerosol Chamber Study of the α-Pinene/O3 Reaction: Influence of Particle Acidity in Aerosol Yields and Products . Atmos. Environ. , 38 : 761 – 773 .
- Jacobson , M. C. , Hansson , H.-C. , Noone , K. J. and Charlson , R. J. 2000 . Organic Atmospheric Aerosols: Review and State of the Science . Rev. Geophys. , 38 : 267 – 294 .
- Jaoui , M. , Kleindienst , T. E. , Lewandowski , M. , Offenberg , J. H. and Edney , E. O. 2005 . Identification and Quantification of Aerosol Polar Oxygenated Compounds Bearing Carboxylic or Hydroxyl Groups: 2. Organic Tracer Compounds from Monoterpenes . Environ. Sci. Technol. , 39 : 5661 – 5673 .
- Jordan , D. E. and Veatch , F. C. 1964 . Spectrophotometric Determination of Trace Concentrations of Carbonyl Compounds . Anal. Chem. , 36 : 120 – 124 .
- Korany , M. A. , Abdel-Hay , M. H. , Bedair , M. M. and Gazy , A. A. 1989 . Colorimetric Determination of Some Penicillins and Cephalosporins with 2-Nitrophenylhydrazine Hydrochloride . Talanta , 36 : 1253 – 1257 .
- Kroll , J. H. and Seinfeld , J. H. 2008 . Chemistry of Secondary Organic Aerosol: Formation and Evolution of Low-Volatility Organics in the Atmosphere . Atmos. Environ. , 42 : 3593 – 3624 .
- Lappin , G. 1951 . Colorimetric Method for Determination of Traces of Carbonyl Compounds . Anal. Chem. , 23 : 541 – 542 .
- Lipmann , F. and Tuttle , L. C. 1945 . A Specific Micromethod for the Determination of Acyl Phosphates . J. Biol. Chem. , 159 : 21 – 28 .
- Matsunaga , A. and Ziemann , P. J. 2009 . Yields of β-Hydroxynitrates and Dihydroxynitrates in Aerosol Formed from the OH Radical-Initiated Reactions of Linear Alkenes in the Presence of NOx . J. Phys. Chem. A , 113 : 599 – 606 .
- Meyer , S. and Rebrovic , L. 1995 . The Spectroscopic Quinoidal Ion Method for the Analysis of Carbonyl Compounds . J. Am. Oil. Chem. , 72 : 385 – 387 .
- Miwa , H. , Hiyama , C. and Yamamoto , M. 1985 . High Performance Liquid Chromatography of Short-and Long-Chain Fatty Acids as 2-Nitrophenylhydrazides . J. Chromatogr. , 321 : 165 – 174 .
- Miwa , H. , Yamamoto , M. and Momose , T. 1980 . Colorimetric Detection and Determination of Carboxylic Acids with 2-Nitrophenylhydrazine Hydrochloride . Chem. Pharm. Bull. , 28 : 599 – 605 .
- Morgan , D. M. and Kingsbury , K. J. 1959 . A Modified Hydroxamic Acid Method for Determinating Total Esterified Fatty Acids in Plasma . The Analyst , 84 : 409 – 414 .
- Morgan , K. J. 1958 . The Ferric Hydroxamate Colour Reaction II. The Spectrophotometric Determination of Acetic Anhydride . Anal. Chem. Acta. , 19 : 27 – 31 .
- Munson , J. W. 1974 . 2-Nitrophenylhydrazine: A Selective Reagent for Colorimetric Determination of Carboxylic Acid Anhydrides and Chlorides . J. Pharm. Sci. , 63 : 252 – 257 .
- Nachtmann , F. 1976 . Some Properties of 4-Nitrobenzoates of Saccharides and Glycosides: Application to High-Pressure Liquid Chromatography . Z. Anal. Chem. , 282 : 209 – 213 .
- Presser , A. and Hüfner , A. 2004 . Trimethylsilyldiazomethane—A Mild and Efficient Reagent for the Methylation of Carboxylic Acids and Alcohols in Natural Products . Monatshefte für Chemie , 135 : 1015 – 1022 .
- Reinnig , M-C. , Warneke , J. and Hoffmann , T. 2009 . Identification of Organic Hydroperoxides and Hydroperoxyacids in Secondary Organic Aerosol Formed During the Ozonolysis of Different Monoterpenes and Sesquiterpenes by On-Line Analysis Using Atmospheric Chemical Ionization Ion Trap Mass Spectrometry . Rapid. Commun. Mass Spectrom. , 23 : 1735 – 1741 .
- Russell , L. M. , Bahadur , R. and Ziemann , P. J. 2011 . Identifying Organic Aerosol Sources by Comparing Functional Group Composition in Chamber and Atmospheric Particles . Proceed. Natl. Acad. Sci. USA , 108 : 3516 – 3521 .
- Shilling , J. E. , Chen , Q. , King , S. M. , Rosenoern , T. , Kroll , J. H. Worsnop , D. R. 2009 . Loading-Dependent Elemental Composition of α-Pinene SOA Particles . Atmos. Chem. Phys. , 9 : 771 – 782 .
- Shriner , R. L. , Fuson , R. C. and Curtin , D. Y. 1964 . The Systematic Identification of Organic Compounds , (5th ed. , New York : John Wiley & Sons .
- Stern , I. and Shapiro , B. 1953 . A Rapid and Simple Method for the Determination of Esterified Fatty Acids and for Total Fatty Acid in Blood . J. Clin. Path. , 6 : 158 – 159 .
- Thompson , A. R. 1950 . A Colorimetric Method for the Determination of Esters . Australian J. Sci. Research , 3A : 128 – 135 .
- Verheyden , J. and Nys , J. 1962 . The Estimation of Fatty Acid Esters in Serum by the Hydroxamate Method . Clin. Chim. Acta , 7 : 262 – 269 .
- Yasmeen , F. , Vermeylen , R. , Szmigielski , R. , Iinuma , Y. , Böge , O. , Herrmann , H. , Maenhaut , W. and Claeys , M. 2010 . Terpenylic Acid and Related Compounds: Precursors for Dimers in Secondary Organic Aerosol from the Ozonolysis of α- and β Pinene . Atmos. Chem. Phys. , 10 : 9383 – 9392 .
- Yu , J. , Cocker , D. R. , Griffin , R. J. , Flagan , R. C. and Seinfeld , J. H. 1999 . Gas-Phase Ozone Oxidation of Monoterpenes: Gaseous and Particulate Products . J. Atmos. Chem. , 34 : 207 – 258 .
- Yukawa , N. , Takamura , H. and Matoba , T. 1993 . Determination of Total Carbonyl Compounds in Aqueous Media . J. Am. Oil. Chem. , 70 : 881 – 884 .