Abstract
Respiratory illnesses are a significant cause of morbidity for individuals who work within concentrated animal feeding operations (CAFOs); however, most available information about CAFO aerobiology has derived from culture-based studies, which may detect only a small fraction of microbial diversity present. In this study, we characterized the identity, spatial distribution, and abundance of airborne microorganisms present in swine and dairy CAFOs using direct microscopy, broad-range rRNA PCR, and sequence analysis of air samples collected from within and nearby swine and cattle operations in the western United States. We report that indoor airborne bacterial loads were not elevated above those measured immediately outdoors. The microbial assemblage of these indoor environments was considerably more diverse than reported in previous CAFO aerosol studies. Members of bacterial genera associated with animal gut microbiota, including Bacillus spp., Clostridium spp., and Lachnospira spp., were most frequently observed. We detected no recognized, acute respiratory pathogens, but identified the common opportunistic pathogen Aerococcus viridans in several samples. Fungal species were not recovered in any of the indoor clone libraries. Specific PCR assay for porcine circovirus demonstrated that this pathogen is prevalent in the atmosphere of swine environments sampled, but was not detected in the bovine dairy facilities.
Copyright 2013 American Association for Aerosol Research
INTRODUCTION
Since the end of the Second World War, the commercial livestock industry has undergone increased industrialization. One result of this modernization has been a decrease in the number of traditional animal farms in the United States, along with an increase in the numbers of animals raised (Tellier Citation2006; Thorne Citation2007). To meet consumer demand and to compensate for reduced farm space, livestock are frequently raised indoors in high numbers and densities under controlled environmental conditions. While such concentrated animal feeding operations (CAFOs) reduce labor costs and ensure consistent growth conditions and product quality, they also promote prolonged confinement at high population densities, which can increase susceptibility to infectious disease among livestock and workers. Additionally, livestock and workers are routinely exposed to relatively high concentrations of dusts, gases, and endotoxin as a result of indoor confinement practices (Radon et al. Citation2002; Heederik et al. Citation2007).
Concentrated animal feeding operation (CAFO) employment is associated with increased incidences of multiple respiratory disorders, including asthma, chronic bronchitis, organic dust toxic syndrome, chronic obstructive pulmonary disease, rhinitis, hypersensitivity, hydrogen sulfide and carbon dioxide poisoning, and acute lower respiratory tract inflammation (Donham et al. Citation1989a; Crook et al. Citation1991; Schenker et al. Citation1998; Von Essen and Donham Citation1999; Cole et al. Citation2000; Sprince et al. Citation2000; Danuser et al. Citation2001; Radon et al. Citation2001, Citation2002; Monso et al. Citation2003, Citation2004). Chronic airway inflammation possibly is exacerbated by exposure to microbes and their products in bioaerosols. For instance endotoxin, a highly toxigenic, bacterial lipopolysaccharide component of gram-negative bacterial cell walls, has been associated with reduced lung function in swine farmers (Donham et al. Citation1989b; Vogelzang et al. Citation1998). Correlations between biotic and abiotic exposures and respiratory disease have been the focus of many aerosol studies and have led to the establishment of dust, gas, and endotoxin exposure levels (Crook et al. Citation1991; Seedorf et al. Citation1998; Predicala et al. Citation2001; Avery et al. Citation2004; Pomorska et al. Citation2007; Ni and Heber Citation2008).
CAFO workers, particularly those working with swine and poultry, are at the forefront of risk for emerging zoonotic diseases of substantial public health concern, including avian influenza (Nicholson et al. Citation2003; Koopmans et al. Citation2004), swine influenza (Olsen et al. Citation2002; Myers et al. Citation2006), and multi-drug resistant pathogens (Chapin et al. Citation2005; Scott et al. Citation2005). Consequently, a better understanding of CAFO airborne microbiology and development of sensitive surveillance protocols are needed. Any relationship between CAFO-related infectious diseases and the microbial assemblages present within these environments remains poorly understood. Except for the most acute infections, the etiological agents of lung disorders in CAFO workers are not well characterized, in part because the current understanding of the aerobiology in these environments is based on traditional culture analyses, which typically isolate only a small fraction of microbes (Amann et al. Citation1995; Pace Citation1997). Additionally, past studies have been limited to descriptions of mesophilic aerobic bacteria and/or fungi (Cormier et al. Citation1990; Crook et al. Citation1991; Chang et al. Citation2001; Predicala et al. Citation2002; Jo and Kang Citation2005; Kim et al. Citation2008) isolated from these environments. As a result of these constraints, little is known about the bulk composition of CAFO aerosol microbiology.
The aims of this study were to determine airborne biological loads and to survey the major airborne microbial assemblages found within swine and dairy cattle CAFOs. We used culture-independent molecular methods based on ribosomal RNA (rRNA) gene sequences for characterization of microbes, which potentially provide a broader perspective on the microbial diversity of CAFOs than do culture analyses. Additionally, we demonstrate that the analytical protocols used for rRNA gene sequencing also can be applied for airborne virus detection. Detection and monitoring of viral pathogens is complex and usually requires submission of swab or pathological samples to tissue culture or immunological assays for target viruses. To assess the efficacy of detecting viral signatures in bioaerosols, we assayed Porcine Circovirus-2, which commonly occurs in swine CAFOs (Verreault et al. Citation2010), and sometimes is pathogenic (Madec et al. Citation2008). Overall, the results of this study provide a model for obtaining more robust descriptions of airborne microorganisms in CAFO environments than previously available, including concomitant characterization of selected viral and bacterial genes.
METHODS
Aerosol Sampling
Aerosol samples were collected in and near one dairy and two swine facilities located in the Rocky Mountain region of the United States. The prevailing winds during these sampling campaigns were westerly and light. Background samples were collected approximately 5 miles west (northwest) of the dairy and swine facilities observed in this study. All background samples were taken on the same days when aerosols were collected from the respective animal housing sites. The dairy consisted of 24 stalls and was opened at one end for the animals to enter and exit. Aerosol sample collection took place at the closed end of the building and as near to the milking operations as possible. The average time spent by a cow within the building was 12 min. Following milking, the cows returned to holding pens until the next milking session. Seven samples were collected at this facility during Spring 2007 and four were randomly selected for sequencing analyses.
Swine facility A (Swine A) consisted of three growth-phase barns: nursery, grower, and finisher. Samples were collected from each type of barn. The nursery was divided into six rooms separated by doors and was entirely enclosed. Each room was approximately 25′ wide and 32′ long with fan units in each room to maintain a temperature of approximately 25°C. The grower and finisher barns were approximately 50′ wide and 100′ long and contained 40 pens. Each pen held 10 to 12 pigs. These barns were partially enclosed with 0.6-m gaps between the roof and walls to allow air circulation, and a 0.3-m gap between the roof slats. Manure pits were located under slated concrete floors in both barns. Sample collection took place as close to the center of each barn as was possible. Six samples were collected for sequence analyses during Summer 2007.
Swine facility B (Swine B) was an entirely enclosed barn with 18 rooms (each approximately 33′ wide and 60′ long) separated by doors. The pigs were brought to the building from a farrowing barn and remained within the sampled barn for the duration of their lives. At the time of sampling, the pigs were at the finishing stage (20–30 weeks old). Air circulation within the barn was controlled automatically by motors, which opened and closed the barn walls to maintain a temperature of approximately 25°C. Three samples were collected at one end of the barn in Fall 2007 and analyzed.
Aerosol samples were concurrently collected with SKC Biosamplers (BioSampler SKC Inc., Eighty Four, PA, USA) and an Omni 3000 Portable Sampler (Sceptor Industries, Inc., Kansas City, MO). Both samplers used in this study are designed as swirling liquid impingers; they capture particles from an airstream introducing a tangential shear stress to the reservoir contents. While they have markedly different flow rates, the physical details underlying the operations of these samplers, have been comprehensively described in the literature (Xuejun et al. Citation1999; Zhao et al. Citation2011). SKC Biosamplers were operated approximately 1 m above the ground. The collection medium in the SKC samplers was sterile 10 mM Tris-Cl pH 8.0, 1 mM EDTA (Thermo Fisher Scientific, Pittsburg, PA, USA). Two Biosamplers were operated in parallel at a flow rate of 12.5 L/min and they were allowed to collect continuously for at least 30 min at each sampling site. Collection fluids from the parallel impinger runs were pooled for processing. The Omni collected samples in parallel with the SKC Biosamplers at a height of 1.8 m above ground and a flow rate of 270 L/min. The collection medium in the Omni was sterile 1X PBS supplemented with 0.005% Tween (Thermo Fisher Scientific, Pittsburg, PA, USA). Samples were collected at 5-min intervals and pooled to comprise a 30-min collection period. Sterile water was added during collection to both impingers to compensate for evaporative losses. All samples were held on ice until processed, no more than a few hours. Negative controls of impinger media were processed in parallel with other samples.
Sample aliquots for microscopic counting were preserved with 2% (v/v) formaldehyde. Samples for molecular analysis were collected onto 0.2 micron nitrocellulose filters (Nalgene Analytical Test Filter Funnel, Nalge Nunc International Rochester, NY, USA). Processed samples were stored at 4°C (microscopy) or −80°C (DNA analyses) until analyzed.
Airborne Microbe Enumeration
Aerosol samples were stained and enumerated using 4′6-diamidino-2-phenylindole (DAPI) (Sigma Chemicals, St. Louis, MO, USA) and previously described methods (Hernandez et al. Citation1999). Samples were incubated with DAPI (20 μg/mL) for 5 min at 4°C then filtered onto 25 mm, 0.22 micron pore, black polycarbonate filters (Poretics, Inc., Livermore, CA, USA). Filters were mounted in low fluorescence immersion oil containing an antifadent (CitiFluor, Leiscester, UK) and examined at 1100× using a Nikon Eclipse E400 epifluorescence microscope.
A minimum three independent aliquots from each impinger were used for direct counts each of which required that at least 10 random fields were counted per slide; only intact, brightly stained cells with obvious bacterial or fungal morphology were counted. Direct counts were calculated based on the average of all fields counted; in all cases, the coefficient of variation observed was less than 20% (Hernandez et al. Citation1999). After testing for normality, a paired t-test, with a one-tailed distribution was applied at a 90% confidence level to test for significant differences between the direct counts recovered from each environment.
Genomic DNA Extraction
Concentrated samples and negative controls were extracted from filters using a bead-beating and solvent extraction protocol modified from Frank et al. (Citation2003). Extracted DNA was dissolved in 20 μL of Tris-EDTA, pH 8.0 (TE), and stored at −80°C.
PCR of rRNA Genes
DNAs were submitted to rRNA gene PCR amplification using universally conserved (515 Forward/1391 Reverse) primers and HotMasterMix® PCR master mix (Eppendorf North America, Inc., Westbury, NY, USA). A typical 20μL PCR reaction included: 8 μL 2X HotMasterMix, 0.5 μL Forward Primer (25 ng/μL), 0.5 μL Reverse Primer (25 ng/μL), 8 μL water, 2 μL bovine serum albumin (10 μg/μL), and 1 μL DNA template. Amplification was performed on an Eppendorf Mastercycler® (Eppendorf North America, Inc., Westbury, NY, USA) using a protocol of: 94°C for 2 min, 94°C for 20 s, 52°C for 20 s, 65°C for 90 s, 30 cycles total. PCR controls with no DNA template were included with each reaction. To confirm rRNA gene amplification, PCR products were examined by agarose gel electrophoresis (1% agarose with ethidium bromide). Extraction and negative controls never showed evidence of amplification due to methodological contamination.
Clone Library Construction
Amplicons from four independent PCR reactions with the same template DNA were pooled and purified by agarose gel electrophoresis using a Montage® DNA Gel Extraction Kit (Milipore, Billerica, MA, USA). Purified product was then ligated into pCR4.0 TOPO plasmid vectors and transformed using Top10 electro competent cells (Invitrogen Corporation, Carlsbad, CA, USA). Transformed cells were plated and grown overnight on Luria-Bertani (LB) agar. For each library (multiple libraries were made with some samples), a total of 96 clones were picked randomly into 10 μL TE, heated at 85°C for 10 min for lysis and centrifuged at 4000xG for 10 min to pellet cellular debris.
DNA Sequencing
Sequencing was performed as previously described by Papineau et al. (Citation2005). Briefly, sequencing template was prepared from rRNA gene clones by PCR amplification with T3 and T7 vector primers, and treatment of amplicons with ExoSAP-IT (GE Healthcare, Piscataway, NJ, USA). Templates were sequenced with the DYEnamic ET Terminator Cycle Sequencing Kit (GE Healthcare, Piscataway, NJ, USA) on a Mega-BACE 1000 DNA Sequencer using T3 and T7 primers. Base calling and assembly of raw sequence data were performed with the phred and phrap software packages (Ewing et al. Citation1998), using the software XplorSeq (Frank Citation2008).
SSU rRNA Phylogenetic Analysis
The sequence collection was screened for chimeric sequences (Huber et al. Citation2004), which were discarded. The closest known relative of each rRNA gene clone was identified by BLAST (Altschul et al. Citation1990). Sequences were aligned with the NAST software package (DeSantis et al. Citation2006) and phylogenetic assignments were inferred with the ARB software package (Ludwig et al. Citation2004) by parsimony insertion. Sequences were clustered into operational taxonomic units (OTUs) at 97% sequence identity bins using the program sortx (Frank Citation2008).
Richness Estimates and Community Relatedness
Sample coverage and species richness estimates were estimated using biodiv (Frank Citation2008), which calculates coverage statistics (Goods, CACE), species richness estimates (Schao1 and SACE), and diversity indices (Simpson, Shannon) based on user defined sequence identity levels. Sequence libraries were compared using UniFrac (Lozupone and Knight Citation2005), which compares related sets of sequences (samples) through a sequence distance index based on the fraction of the total branch-length in a phylogenetic tree that is unique to a particular sample.
PCV2-Specific Quantitative PCR
Sevevteen aerosol samples were analyzed for the presence of porcine circovirus type 2 (PCV2) using specific quantitative PCR (Q-PCR) with PCV2-specific primers (Verreault et al. Citation2010). As a control template for the PCV2 survey, we used a plasmid carrying the PCV2 gene, into which we introduced a 10 base-pair deletion (PCV2Δ). Use of this control ensured that any potential contamination of experimental samples by control DNA, a common problem in QPCR, could be detected by QPCR product size. To construct the deletion plasmid, the plasmid pCR4/PCV2a, which contained the entire genome of PCV2 genotype 2a (Gagnon et al. Citation2008), was amplified by PCR with a primer incorporating a 10 base pair deletion in positions 591–600 in the PCV2a genome (AY094619). Amplicons were cloned into pCR®4-TOPO® vector (TOPO® TA Cloning kit, Invitrogen Corp., Carlsbad, CA, USA), and sequenced to verify the deletion. The control plasmid was purified from E. coli with a QIAprep® Spin Miniprep Kit (Qiagen Inc.) and quantified by spectrofluorometry. Q-PCR was performed on a DNA Engine Opticon System (MJ Research Inc., Incline Village, NE, USA) in 25-μL reaction volumes composed of 12.5 μL Power SYBR® Green PCR Master Mix (Applied Biosystems, Warrington, UK), 150 nM of each primer, 1 μL of 10 mg/mL bovine serum albumin (BSA) (Sigma-Aldrich Inc., St. Louis, MO, USA), 8.62 μL water, and 1 μL of sample DNA. The amplification and detection program was as follows: 95°C for 10 min followed by 38 cycles of 95°C for 15 s, 65°C for 20 s, 60°C for 45 s, a fluorescence read, then 1 s at 80°C, and a second plate read. Three replicate QPCR reactions were performed for each sample. Multiple negative-template controls were included in all assays. A melting curve and agarose gel electrophoresis of PCR amplicons were used to ensure specificity of amplification. A set of QPCR products was sequenced directly to verify that amplified sequences corresponded to PCV2 and not a spurious sequence. Quantification of template concentrations was made by linear extrapolation of baseline-subtracted data from the plasmid dilution series standard curves, and calculated as the number of PCV2 genomes per cubic meter of air (PCV2/m3). We estimate a sensitivity of these Q-PCR assays to be ≈100–200 PCV2 genomes/m3.
PCV2 DNA Sequencing
Impinger sample DNAs were amplified using PCV2-specific primers that span a highly variable region of the PCV-2 genome based on comparison of GenBank sequences and correspond to: forward primer, 5′-GGGCCAGAATTCAACCTTAA-3′ (location in PCV2a genome [AY094619]: 1415–1434); reverse primer 5′-AGGAGGCGTTACCGSAGAAG-3′ (S = C or G) (location in PCV2a genome [AY094619]: 1723–1704). Amplification products were cloned and sequences determined as above.
PCV2 Phylogenetic Analysis
The Swine facility PCV2 sequences were augmented with genomic PCV2 sequences from GenBank trimmed to correspond to the PCR priming sites and aligned with ClustalX version 2.1 (Larkin et al. Citation2007). One thousand bootstrap trees were cast from the aligned sequence set and the bootstrap scores were annotated onto to the best scoring maximum likelihood tree found with RAxML (Stamatakis Citation2006). The annotated tree was drawn with Phylovis, as described previously (Robertson et al. Citation2009).
Sequence Accession Numbers
All sequences from this study have been deposited in GenBank, with the accession numbers JN566417 - JN566807 (Circovirus 2) and JN56608-N568240 (16S rRNA sequences).
RESULTS
Airborne Microbial Concentrations
Total airborne microorganism concentrations, as determined by microscopic counting of DAPI-stained aliquots of impinger samples, are summarized in . The airborne microbe concentrations observed within the different CAFOs were in the range of 6 × 105–2 × 107 cells/m3, only a few-fold higher than the corresponding outdoor environments (2 × 106–3 × 106 cells/m3). These observations were similar to bioaerosol loads (1 × 106–7 × 107 cells/m3) reported for other industrial animal farming environments measured using direct microscopic methods (Agranovski et al. Citation2004; Chi and Li Citation2005).
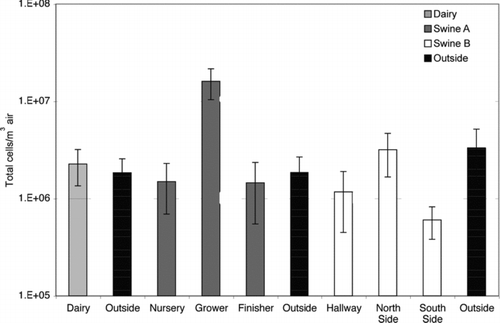
Table 1 TABLE 1 Ecological indices associated with clone libraries
Microbial Diversity of Sampled Environments
Aerosol samples were subjected to rRNA gene sequence analysis in order to identify microorganisms in the aerosols. As detailed in Materials and Methods, DNAs were extracted from samples, used as templates for PCR with universally conserved rRNA gene primers (515F/1391R) and PCR products were cloned; at least 96 clones were randomly selected and sequenced for each sample. A total of 17 rRNA gene libraries (16 bacterial and 1 eukaryotic) were analyzed and 1433 sequences determined. Because of high microbial loads, this depth of sequence analysis undersamples the total assemblage, but we identify the abundant organisms and provide sufficient information for community comparisons (Kuczynski et al. Citation2010).
The majority of sequences, >90%, were highly similar (≥97% identity, species-level variation [Goebel and Stackebrandt Citation1994]) to sequences in GenBank. Approximately 5% of sequences had less than 95% sequence identity (genus-level variation) to database sequences. Collectively, the sequences fell into ∼750 different 97% identity operational taxonomic units (OTUs). OTU richness and diversity indices (SChao1, SACE, Shannon, and Simpson) for the samples, using rarefaction to control for unequal sample sizes, are summarized in . By these metrics, the dairy parlor samples had the highest species richness of the indoor environments followed by the grower and finisher barns (624-1762 OTUs as predicted by SChao1). Outdoor environments presented markedly lower richness (197-592 OTUs as predicted by SChao1). Sample coverage, the observed fraction of predicted OTUs, ranged between 7 and 21%, indicating that much biodiversity remains uncharacterized in these environments.
Microbial Composition of the Sampled CAFO Environments
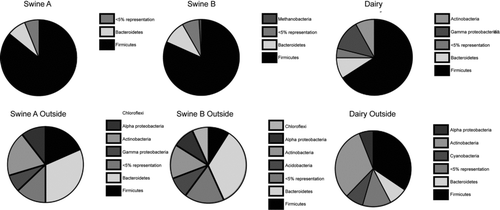
The phylogenetic distributions of rRNA gene sequences recovered from the indoor CAFO air samples and the corresponding local outdoor environments are summarized in . lists those bacterial families detected more than twice. Approximately 80% of all sequences obtained from the CAFO indoor environments were represented by members of the Firmicutes phylum and 21% of these had high similarity (>97%) to sequences previously reported in animal or human gut, as determined by comparison to datasets of published studies. An additional 6% of the Firmicutes sequences were closely related to human microbiota, for instance species detected in subgingival plaque. The remaining Firmicutes sequences were diverse with many observed only once. Sequences indicative of the opportunistic pathogen Aerococcus viridans was observed in all environments, except the swine nursery.
Table 2 TABLE 2 Phylogenetic distribution of 16S rRNA sequences in CAFO bioaerosols
Other notable phyla observed in the CAFO environments included Bacteroidetes, Proteobacteria, and Methanobacteria. Representatives of the Bacteroidetes, common fecal bacteria, comprised the second most abundant phylum (9%) observed indoors. Most of these sequences (67%) belonged to the family Prevotellaceae and were closely related to microorganisms previously detected in the human gut. Proteobacteria composed 5% of the total sequences observed with Gammaproteobacteria being the largest phylum. The Gammaproteobacteria observed were primarily from indoor dairy air and were related to microbes from a wide variety of environments. Representatives of archaeal methanogens closely related to rumen gut clones were observed in all CAFO environments (3% of total sequences).
Microbial Composition of Corresponding Nearby Local Outdoor Air
The airborne microbial composition of the respective outdoor environments was collectively more diverse than inside the CAFOs. Groups with the largest representation in the outdoor samples included Actinobacteria (26%), Firmicutes (26%), Bacteroidetes (19%), and Proteobacteria (15%), which were generally distinct from the indoor air microbiota (). Consistent with this high diversity, most outdoor sequences were encountered only once and generally were related to other environmental sequences (data not shown).
Community Comparisons
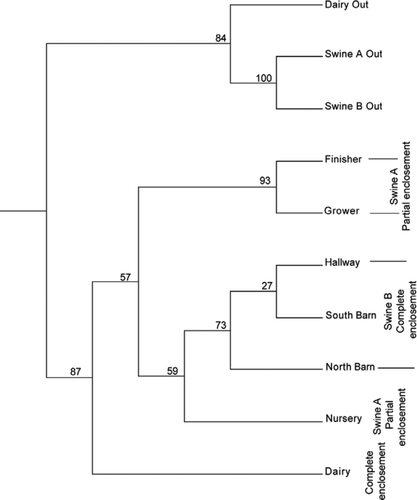
In order to test more rigorously relationships between the different sites and the outdoors, we used UniFrac (Lozupone and Knight Citation2005) analysis, which quantifies phylogenetic relationships between sets of taxa. Results indicated that airborne microbes recovered from indoors were markedly different from those recovered outdoors, regardless of the animals being housed (). Airborne microbial assemblages inside the dairy and swine houses also differed. Within the swine houses, significant differences in airborne microbe populations could be associated with housing practices (partial vs. complete enclosure). Similarities within the CAFO environments are dominated by sequences representative of fecal bacteria. UniFrac-based principal coordinates analysis (not shown) suggests the variation observed was best explained by aerosol sample source—indoor versus outdoor and animal association.
Analysis of Aerosolized Viruses
In order to extend this method beyond detection of bacteria, we explored detection of viruses captured in impinger fluids, using as a proxy porcine circovirus 2 (PCV2), which is common in swine facilities (Verreault et al. Citation2010). Accordingly, DNAs from the aerosol samples analyzed for bacterial contents were also assayed for PCV2 viral contents using QPCR as described (Verreault et al. Citation2010). PCV2 loads were detected in all swine facilities with mature pigs, but were not detected in the nursery, the dairy facilities or in outdoor samples. Where detected, PCV2 aerosolized viral loads were significant (103–105 viral DNAs/m3). These findings are consistent with other reports of the widespread occurrence of PCV2 in swine facilities and indicate that the virus is a commensal of swine as well as an opportunistic pathogen.
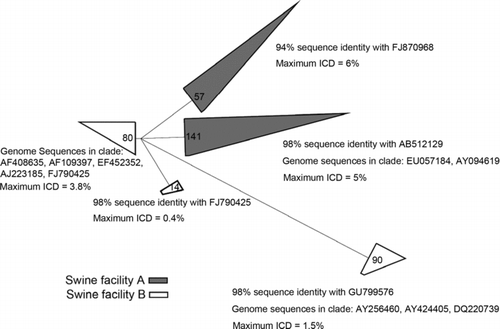
PCV2 virus is a “quasispecies,” a collection of closely related viruses that have undergone rapid variation (Hughes and Piontkivska Citation2008; Rosario et al. Citation2009) Consequently, sequence variation in rapidly changing regions of the PCV2 genome can be used for precise typing of the virus. In an effort to identify the PCV2 subtypes associated with the swine CAFOs, we amplified from the sample DNAs by PCR a ∼300 bp highly variable (capsid protein) region of the PCV2 genome, cloned the products and sequenced ∼400 representative clones. Phylogenetic analysis of the sequences () indicates that several PCV2 relatedness groups, subspecies, occurred in the two swine facilities, including subspecies variously termed PCV2, PCV2a, PCV2b, PCV1/2a, and PCV2e (Morozov et al. Citation1998; Olvera et al. Citation2007; Gagnon et al. Citation2010; Beach et al. Citation2010; Takahagi et al. Citation2010). No novel PCV2 clades were detected. The phylogenetic analysis also shows that different subspecies of PCV2 predominated at the two swine facilities, almost mutually exclusively. Swine facility A had a relatively narrow range of PCV2 sequence variation compared to the broader range of swine facility B. Each of the PCV2 relatedness groups is itself variable, with a few percent sequence variation within the groups (ICD, intraclade distance, in ).
DISCUSSION
Until very recently, CAFO aerobiology has been characterized mainly with culture-based techniques that emphasize species that grow best at moderate temperature under oxic conditions (Cormier et al. Citation1990; Crook et al. Citation1991; Chang et al. Citation2001; Predicala et al. Citation2002; Jo and Kang Citation2005; Kim et al. Citation2008). Our study sidestepped these constraints by use of culture-independent technology. Microbial loads were quantified by microscopy and microbial species were identified by broad-spectrum rRNA gene PCR and sequencing.
Relatively few published studies report the total (both viable and nonviable) numbers of airborne microorganisms present within CAFO environments. Total airborne microbe concentrations observed in all three facilities were similar to each other and to their corresponding outdoor environments. Airborne bacterial loads within both swine facilities were comparable to those reported in previous studies using similar counting methods (Agranovski et al. Citation2004; Chi and Li Citation2005), but were greater (10–1000×) than levels previously reported for airborne fungi (Adhikari et al. Citation2004a; Lee et al. Citation2006). Significant differences in total airborne microbial loads measured at Swine Facility A () may be attributed to animal density within the sampled barns: the grower barn, with ten-fold higher microbial load, housed three times more pigs than the sampled nursery room, with much smaller pigs. With the exception of the grower barn, the loads measured at Swine Facility A were statistically indistinguishable from those measured outdoors.
The results of this study suggest that the microbial diversity aloft within swine and dairy CAFO environments is substantially more extensive than previously recovered by culture (). Overall more than 750 species-level OTUs (97% sequence identity bins) were observed among the 1433 sequences analyzed, with most observed only once or twice. OTU richness estimates (SACE and SChao1) predicted large numbers of species (between 350–1762) present in each of the CAFO aerosol samples. The most diverse environments in this sampling campaign, measured by both Shannon and Simpson diversity indices, were predicted in those places where some mixing with or stirring by outdoor air took place, such as the dairy parlor and the swine grower and finisher barns. Based on these diversity and richness estimates, and the number of OTUs observed, the estimated clone library coverage was typically 10%–20%. Thus, CAFO biodiversity was undersampled, although the more abundant kinds of organisms were captured (). Deeper sequence analysis will certainly reveal considerable further microbial diversity in these settings.
Like the observations reported by Ravva in a US Department of Agriculture study of dairies in Central California (Ravva et al. Citation2011), and pig and poultry confinements in the Midwest US (Hong et al. Citation2012), the most frequently observed species in these CAFO environments were primarily representatives of the phylogenetic group Firmicutes, of the kinds associated with nonpathogenic gut or rumen microorganisms (). Major Firmicute lineages included Bacillus spp. (33% of Firmicute sequences), Lachnospiraceae spp. (30%), and Clostridium spp. (20%). The relative abundances of some phylotypes (Aerococcus spp., Lactobacillus spp., Streptococcus spp., and Clostridium spp.) observed in both swine facilities were similar to those of swine gastrointestinal tracts (Leser et al. Citation2002; Snell-Castro et al. Citation2005) and swine confinement buildings (Cormier et al. Citation1990; Crook et al. Citation1991; Predicala et al. Citation2002; Nehme et al. Citation2008). The phylogenetic types of organisms observed in the dairy samples were similar to those reported in the feces of dairy cows (Dowd et al. Citation2008) and cultured in a previous aerosol study of cattle feedlots (Wilson et al. Citation2002). Particular species of organisms and their proportions, however, differ among the various dairy studies. Nonetheless, the general conclusion from this and other studies is that most airborne bacteria in the CAFO setting are associated with the aerosolization of manure, and that there is a relatively low incidence of airborne bacterial pathogens either immediately near dairy confinements or downwind (Dugan Citation2012).
Few microbes encountered might be considered to be of known health-related concern. Sequences closely related to the common opportunistic pathogen Aerococcus viridans, which is associated with arthritis in swine (Martin et al. Citation2007), were found in all environments except the Swine A nursery. A. viridans has been cultured from environmental aerosol samples in previous studies (Williams et al. Citation1953; Kerbaugh and Evans Citation1968). Sequences closely related to that of Propionibacterium acnes, a common human commensal, were also recovered from all environments. While generally considered harmless, P. acnes is associated with corneal ulcers, acne, and other illness (Bruggemann et al. Citation2004). P. acnes is apparently ubiquitous in the environment and has been observed in other outdoor aerosol studies (Brooks et al. Citation2007; Tringe et al. Citation2008; de Evgrafov et al. Citation2010). Whether deposition of P. acnes in the lungs through aerosol inhalation represents a health threat is unknown.
At the limited depth to which we sequenced (1433 sequences), no fungal rRNA sequences were detected. In contrast, previous CAFO bioaerosol studies based on culture counts concluded that fungi contributed a sizable portion of the overall observed (via culture) airborne ecology (Hanhela et al. Citation1995; Chang et al. Citation2001; Adhikari et al. Citation2004a,b; Jo and Kang Citation2005; Chi and Li Citation2005; Lee et al. Citation2006; Kim et al. Citation2008). This conclusion based on culture, however, is not supported by our results. The universal primers used in this study readily amplify fungal DNA from other environments, so our results indicate that in fact fungal genes are rare compared to bacterial genes. This is consistent with the results of sequence-based surveys in general, which find that bacterial rRNA genes numerically overwhelm fungal genes in the environment by orders of magnitude. Moreover, the kinds of organisms detected by sequencing, gut and rumen bacteria, tend to be anaerobic and therefore not detected by routine oxic cultivation. We conclude that any meaningful survey of the microbiology of the CAFO aerosol environment needs to include culture-independent analysis.
Aerosol transmission of viral diseases is a major concern in global health and international monitoring of recognized and emerging diseases is an ongoing effort. Surveillance for disease viruses is expensive and usually involves point samples such as swabs or clinical specimens. We demonstrate here that fluid impinger aerosol sampling, coupled with molecular analysis, readily detects the commonly occurring swine CAFO virus, PCV2. The method likely extends to all viruses, the desired detection targets selected by the choice of primers used in QPCR or other molecular assays. This aerosol protocol potentially offers a more efficient and economical way than point sampling to obtain facility-wide surveillance for disease agents.
In summary, the results of this study show that the microbial diversity of CAFO atmospheric environments has the potential to be far greater than previously reported based on culture studies, and confirms that viruses, which are germane to health of animals within these impoundments, are airborne in significant numbers. The results also provide baseline information for the deeper sequencing and longitudinal studies that will be necessary to better understand the atmospheric environment of CAFOs.
REFERENCES
- AdhikariA.ReponenT.LeeS.A.GrinshpunS.A.2004a11269277
- AdhikariA.SenM.M.Gupta-BhattacharyaS.ChandaS.2004b2910711078
- AgranovskiV.RistovskiZ.BlackallP.J.MorawskaL.20043838933901
- AltschulS.F.GishW.MillerW.MyersE.W.LipmanD.J.1990215403410
- AmannR.I.LudwigW.SchleiferK.H.199559143169
- AveryR.C.WingS.MarshallS.W.SchiffmanS.S.200459101108
- BeachN.M.RamamoorthyS.OpriessnigT.WuS.Q.MengX.J.201029221232
- BrooksJ.GerbaC.PepperI.200710317791790
- BruggemannH.HenneA.HosterF.LiesegangH.WiezerA.StrittmatterA.et al.2004305671673
- ChangC.W.ChungH.HuangC.F.SuH.J. J.200167155161
- ChapinA.RuleA.GibsonK.BuckleyT.SchwabK.2005113137142
- ChiM.C.LiC.S.20053911011110
- ColeD.ToddL.WingS.2000108685699
- CormierY.TremblayG.MeriauxA.BrochuG.LavoieJ.199051304309
- CrookB.RobertsonJ.GlassS.BotheroydE.LaceyJ.ToppingM.199152271279
- DanuserB.WeberC.KunzliN.SchindlerC.NowakD.200139410418
- de EvgrafovM.R.WalkerJ.J.PaceN.R.HernandezM.T.201044236245
- DeSantisT.Z.HugenholtzP.KellerK.BrodieE.L.LarsenN.PicenoY.M.et al.200634W394W399
- DonhamK.HaglindP.PetersonY.RylanderR.BelinL.1989a463137
- DonhamK.HaglindP.PetersonY.RylanderR.BelinL.1989b463137
- DowdS.E.CallawayT.R.WolcottR.D.SunY.McKeehanT.HagevoortR.G.et al.200888
- DuganR.S.201241814
- EwingB.HillierL.WendlM.C.GreenP.19988175185
- FrankD.N.2008916
- FrankD.N.SpiegelmanG.B.DavisW.WagnerE.LyonsE.PaceN.R.200341295303
- GagnonC.A.del CastilloJ.R. E.MusicN.FontaineG.HarelJ. e.TremblayD.200820545558
- GagnonC.A.MusicN.FontaineG.TremblayD.HarelJ.20101441823
- GoebelB.M.StackebrandtE.19946016141621
- HanhelaR.LouhelainenK.PasanenA.L.199521223228
- HeederikD.SigsgaardT.ThorneP.S.KlineJ.N.AveryR.BonlokkeJ.H.et al.2007115298302
- HernandezM.MillerS.L.LandfearD.W.MacherJ.M.199930145160
- HongP.-Y.LiX.YangX.ShinkaiT.ZhangY.WangX.et al.201214614201431
- HuberT.FaulknerG.HugenholtzP.20042023172319
- HughesA.L.PiontkivskaH.20088130138
- JoW.K.KangJ.H.200560140146
- KerbaughM.EvansJ.196816519523
- KimK.Y.KoH.J.KimH.T.KimC.N.KimY.S.200899565572
- KoopmansM.WilbrinkB.ConynM.NatropG.van der NatH.VennemaH.et al.2004363587593
- KuczynskiJ.LiuZ.LozuponeC.McDonaldD.FiererN.KnightR.20107813819
- LarkinM.A.BlackshieldsG.BrownN.P.ChennaR.McGettiganP.A.McWilliamH.et al.20072329472948
- LeeS.A.AdhikariA.GrinshpunS.A.McKayR.ShuklaR.ReponenT.20063118130
- LeserT.D.AmenuvorJ.Z.JensenT.K.LindecronaR.H.BoyeM.MollerK.200268673690
- LozuponeC.KnightR.20057182288235
- LudwigW.StrunkO.WestramR.RichterL.MeierH.KumarY.et al.20043213631371
- MadecF.RoseN.GraslandB.CarioletR.JestinA.200855273283
- MartinV.VelaA.I.GilbertM.CebollaJ.GoyacheJ.DominguezL.et al.20074530533057
- MonsoE.RiuE.RadonK.MagarolasR.DanuserB.IversenM.et al.200446357362
- MonsoE.SchenkerM.RadonK.RiuE.MagarolasR.McCurdyS.et al.200321323331
- MorozovI.SirinarumitrT.SordenS.D.HalburP.G.MorganM.K.YoonK.J.et al.19983625352541
- MyersK.P.OlsenC.W.SetterquistS.F.CapuanoA.W.DonhamK.J.ThackerE.L.et al.2006421420
- NehmeB.LetourneauV.ForsterR.J.VeilletteM.DuchaineC.200810665675
- NiJ.Q.HeberA.J.Sampling and Measurement of Ammonia at Animal Facilities, in Advances in Agronomy Elsevier Academic Press Inc. San Diego 2008 98 201 269
- NicholsonK.G.WoodJ.M.ZambonM.200336217331745
- OlsenC.W.BrammerL.EasterdayB.C.ArdenN.BelayE.BakerI.et al.20028814819
- OlveraA.CorteyM.SegalesJ.2007357175185
- PaceN.R.1997276734740
- PapineauD.WalkerJ.J.MojzsisS.J.PaceN.R.20057148224832
- PomorskaD.LarssonL.SkorskaC.SitkowskaJ.DutkiewiczJ.200714291298
- PredicalaB.Z.MaghirangR.G.JerezS.B.UrbanJ.E.GoodbandR.D.20014412911298
- PredicalaB.Z.UrbanJ.E.MaghirangR.G.JerezS.B.GoodbandR.D.200244136140
- RadonK.DanuserB.IversenM.JorresR.MonsoE.OpravilU.et al.200117747754
- RadonK.DanuserB.IversenM.MonsoE.WeberC.HartungJ.et al.200294148
- RavvaS.V.SarrealRobertC.Z.MandrellE.201162e17281
- RobertsonC.E.SpearJ.R.HarrisJ.K.PaceN.R.20097518011810
- RosarioK.DuffyS.BreitbartM.20099024182424
- SchenkerM.B.ChristianiD.CormierY.Dimich-WardH.DoekesG.DosmanJ.et al.1998158S1S76
- ScottH.M.CampbellL.D.HarveyR.B.BischoffK.M.AlaliW.Q.BarlingK.S.et al.200522437
- SeedorfJ.HartungJ.SchroderM.LinkertK.H.PhillipsV.R.HoldenM.R.et al.19987097109
- Snell-CastroR.GodonJ.-J.DelgenesJ.-P.DabertP.200552229242
- SprinceN.L.LewisM.Q.WhittenP.S.ReynoldsS.J.ZwerlingC.200038455462
- StamatakisA.20062226882690
- TakahagiY.TokiS.NishiyamaY.MorimatsuF.MurakamiH.2010723541
- TellierR.20061216571662
- ThorneP.S.2007115296297
- TringeS.G.ZhangT.LiuX.YuY.LeeW.H.YapJ.et al.20083e1862
- VerreaultD.LÈtourneauV.GendronL.MassÈD.GagnonC.A.DuchaineC.2010141224230
- VogelzangP.F.van der GuldenJ.W.FolgeringH.KolkJ.J.HeederikD.PrellerL.et al.19981571518
- Von EssenS.DonhamK.199914337350
- WathesC.M.19987097109
- WilliamsR.HirschA.CowanS.19538475480
- WilsonS.C.Morrow-TeschJ.StrausD.C.CooleyJ.D.WongW.C.MitlohnerF.M.et al.20026832383242
- XuejunL.ReponenT.A.WillekeK.GrinshpunS.A.FoardeK.K.EnsorD.S.19993342914298
- ZhaoY.AarninkA.DoornenbalP.HuynhT.T.Groot KoerkampP.W.LandmanW.J.et al.201145432442