Abstract
Chemically resolved submicron (PM1) particle mass fluxes were measured by eddy covariance with a high resolution time-of-flight aerosol mass spectrometer over temperate and tropical forests during the BEARPEX-07 and AMAZE-08 campaigns. Fluxes during AMAZE-08 were small and close to the detection limit (<1 ng m−2 s−1) due to low particle mass concentrations (<1 μg m−3). During BEARPEX-07, concentrations were five times larger, with mean mid-day deposition fluxes of −4.8 ng m−2 s−1 for total nonrefractory PM1 (Vex,PM1 = −1 mm s−1) and emission fluxes of +2.6 ng m−2 s−1 for organic PM1 (Vex,org = +1 mm s−1). Biosphere–atmosphere fluxes of different chemical components are affected by in-canopy chemistry, vertical gradients in gas-particle partitioning due to canopy temperature gradients, emission of primary biological aerosol particles, and wet and dry deposition. As a result of these competing processes, individual chemical components had fluxes of varying magnitude and direction during both campaigns. Oxygenated organic components representing regionally aged aerosol deposited, while components of fresh secondary organic aerosol (SOA) emitted. During BEARPEX-07, rapid in-canopy oxidation caused rapid SOA growth on the timescale of biosphere-atmosphere exchange. In-canopy SOA mass yields were 0.5–4%. During AMAZE-08, the net organic aerosol flux was influenced by deposition, in-canopy SOA formation, and thermal shifts in gas-particle partitioning. Wet deposition was estimated to be an order of magnitude larger than dry deposition during AMAZE-08. Small shifts in organic aerosol concentrations from anthropogenic sources such as urban pollution or biomass burning alters the balance between flux terms. The semivolatile nature of the Amazonian organic aerosol suggests a feedback in which warmer temperatures will partition SOA to the gas-phase, reducing their light scattering and thus potential to cool the region.
Copyright 2013 American Association for Aerosol Research
1. INTRODUCTION
Understanding the sources and sinks of atmospheric aerosol particles is necessary for accurately modeling regional and global aerosol concentrations, and thus for controlling air quality and predicting radiative forcing for both past and future climates. Although global models estimate that biogenic secondary organic aerosol (BSOA) particles represent a substantial fraction of the global organic aerosol particle burden, BSOA is a challenging component to constrain, possibly due to the difficulty in defining and measuring BSOA separately from anthropogenic SOA and other OA sources (Spracklen et al. Citation2011). Recent global-scale BSOA estimates from bottom-up and top-down approaches span two orders of magnitude (Chung and Seinfeld Citation2002; Goldstein and Galbally Citation2007; Hallquist et al. Citation2009; Spracklen et al. Citation2011). BSOA is comprised of low- and semivolatile biogenic organic compounds that are the products of oxidation reactions of precursor biogenic volatile organic compounds (BVOCs). The most abundant BVOCs are isoprene, monoterpenes, and sesquiterpenes, which are oxidized in the atmosphere by OH, O3, and NO3 on timescales of seconds to days. Depending on their vapor pressure and concentration, some fraction of the oxidized products can partition to the particle phase, either by new particle formation or by condensation onto existing particles (Seinfeld and Pankow Citation2003).
Current models suggest that forests are a key source of gaseous SOA precursors, emitting approximately 70% of global biogenic volatile organic compound (BVOC) emissions (Guenther et al. Citation1995). However, forests are expected to provide a net sink with respect to particle number and total mass, with dry deposition being most efficient for such aerodynamically rough surfaces (Fowler et al. Citation1999). This has been confirmed by measurements of submicron particle number flux over both tropical and temperate forests (Ahlm et al. Citation2009; Vong et al. Citation2010). Nevertheless, the deposition rates can vary significantly for different chemical components (Ruijgrok et al. Citation1997; Fowler et al. Citation2009; Farmer et al. Citation2011; Gordon et al. Citation2011). Some observations of upward fluxes of submicron particles from forests have hinted that biogenic SOA may be formed rapidly enough in forest canopies for forests to occasionally act as a net particle source (Buzorius et al. Citation1998). Furthermore, comparisons between direct plant VOC emission rates and observed concentrations provide evidence that some VOCs are being oxidized on rapid timescales, as determined from measurements of fluxes and vertical gradients of oxidized VOCs, O3, and reactive nitrogen oxides (Goldstein et al. Citation2004; Holzinger et al. Citation2005; Farmer and Cohen Citation2008; Bouvier-Brown et al. Citation2009; Wolfe et al. Citation2009). Due to their rapid oxidation, these BVOCs are candidates for in-canopy BSOA formation. A recent estimate of SOA fluxes based on measurements of SOA precursor gas concentrations during BEARPEX-07 (Biosphere Effects on Aerosols and Photochemistry Experiment) in Blodgett Forest, California, USA, combined with laboratory SOA particle mass yields, suggests that rapid in-canopy oxidation of sesquiterpenes can account for the build-up of an SOA particle mass concentration of 0.2–1.0 μg m−3 within the canopy, or approximately 6–32% of the total organic particle mass concentration observed above the forest canopy (Bouvier-Brown et al. Citation2009). In the presence of typically daytime turbulence, such a large SOA source within the forest canopy would create a strong upward flux term. Wolfe et al. (Citation2011) used a vertically resolved canopy model to constrain the chemistry during BEARPEX-07; their findings suggest that the oxidation products of very reactive BVOCs, following ventilation of gas-phase species from the canopy into the boundary layer, ultimately contribute 0.5–0.7 μg m−3 of SOA above the forest canopy. These studies suggest that forest emissions, particularly from strong terpene-emitting plant species, may lead to substantial and rapid SOA production. However, the potential role of rapid, in-canopy BSOA formation in forest particle fluxes has remained unconstrained due to a lack of chemically resolved particle flux measurements.
Forests are also direct sources of primary biological aerosol particles (PBAP) (Pöschl et al. Citation2010). PBAPs include fungal spores, pollen, bacteria, and other airborne biological material, and potentially constitute a substantial aerosol source by mass, perhaps even to the submicron fraction. At the global scale, fungal spore emissions are predicted to contribute 28 Tg yr−1 with about 25% of those emissions <2.5 μm, though uncertainties are large due to the small number of observational studies (Heald and Spracklen Citation2009).
Dry deposition modules in chemical transport models typically use a resistance approach (Hicks et al. Citation1987; Wesely and Hicks Citation2000). A few empirical parameterizations consider chemically dependent deposition (Ruijgrok et al. Citation1997). None of these parameterizations account for upward fluxes of particles. However, many components of particles are semivolatile and thus sensitive to temperature gradients. Vertical thermal gradients between the ground and sensor height can cause shifts in the NH3-NH4NO3-HNO3 equilibrium, for example, and the kinetic approach to the new equilibrium can occur on rapid enough timescales to induce gradients in particle concentration (Nemitz et al. Citation2004; Trebs et al. Citation2006). Similar effects may be operative for semivolatile organic species, some of which have volatilities similar to that of NH4NO3 (Huffman et al. Citation2009). However, aside from studies of NH4NO3, little is known as to how thermal and chemical gradients affect aerosol particle fluxes, in particular whether SOA formation from BVOC oxidation occurs sufficiently rapidly to affect ecosystem-scale particle fluxes.
For the study presented in this article, chemically resolved aerosol particle flux measurements taken over both tropical and temperate forests are used to investigate organic particle mass fluxes, including fluxes of specific chemical components. These measurements were made over a monoterpene-dominated ponderosa pine plantation (BEARPEX-07) and an isoprene-dominated tropical rainforest (AMAZE-08).
2. EXPERIMENTAL
2.1. Measurement Sites
Measurements described herein were made during two field campaigns: the Biosphere Effects on Aerosols and Photochemistry Experiment in 2007 (BEARPEX-07) and the Amazonian Aerosol Characterization Experiment in 2008 (AMAZE-08). The BEARPEX-07 experiment took place between 10 August and 3 October 2007 at the University of California-Berkeley Blodgett Forest Research Station, a ponderosa pine plantation in the mid-elevation Sierra Nevada Mountains (38.90°N, 120.63°W, elevation 1315 m). Blodgett Forest is characterized by daytime upslope flows and nighttime downslope flows. The upslope flows carry air from the surrounding mid-elevation coniferous forest in the morning, followed in the mid-day by air advected from lower elevation oak forests. In the late afternoon, air is additionally influenced by urban area around Sacramento (Goldstein et al. Citation2000; Day et al. Citation2009). Ambient air was sampled from a tower 25 m above the ground through ½″ OD (1.27 cm) copper tubing at 6 m s−1. Vegetation at the Blodgett Forest site is dominantly ponderosa pine (mean height of 7.9 m during BEARPEX-07). There is an understory (<2 m height) of whitethorn and manzanita and a one-sided leaf area index (LAI) of 5.1 m2 m−2. Several papers have presented results from BEARPEX-07 for VOCs and SOA (Bouvier-Brown et al. Citation2009; Huisman et al. Citation2011; Wolfe et al. Citation2011; Worton et al. Citation2011).
The AMAZE-08 campaign took place between 7 February and 13 March 2008 in the wet season. The field site (02°35.68′S, 60° 12.56′W, elevation 110 m) was 60 km NNW of Manaus, Brazil (Martin et al. Citation2010). Back-trajectories showed that air traveled over the Atlantic Ocean and then over 1000 km of pristine forest before reaching the site (Martin et al. Citation2010). Ambient air was sampled above a 33 m forest canopy from the top of the 38.75 m TT34 tower on the ZF2 reserve through ½′′ OD stainless steel tubing at 7–14 m s−1. Instruments were housed in an airconditioned container at the bottom of the tower. The tower was located in primary tropical terra firme rainforest comprised of broadleaf hardwoods and some understory palms. One-sided leaf area index (LAI) at a nearby site was 5.7 m2 m−2 (McWilliam et al. Citation1993).
2.2. Methods
Chemically resolved particle fluxes were determined by eddy covariance analysis using an Aerodyne High Resolution Time-of-Flight Aerosol Mass Spectrometer (HR-AMS) (DeCarlo et al. Citation2006). Eddy covariance measurements derive the vertical flux of a scalar over a surface using the correlation between vertical wind speed and concentration, typically on a half-hour timescale (Baldocchi et al. Citation1988). Three-dimensional wind speed measurements were made with a sonic anemometer (Applied Technologies Inc., Longmont, CO, USA) that was co-located with the aerosol inlet on a boom 2 m from the tower. The HR-AMS quantitatively measured nonrefractory components of aerosol particles that have vacuum aerodynamic diameters between 50 nm and 1000 nm (DeCarlo et al. Citation2006; Canagaratna et al. Citation2007). To obtain eddy covariance fluxes, the HR-AMS was alternated between the standard acquisition mode (Canagaratna et al. Citation2007), which collected concentration and size-dependent mass spectra and a recently described flux mode (Kimmel et al. Citation2010; Farmer et al. Citation2011). Mass spectra were collected at 10 Hz and fluxes were averaged for 30 min of every hour (Farmer et al. Citation2011). The inlet and sampling set-ups for the AMS were similar for the two campaigns and are described in detail elsewhere (Chen et al. Citation2009; Martin et al. Citation2010; Farmer et al. Citation2011). Due to the high ambient humidity, sampled air was dried (Nafion dryer) to <70% RH prior to detection by HR-AMS during the AMAZE-08 project. As ambient RH was typically <60%, sampled air was not dried during BEARPEX-07. Lag-times between sonic anemometer and HR-AMS data were 3.8 s and 5 s for BEARPEX-07 and AMAZE-08, respectively.
Eddy covariance fluxes of particle mass were determined from the HR-AMS data in three different ways: (1) fluxes of unit resolution mass-to-charge ratios (m/z), (2) fluxes of high-resolution signals integrated over a defined m/z window, and (3) fluxes of chemical components of PM1 as deduced from fragmentation patterns of, for example, organic aerosol (Allan et al. Citation2004; Farmer et al. Citation2011). As described in detail elsewhere (Farmer et al. Citation2011), multiple species may fragment to the same ion and thus be observed by the HR-AMS at the same m/z. For example, both ammonium nitrate and organic nitrates produce NO+ and NO2+ ions in the HR-AMS (Farmer et al. Citation2010). AMS species are calculated as a combination of unit mass resolution m/z. Thus, the three flux approaches used in combination provide information on the chemistry of the particle flux. This study follows the convention that a positive flux is upward from the canopy, corresponding to a greater concentration below the sensor height than above. A negative flux is a movement downward from the atmosphere to the surface. Details of the flux calculations, including discussion of uncertainties, lag time corrections, and filtering, were described previously (Farmer et al. Citation2011). To compare results from the two datasets and other experimental and modeling studies more easily, local (i.e., at the measurement height) exchange velocities (Vex) are calculated as the flux divided by the concentration:
Exchange velocities have the same sign convention as the flux measurements, meaning that positive Vex correspond to upward fluxes (emission) and negative Vex correspond to downward fluxes (deposition).
Results described herein are averages taken from a subset of the campaign data. Unless otherwise stated, data presented are from 12 September to 27 September 2007 of BEARPEX-07 and from 24 February to 13 March 2008 for AMAZE-08. These periods represent as the most stable ones for instrument parameters and performance during the measurement campaigns. Figures and data are presented in local standard time (for BEARPEX-07, PST = UTC–08:00; for AMAZE-08, AMT = UTC–04:00). Concentrations and fluxes are presented under local temperature and pressure conditions. Temperature and pressure are normalized in the calculation of Vex, thus allowing comparison of these values across different sites. Temperatures at the measurement height were 18–27°C during BEARPEX-07 and 22–27°C during AMAZE-08. The pressure was typically 870 mbar during BEARPEX-07 and 1000 mbar during AMAZE-08.
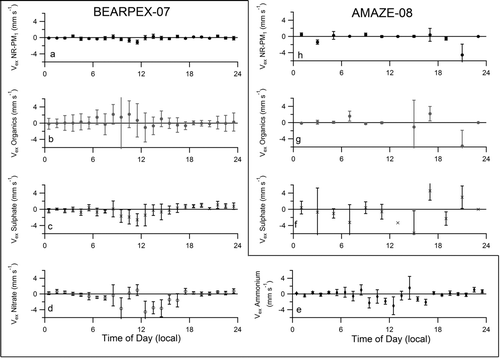
![Figure 1 FIG. 1 Diel cycles (local time: PDT for BEARPEX-07, AST for AMAZE-08) of mass concentrations for NR-PM1 and the organic, sulfate, nitrate and ammonium components for the entire BEARPEX-07 campaign [(a and b)18 August to 28 September 2007] and the AMAZE-08 campaign [(c and d) 24 February to 13 March 2008]. Vertical bars indicate the standard error of the mean for the dataset. The diel cycles for NR-PM1 and ammonium from BEARPEX-07 are reproduced from Farmer et al. (Citation2011). Elevated concentrations at midnight may be due to local influences. Note the different scales between the various panels.](/cms/asset/b29351d2-1306-439e-a142-31eb263be67f/uast_a_791022_f0001_b.gif)
3. OBSERVATIONS
3.1. Concentrations
Observations of aerosol particle mass concentrations during both campaigns are described in detail elsewhere (Chen et al. Citation2009; Martin et al. Citation2010; Farmer et al. Citation2011). The averages during the time periods used for the flux analysis subset were similar to campaign averages. Nonrefractory submicron aerosol (NR-PM1) during BEARPEX-07 had higher concentrations (4 ± 2 μg m−3 average) than during AMAZE-08 (0.8 ± 0.4 μg m−3). In both cases, the submicron aerosol was dominated by organic material (average 70% of NR-PM1 for BEARPEX-07; 80% for AMAZE-08). Unless stated otherwise, the uncertainties quoted are one standard error. Sulfate was the dominant inorganic ion for both campaigns, accounting for 14% of the total NR-PM1 during BEARPEX-07 and 17% during AMAZE-08. Ammonium was a larger component during BEARPEX-07 than AMAZE-08 (7% in BEARPEX-07 compared with 3% in AMAZE-08) (Farmer et al. Citation2011). Nitrate accounted for an average of 5% of the aerosol mass during BEARPEX-07 and <1% during AMAZE-08. The inorganic acids were neutralized by ammonium at BEARPEX-07, while the aerosol during AMAZE-08 was acidic, with ammonium neutralizing about one-third of the anions, assuming that all nitrate, ammonium, and sulfate observed by the HR-AMS were inorganic.
Diel cycles in concentration for both organic and inorganic PM components during BEARPEX-07 () were strongly influenced by meteorology and transport. This observation is consistent with previous observations of both organic and reactive nitrogen oxide trace gases. Meteorology at Blodgett Forest is regular and well documented, with daytime upslope flow (west to east) and nighttime downslope flow (east to west) (Murphy et al. Citation2006; Day et al. Citation2009). Following the morning break-up of the boundary layer (08:00), NR-PM1 concentrations increased, reflecting the additive effects of local emissions, transport of oak forest-influenced air at noon, and the Sacramento plume from mid-afternoon to early evening. The morning period during BEARPEX-07 is taken as the time period during which only local pine forest emissions affect the aerosol composition (08:00–13:00). The oxygen-to-carbon (O/C) ratios of the organic component of the mass spectrum ranged from 0.66 to 0.77 (not shown), which is typical of aged, oxygenated organic components of aerosol particles (Jimenez et al. Citation2009), consistent with the importance of SOA at this location.
Pöschl et al. (Citation2010) characterized individual particles within the accumulation mode during AMAZE-08 and reported that they consisted of an external mixture of SOA-dominated particles (>85% of the particle number), mixed SOA-inorganic particles (∼10%), and pyrogenic carbon particles (<5%). During periods of minimal influence by sources external to the Amazon rainforest, the organic mass spectra resembled lab-generated SOA, with an O/C ratio of 0.4, a value that is typical of freshly formed SOA (Chen et al. Citation2009). Organic aerosol mass concentrations increased through the day after sunrise, peaking in the mid-afternoon (). During these periods, organic material accounted for 91% of the NR-PM1 mass, with sulfate (7%), nitrate (1%), and ammonium (1%) as minor components (Chen et al. Citation2009). The sulfate concentration was typically higher at midnight, potentially due to emissions from a local generator into the stratified nocturnal boundary layer ().
3.2. Fluxes
There was net deposition of total NR-PM1 during BEARPEX-07 and slight near-zero deposition during AMAZE-08. Both upward and downward fluxes were observed for the chemically resolved components during both campaigns ( and ).
The average mid-day Vex of NR-PM1 during BEARPEX-07 was −1 mm s−1. The average noon NR-PM1 flux was −2.5 ± 1.0 ng m−2 s−1. An important observation is that the inorganic and organic components had opposite diel cycles. Sulfate, ammonium, and nitrate deposited in the day, with average Vex of −1.3 ± 0.8, −1.8 ± 0.8, and −1.4 ± 1.0 mm s−1, respectively, from 08:00 to 13:00. Organic components had an upward flux during this same time window (Vex of +1.0 ± 0.6 mm s−1). The organic flux was nearly zero in the afternoon, at least within measurement capability. Thus, the lower concentration inorganic components were driving the net NR-PM1 flux with relatively efficient deposition, even as the organic components were showing an upward flux.
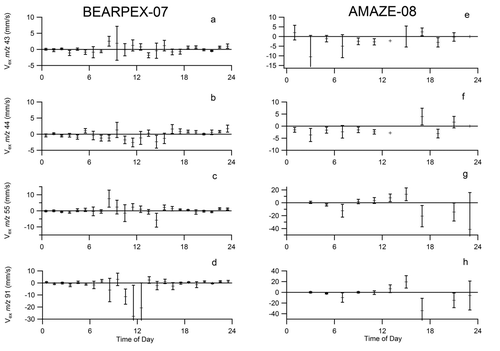
Individual organic m/z had resolvable fluxes in both directions, with a diel cycle. shows exchange velocities of four m/z ratios that are commonly observed in ambient organic aerosol: m/z 43, 44, 55, and 91. The diel cycles in the Vex of these four m/z ratios are typical of the variation observed for organic-influenced m/z ratios in the HR-AMS. For example, while m/z 44 and 91 deposited throughout the day, m/z 43 and 55 had upward fluxes in the morning and near-zero or downward fluxes in the afternoon. Nighttime fluxes were not substantially different from zero.
During AMAZE-08, NR-PM1 had a slight net deposition, with an average Vex of −0.1 mm s−1. Nitrate and ammonium fluxes were negligible within uncertainty. Sulfate had a downward flux in the morning (8:00–12:00) and the flux was not significantly different from zero during the afternoon (12:00–18:00). The total organic flux was highly variable and a diel pattern was not discernible. However, fluxes of individual organic m/z did have diel cycles. For example, m/z 43 and 44 deposited throughout the day, with the strongest downward fluxes occurring in the mid-afternoon. In contrast, m/z 55 and 91 had slight upward fluxes in the early afternoon, transitioning to downward fluxes in the late afternoon and early evening (). Fluxes for the early evening did not fit an identifiable pattern, with periods of upward and downward fluxes, possibly due to variable meteorology, atmospheric turbulence, and frequent rains.
4. DISCUSSION
The mid-day planetary boundary layer (PBL) flux budget of submicron organic aerosol is useful for interpreting the organic aerosol flux observations described herein. Aerosol mass fluxes that remove organic aerosol from the PBL include advection of aerosol to downwind areas and dry deposition. Aerosol mass fluxes into the PBL above the forest include advection of primary and secondary aerosol from upwind sources, emission of PBAP, and in-canopy SOA production. In addition, partitioning of semivolatile organic compounds (SVOC) between the gas and particle phases also influences the particle mass flux budget: condensation of SVOC constitutes an aerosol source, while volatilization is an aerosol sink. The net effect of partitioning on the flux budget depends mainly on thermal gradients, the SVOC concentration and its volatility distribution. Oxidation chemistry of organic compounds within the PBL alters the SVOC concentration and volatility distribution and may also lead to new particle formation and growth into the accumulation-mode size range observed by the HR-AMS. In order to accurately constrain the organic aerosol flux budget, deposition, emission, and formation of SVOC must be included, though these processes are unconstrained in this analysis due to the lack of SVOC flux measurements.
4.1. BEARPEX-07 Diel Patterns
Aerosol composition at Blodgett forest was consistent with the diel cycle indicated from gas-phase observations (Lamanna and Goldstein Citation1999; Holzinger et al. Citation2006; Murphy et al. Citation2006; Day et al. Citation2009; Worton et al. Citation2011): successive influences of the local pine forest, the lower elevation oak forests, and finally the Sacramento urban area. Consistent with these successive influences, the average organic fraction of NR-PM1 increased throughout the day, from a minimum of 67% at 07:30 to 73% at 17:30, as did the oxygenated organic aerosol components derived from positive matrix factorization (Worton et al. Citation2011).
Net deposition of total NR-PM1 mass was observed over Blodgett Forest during BEARPEX-07, consistent with particle number flux measurements carried out contemporaneously (Vong et al. Citation2010). The chemically resolved fluxes () suggest that this net flux is not merely the result of deposition of advected aerosol. Competing upward aerosol fluxes must simultaneously occur. The morning upward fluxes at BEARPEX-07 coincided with airmasses dominated by local pine forest emissions, while the afternoon near-zero fluxes coincided with the arrival of processed airmasses. A strong canopy source of particles outcompeted deposition of regional and background particle sources in the morning, but did not overwhelm the deposition of transported aerosol in the afternoon. To constrain this upward flux, organic aerosol deposition is determined from the product of organic aerosol concentration and Vex,sulfate (). Sulfate is assumed to be a proxy for regional deposition of organic aerosol, since local sources are minimal and Vex, m/z 44 and Vex,sulfate are similar. The depositional flux of organic aerosol can thus be estimated and, by difference, the net in-canopy source is constrained to 10–12 ng m−2 s−1 in the morning ().
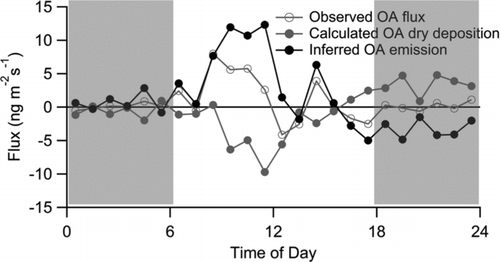
The diel cycles in individual m/z fluxes suggest that this in-canopy source of organic aerosol has a chemical signature that includes m/z 43 and 55 (). In contrast, m/z 44 and 91 deposited throughout the day. m/z 43 is a general organic aerosol marker that is present in similar proportions in hydrocarbon-like organic aerosol, semivolatile oxidized organic aerosol, and other organic aerosol types. m/z 55 has contributions from both hydrocarbon-like and semivolatile oxidized organic aerosol. In contrast, m/z 44 is typically considered to be more influenced by low-volatility, more processed oxidized organic aerosol (Ng et al. Citation2011). This suggests that the fluxes of organic components are consistent with deposition of regional, more oxidized low-volatility organic aerosol and emission of more local semivolatile oxidized organic aerosol, as has been observed before at a site with mixed urban/biogenic influences (Nemitz et al. Citation2008).
4.2. BEARPEX-07 Organic Aerosol Mid-Day Flux Budget
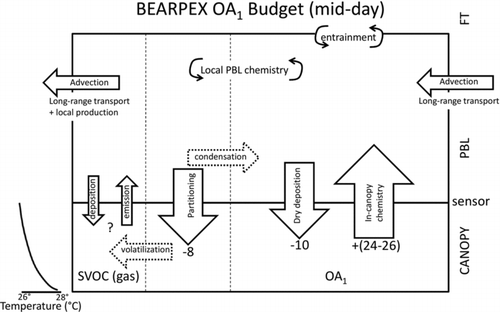
summarizes the mid-day flux budget of organic aerosol during BEARPEX-07. Deposition, in-canopy chemistry, and shifts in gas-particle partitioning due to thermal gradients all contributed to the observed fluxes. Two potential fluxes into the PBL from the forest are ignored in this analysis: (1) venting of aerosols stored within the canopy air space at night to either the mixed layer or free troposphere and (2) emissions of primary biological aerosol particles. Venting is not included in the mid-day flux budget as mixing at Blodgett forest occurs early in the morning (06:00–08:00) and is complete well before the calculated maximum upward flux (10:00–12:00). Furthermore, there is no evidence that PBAP contributed to submicron organic aerosol mass or fluxes during BEARPEX-07. PBAP markers for amino acids, including C3H6N+ and C2H4N+ (Schneider et al. Citation2011), were not clearly observed above their noise levels by the HR-AMS during BEARPEX-07. Fluxes of HR-AMS ions associated with carbohydrates were either indistinguishable from zero (C2H4O2+ at m/z 60, C3H5O2+ at m/z 73) or downward (C2H5O+ at m/z 61, Vex = −2 to −4 mm s−1).
4.2.1. Downward Canopy-PBL Fluxes (BEARPEX-07)
As described above, the mid-day organic aerosol dry deposition flux accounts for an estimated −10 ng m−2 s−1. Wet deposition does not impact the PBL flux budget, as no precipitation occurred during the subset of the BEARPEX-07 campaign described in this manuscript. Entrainment of air from the free troposphere is expected to dilute organic aerosol in the PBL as aerosol concentrations in the free troposphere are typically lower than in the PBL. Constraints on entrainment fluxes from the free troposphere to the PBL are beyond the scope of this work.
Temperatures at Blodgett Forest were on average 2°C warmer at ground level than above the top of the forest canopy. Such negative thermal gradients (i.e., temperatures colder above the canopy than below, dT/dz < 0) cause semivolatile components to preferentially partition to the particle phase at the top of the canopy and to the gas phase at ground level, resulting in net mass transfer between the gas and particle phases and the establishment of a vertical concentration gradient, independent of SOA formation from in-canopy processing of VOCs. The resulting aerosol concentration gradient would lead to the measurement of downward aerosol mass fluxes. While such fluxes have been observed for ammonium nitrate at other sites (Nemitz and Sutton Citation2004), the inorganic component at our location was dominated by nonvolatile ammonium sulfate (Farmer et al. Citation2011). Thus, while the thermal gradient-induced partitioning during BEARPEX-07 would have little effect on inorganic aerosol fluxes, the organic aerosol mass flux was likely impacted, resulting in an increase in deposition rates observed at the sensor height. Using a kinetic-equilibrium model (Cappa Citation2010) and considering typical residence time for air in the canopy (90 s) (see the online supplemental information for details of calculation), thermal gradients are estimated to cause particles moving from the bottom to the top of the canopy to gain ∼0.7% of the particle mass, resulting in a deposition flux term of −8 ng m−2 s−1.
4.2.2. Upward Canopy-PBL Fluxes (BEARPEX-07).
Evidence for in-canopy oxidation chemistry at Blodgett Forest from previous studies includes observation of emission of reactive VOCs and their oxidation products from the forest canopy (Kurpius and Goldstein Citation2003; Goldstein et al. Citation2004; Holzinger et al. Citation2005) and subsequent large O3 deposition fluxes influenced by VOC chemistry (Kurpius and Goldstein Citation2003; Wolfe et al. Citation2011). Oxidation of these reactive VOCs may produce SVOCs that condense to form SOA, with previous studies estimating that 0.2–6 μg m−3 of biogenic SOA was produced either in or above the forest canopy (Bouvier-Brown et al. Citation2009; Wolfe et al. Citation2011). Such SOA production would cause aerosol emission fluxes on the order of +(56–1700) ng m−2 s−1, far greater than those observed in this study. However, these estimates were based on SOA yields that were obtained in high aerosol mass loading experiments (Lee et al. Citation2006). Studies of SOA yield from monoterpenes and sesquiterpenes show a dependency on aerosol concentrations, with SOA yields on the order of 10% for the much lower organic aerosol concentrations observed at this site (Ng et al. Citation2007; Chen et al. Citation2011). These updated yields, in combination with observed VOC fluxes, suggest an organic aerosol flux of +(3.5–4.8) ng m−2 s−1 from in-canopy chemistry of sesquiterpenes and monoterpenes at Blodgett Forest (see the online supplemental information). The very-reactive biogenic VOCs previously reported for Blodgett Forest (Holzinger et al. Citation2005) increase this in-canopy SOA flux estimate to +(10–234) ng m−2 s−1. In comparison, the observed noon-time upward organic aerosol flux is calculated as the difference between the observed flux and downward flux derived from deposition and thermal partitioning estimates, to be +(24–26) ng m−2 s−1. Thus the observed flux is consistent with the lower end of the in-canopy chemistry estimate. Assuming a typical oxidized VOC molecular weight of 180–250 g mol−1 (King et al. Citation2009; Li et al. Citation2011), the observed flux suggests an overall in-canopy SOA yield of 0.5–4% of the total VOC emissions at Blodgett Forest. This yield calculation includes 17–70 μmol m−2 hr−1 for very reactive biogenic VOCs determined by Holzinger et al. (Citation2005) plus monoterpenes observed during BEARPEX-07. This reasoning suggests that reactive biogenic VOCs emitted by the ponderosa pine ecosystem can be rapidly oxidized, producing semivolatile compounds that partition to the particle phase and cause vertical gradients in concentration in the time scale of canopy/atmosphere exchange. These observations further suggest that reactive BVOCs can produce SOA in the canopy. This in-canopy production of BSOA can be sufficiently strong to out-compete the deposition flux of regional organic aerosol and to produce net upward fluxes when concentrations, and thus deposition rates, of organic aerosol are low.
These observations suggest a caveat to interpret nonchemically resolved particle flux data: simultaneous upward and downward components are not only possible, but likely due to a combination of aerosol deposition, direct emissions, in-canopy chemistry, and thermal gradients. Blodgett Forest may thus be considered an emission source of SOA to the boundary layer, although it is a net sink for total nonrefractory PM1 on the timescale of forest/atmosphere exchange. Because substantial fractions of terpenes can escape the canopy and react downwind to form SOA over longer timescales, Blodgett Forest can be a net contributor to regional PM1 concentrations. For forests having similarly strong emissions of reactive BVOCs, the relative balance between in-canopy SOA production and deposition is anticipated to depend on regional sources of PM1. For example, the fractional contribution of in-canopy production to the total organic aerosol and PM1 mass flux can be smaller for sites downwind of large urban or biomass burning sources where the regional deposition term is substantial. However, in-canopy SOA production could be significant for cleaner and more remote sites, although this contribution depends on the oxidant levels available for VOC oxidation, as presented below for AMAZE-08.
4.4. AMAZE-08 Diel Patterns
Particle number fluxes over the tropical forest typically correspond to net deposition, with potential forest sources of PBAP in the coarse mode (>1 μm) just after sunrise (Ahlm et al. Citation2009; Ahlm et al. Citation2010; Rizzo et al. Citation2010). The observation of net deposition of NR-PM1 aerosol mass with an exchange velocity of −0.1 mm s−1 is consistent with particle number flux measurements at a nearby tower site with a modal exchange velocity of −0.3 mm s−1 (Ahlm et al. Citation2009) (see the online supplemental information for comparison of particle number versus mass fluxes).
Similar to the BEARPEX-07 observations, different PM1 chemical components had fluxes of varying magnitudes and signs. Organic aerosol components exhibited both upward and downward fluxes (). Fluxes at m/z 44, characteristic of oxidized organic aerosol components, had downward fluxes, more so than less-oxidized components (m/z 55), again consistent with deposition of regional, aged SOA. In contrast to BEARPEX-07, m/z 43 has a similar profile to m/z 44 during AMAZE-08, suggesting that different types of organic aerosol components can contribute to these two m/z's at the two sites. The net effect for the organic aerosol component is a near-zero mid-day flux of +2 ng m−2 s−1 (average from 11:00 to 13:00, local time). The fluxes measured by the HR-AMS are close to the detection limits, which are typically on the order of 0.4–1 ng m−2 s−1 (Farmer et al. Citation2011).
4.5. AMAZE-08 Mid-Day Organic Aerosol Flux Budget
The mid-day organic aerosol flux budget in the Amazon is driven by wet and dry deposition, PBAP emission, vertical partitioning gradients, and in-canopy chemistry (). In contrast to Blodgett Forest, the Amazon is less impacted by anthropogenic pollution, and the mass concentrations are a factor of five lower than observed at BEARPEX-07 (Martin et al. Citation2010). Similar to the BEARPEX-07 campaign, the impacts of entrainment, advection, and SVOC fluxes are likely important, but unconstrained, in the AMAZE-08 mid-day PBL flux budget.
![Figure 6 FIG. 6 The mid-day submicron organic aerosol mass flux budget (ng m−2 s−1) between the canopy and planetary boundary layer (PBL) over the Amazon during the AMAZE-08 experiment. Submicron primary biological aerosol particles (PBAP1) are emitted (+3 ng m−2 s−1) from the forest, but also deposit at equal or lesser rates. In-canopy production of SOA is minor and challenging to constrain, but constitutes an emission source [<(4–41) ng m−2 s−1] from the forest. Dry deposition of organic aerosol (transported, locally and canopy-produced) is also predicted to be small (−2 ng m−2 s−1), though wet deposition may be substantial. The vertical thermal gradients typically observed in the Amazon canopy coupled with observed aerosol thermograms suggest that gas-particle partitioning along the vertical gradient produces an apparent upward flux (<8 ng m−2 s−1). Wet deposition estimates are substantial, but highly uncertain [<|−(11–98)| ng m−2 s−1]. Long-range transported organic aerosol (i.e., not originating from the Amazon and/or nonbiogenic sources) is advected into the Amazon PBL. Advection removes any transported or locally produced aerosol that has not been removed by wet or dry deposition. Unknown components of the OA1 flux budget include entrainment and fluxes of gas-phase semivolatile organic compounds.](/cms/asset/690cc4a3-0b05-4d34-8fce-59e1ed2386ce/uast_a_791022_f0006_b.gif)
4.5.1. Upward Canopy-PBL Fluxes (AMAZE-08)
PBAP contributes <5% of the observed aerosol mass during the daytime (Chen et al. Citation2009; Schneider et al. Citation2011). For the upper limit of 5% of the organic aerosol emission caused by PBAP (0.03 μg m−3), the associated submicron organic aerosol flux term is +3 ng m−2 s−1.
Previous studies at terra firme forest sites near Manaus, during the wet season, observed a thermal gradient of up to 5°C, with the ground-level temperatures being substantially cooler than the above-canopy temperature during the daytime (Kruijt et al. Citation2000). Such vertical thermal gradients can cause condensation at ground-level of gas-phase species to the particle phase, corresponding to an upward flux of organic aerosol from the canopy at the sensor height. Thermograms taken during AMAZE-08 showed that organic aerosol over the Amazon was more volatile than previous observations in the urban areas of Mexico City and Riverside (Martin et al. Citation2010), possibly due to processes involving particle-phase water. Applying these thermograms to a volatility distribution and assuming a linear thermal gradient from 25°C to 30°C from the ground to sensor height, a 12.8% loss in aerosol particle mass concentration is derived because of warming from the bottom of the canopy to the top. This corresponds to an upward aerosol flux of +8 ng m−2 s−1 (see the online supplemental information for details).
Although tropical forest VOC emissions are dominated by isoprene, the more reactive monoterpene fluxes are substantial (Karl et al. Citation2004). However, O3 levels in Amazonia are low, with <10 ppb O3 above the canopy and 2–8 ppb in the canopy (Karl et al. Citation2009)—over five times lower than at Blodgett Forest. The difference between in- and above-canopy O3 is typically attributed to soil NO emissions that react with O3 to form NO2 (Rummel et al. Citation2002) coupled with poor in-canopy mixing, although highly reactive terpenes will also be oxidized (Jardine et al. Citation2011). The SOA yields for in-canopy oxidation derived above for the BEARPEX-07 experiment suggest that in-canopy SOA formation causes an organic aerosol flux of +(4–41) ng m−2 s−1 (see the online supplemental information for details). Due to low light- and oxidant-levels, this is taken as an upper estimate for SOA fluxes in the Amazon.
4.5.2. Downward Canopy-PBL Fluxes (AMAZE-08)
Downward organic aerosol fluxes from the PBL to canopy are driven by wet and dry deposition, which are inferred from the above analysis. The observed flux (+2 ng m−2 s−1) is the sum of upward and downward components. For upward canopy-PBL fluxes estimated as the sum of PBAP emission (+3 ng m−2 s−1), thermal partitioning gradients (+8 ng m−2 s−1), and in-canopy chemistry (4–41 ng m−2 s−1), a total upward flux of 15–52 ng m−2 s−1 is derived. In the absence of additional upward or downward drivers of the observed flux, deposition must be on the order of −(13–50) ng m−2 s−1 to account for the observed +2 ng m−2 s−1. This calculation suggests several possibilities: (1) an implausibly large dry deposition flux for organic aerosol [Vex of −(17–83) mm s−1], (2) substantial wet deposition, (3) an overestimate of canopy chemistry, and/or (4) an overestimate of the thermal partitioning flux due to ignoring SVOC deposition. Alternately, for an assumption that the deposition of m/z 44 (mid-day Vex = −2.8 mm s−1, consistent with sulfate deposition during BEARPEX-07) is representative of organic aerosol deposition during AMAZE-08, dry deposition of organic aerosol is estimated to be a more plausible −1.7 ng m−2 s−1. The associated wet deposition flux is thus inferred to be −(11–48) ng m−2 s−1. This wet deposition estimate is likely an overestimate. A lower percentage of isoprene molecules reacting within the forest canopy, lower SOA yields, or lower average molecular weights for SOA would result in smaller canopy chemistry estimates, and thus smaller wet deposition estimates. Rapid deposition of SVOCs will diminish the impact of thermal gradients, similarly diminishing the wet deposition estimate. In future studies, simultaneous observations of the thermal gradient, volatility, and SVOC flux can further constrain the interpretation of the data.
4.5.3. AMAZE-08: Chemical Signatures of the Flux of Organic Aerosol Components
The chemical profile of the organic aerosol fluxes provides further evidence for a less oxidized component from in-canopy chemistry causing the upward flux. Both m/z 55 and 91, which are typically correlated to less oxidized organic aerosol (Ng et al. Citation2011), had upward fluxes in the morning and early afternoon and downward fluxes in the late afternoon (). m/z 91 has been associated with α-pinene-derived SOA (Shilling et al. Citation2009), not inconsistent with in-canopy terpene oxidation. Downward fluxes are associated with m/z 44, which is indicative of more oxidized and lower volatility organic aerosol. m/z 82 is a useful tracer for isoprene-oxidation SOA over the tropics due to production of isoprene epoxydiols and subsequent rearrangement to form 3-methyltetrahydrofuran-3,4-diols (Robinson et al. Citation2011; Lin et al. Citation2012). Here, m/z 82 typically had fluxes that were either downward or indistinguishable from the noise. This observation is consistent with the hypothesis that regionally produced isoprene-SOA is mostly deposited and only slowly produced within the forest canopy.
The Amazonian wet season flux budget described in represents as pristine a view of tropical rainforest aerosols as currently possible. Precipitation removes most longer distance transported particles, and, for most of the experiment, in-basin biogenic aerosol dominates. In the dry season, biomass burning substantially increases aerosol concentration (Artaxo et al. Citation2002) and thus aerosol deposition. This perturbation may shift the balance between upward and downward organic aerosol fluxes. However, thermal gradients within the forest are also likely to differ due to seasonal differences in climate and biomass-burning aerosols may have a different volatility distribution (Cappa and Jimenez Citation2010), thus creating a different regime for biosphere-atmosphere exchange.
6. CONCLUSIONS
Observations of chemically resolved aerosol mass flux measurements from the BEARPEX-07 and AMAZE-08 campaigns were used to constrain both upward and downward components of the flux. The two field projects presented in this article represent two distinctly defined ecosystems: a temperate pine forest impacted by anthropogenic pollution and a pristine tropical rainforest with minimal anthropogenic influence. Fluxes over both forests are strongly influenced by deposition of regional inorganic PM and biogenic SOA. Less oxidized organic aerosol appears to be more strongly emitted by forests, while more oxidized organic aerosol appears to be more strongly deposited, consistent with previous observations in a more mixed biogenic/urban environment (Nemitz et al. Citation2008).
At Blodgett Forest (), dry deposition of the advected organic aerosol competes with in-canopy SOA formation from reactive BVOCs. In-canopy chemistry results in a smaller flux in Amazonia () because of the lower sesquiterpene emissions, which, on the lower end of the estimate, is comparable to submicron PBAP fluxes. BEARPEX-07 is the first field study to demonstrate that biogenic SOA can be formed on the rapid timescales (min) that govern the residence of particles within the canopy, though not with the upper-end SOA yields previously used for some such estimates. Thus in-canopy chemistry and PBAP are net sources of SOA to the regional PBL, although above-canopy SOA formation is likely the dominant source of regional organic aerosol. Dry deposition affects the flux balance, although wet deposition is likely the major loss process for regional aerosol during the wet season in the Amazon.
The coupling of thermal gradients with the semivolatile nature of the organic aerosol is potentially substantial at both sites and emphasizes the importance of more quantitatively measuring aerosol volatility and SVOC concentration gradients in future campaigns. While the analysis of thermal partitioning gradients is highly uncertain due to a lack of SVOC constraints, it is indicative of the volatile nature of the Amazonian organic aerosol and suggests an intriguing positive feedback: warmer temperatures will partition SVOCs from the particle to the gas phase over the Amazon, lessening the regional aerosol burden and the regional-scale cooling effect of these particles, causing further regional warming. In contrast, the emission of SOA to the PBL above the forest canopy by either in-canopy chemistry or thermal gradient partitioning may provide a negative feedback: aerosols scatter light, proportionally increasing the diffuse radiation that is most efficiently captured by plants for photosynthesis (Gu et al. Citation2002; Oliveira et al. Citation2007). This increased photosynthesis reaches 30–40% during the dry season in Amazonia (Oliveira et al. Citation2007), thereby increasing CO2 uptake, although the effect of in-canopy SOA formation is likely substantially smaller in the wet season.
The observations of chemically resolved aerosol mass fluxes are consistent with the current picture that particles deposit over forests. However, the overall submicron particle flux depends on a complex interplay between competing mechanisms, including wet and dry deposition, chemical production, thermal gradients, and PBAP emission. Furthermore, this study raises caveats to interpreting flux measurements of particles: multiple mechanisms can operate simultaneously to cause upward and downward fluxes of different PM components, confounding the traditional approach to particle flux models and measurement interpretation, especially because size-segregated particle flux measurements are highly sensitive to artifacts due to particle growth/shrinkage (Nemitz and Sutton Citation2004; Nemitz et al. Citation2009). Chemically resolved flux measurements are crucial for elucidating these competing mechanisms, as are measurements in relatively clean environments. With increased air pollution, deposition is expected to increase, further dampening the relative effect of in-canopy chemistry and partitioning on observations.
Supplemental Information_791022.zip
Download Zip (49.8 KB)[Supplementary materials are available for this article. Go to the publisher's online edition of Aerosol Science and Technology to view the free supplementary files.]
This research was funded by NSF ATM-0723582 and ATM-0919189, DOE (BER, ASR Program DE-SC0006035, and DE-FG02-11ER65293), and the UK Natural Environment Research Council (NE/E007309/1). Delphine K. Farmer acknowledges a NOAA Climate & Global Change Postdoctoral Fellowship. Qi Chen acknowledges a NASA Earth and Space Science Fellowship. Paulo A. Artaxo acknowledges funding from FAPESP and CNPq.
REFERENCES
- AhlmL., KrejciR., NilssonE.D., MartenssonE.M., VogtM., ArtaxoP. 20101010237–10253
- AhlmL., NilssonE.D., KrejciR., MartenssonE.M., VogtM., ArtaxoP. 200999381–9400
- AllanJ.D., DeliaA.E., CoeH., BowerK.N., AlfarraM.R., JimenezJ.L., et al. 200435909–922
- ArtaxoP., MartinsJ.V., YamasoeM.A., ProcópioA.S., PauliquevisT.M., AndreaeM.O., et al. 20021078081–8095Doi: 8010.1029/2001JD000666
- BaldocchiD.D., HicksB.B., MeyersT.P. 1988691331–1340
- Bouvier-BrownN.C., GoldsteinA.H., GilmanJ.B., KusterW.C., de GouwJ.A. 200995505–5518
- BuzoriusG., RannikU., MakelaJ.M., VesalaT., KulmalaM. 199829157–171
- CanagaratnaM.R., JayneJ.T., JimenezJ.L., AllanJ.D., AlfarraM.R., ZhangQ., et al. 200726185–222
- CappaC.D. 20103579–592
- CappaC.D., JimenezJ.L. 2010105409–5424
- ChenQ., FarmerD.K., SchneiderJ., ZornS.R., HealdC.L., KarlT.G., et al. 200936L20806Doi: 20810.21029/22009GL039880
- ChenQ., LiuY., DonahueN.M., ShillingJ.E., MartinS.T. 2011454763–4770Doi: 10.1021/es104398s
- ChungS.H., SeinfeldJ.H. 20021074407Doi: 4410.1029/2001JD001397
- DayD.A., FarmerD.K., GoldsteinA.H., WooldridgeP.J., MinejimaC., CohenR.C. 200994879–4896
- DeCarloP.F., KimmelJ.R., TrimbornA., NorthwayM.J., JayneJ.T., AikenA.C., et al. 2006788281–8289
- FarmerD.K., CohenR.C. 200883899–3917
- FarmerD.K., KimmelJ.R., PhillipsG., DochertyK.S., WorsnopD.R., SueperD., et al. 201141275–1289Doi: 1210.5194/amt-1274-1275-2011
- FarmerD.K., MatsunagaA., DochertyK., SurrattJ.D., SeinfeldJ.H., ZiemannP.J., et al. 20101076670–6675
- FowlerD., CapeJ.N., CoyleM., FlechardC., KuylenstiernaJ., HicksK., et al. 19991165–32
- FowlerD., PilegaardK., SuttonM.A., AmbusP., RaivonenM., DuyzerJ., et al. 2009435193–5267
- GoldsteinA.H., GalballyI.E. 2007411514–1521
- GoldsteinA.H., HultmanN.E., FracheboudJ.M., BauerM.R., PanekJ.A., XuM., et al. 2000101113–129
- GoldsteinA.H., McKayM., KurpiusM.R., SchadeG.W., LeeA., HolzingerR., et al. 200431L22106Doi: 22110.21029/22004GL021259
- GordonM., StaeblerR.M., LiggioJ., VlasenkoA., LiS.M., HaydenK. 2011116773–6786
- GuL.H., BaldocchiD., VermaS.B., BlackT.A., VesalaT., FalgeE.M., et al. 2002 107Doi: 10.1029/2001JD001242
- GuentherA., HewittC.N., EricksonD., FallR., GeronC., GraedelT., et al. 19951008873–8892
- HallquistM., WengerJ.C., BaltenspergerU., RudichY., SimpsonD., ClaeysM., et al. 200995155–5236
- HealdC.L., SpracklenD.V. 200936L09806Doi: 09810.01029/02009GL037493
- HicksB.B., BaldocchiD.D., MeyersT.P., HoskerR.P., MattD.R. 198736311–330
- HolzingerR., LeeA., McKayM., GoldsteinA.H. 200661267–1274
- HolzingerR., LeeA., PawK.T., GoldsteinA.H. 2005567–75
- HuffmanJ.A., DochertyK.S., AikenA.C., CubisonM.J., UlbrichI.M., DeCarloP.F., et al. 200997161–7182
- HuismanA.J., HottleJ.R., GallowayM.M., DiGangiJ.P., CoensK.L., ChoiW.S., et al. 20111113655–13691Doi: 13610.15194/acpd-13611-13655-12011
- JardineK., Yanez SerranoA., ArnethA., AbrellL., JardineA., van HarenJ., et al. 2011116 Doi: 10.1029/2011JD016243
- JimenezJ.L., CanagaratnaM.R., DonahueN.M., PrevotA.S. H., ZhangQ., KrollJ.H., et al. 20093261525–1529
- KarlT., GuentherA., TurnipseedA., TyndallG., ArtaxoP., MartinS. 200997753–7767
- KarlT., PotosnakM., GuentherA., ClarkD., WalkerJ., HerrickJ.D., et al. 2004109 Doi: 10.1029/2004JD004738
- KimmelJ.R., FarmerD.K., CubisonM.J., SueperD., TannerC., NemitzE., et al. 201130315–26Doi: 10.1016/j.ijms.2010.12.004
- KingS.M., RosenoernT., ShillingJ.E., ChenQ., MartinS.T. 200992959–2971Doi: 2910.5194/acp-2959-2959-2009
- KruijtB., MalhiY., LloydJ., NorbreA.D., MirandaA.C., PereiraM.G. P., et al. 200094297–331
- KurpiusM.R., GoldsteinA.H. 2003301371Doi: 10.1029/2002GL016785
- LamannaM.S., GoldsteinA.H. 199910421247–21262
- LeeA., GoldsteinA.H., KrollJ.H., NgN.L., VarutbangkulV., FlaganR.C., et al. 2006111D17305Doi: 17310.11029/12006JD007050
- LiY.J., ChenQ., GuzmanM.I., ChanC.K., MartinS.T. 201111121–132Doi: 110.5194/acp-5110-5191-2010
- LinY.H., ZhangZ.F., DochertyK.S., ZhangH.F., BudisulistioriniS.H., RubitschunC.L., et al. 201246250–258
- MartinS.T., AndreaeM.O., AlthausenD., ArtaxoP., BaarsH., BorrmannS., et al. 20101011415–11438Doi: 10.5194/acp-10-11415-2010
- McWilliamA.L. C., RobertsJ.M., CabralO.M. R., LeitaoM.V. B. R., DecostaA.C. L., MaitelliG.T., et al. 19937310–317
- MurphyJ.G., DayA., ClearyP.A., WooldridgeP.J., CohenR.C. 200665321–5338
- NemitzE., DorseyJ.R., FlynnM.J., GallagherM.W., HensenA., ErismanJ.W., et al. 200961627–1645
- NemitzE., JimenezJ.L., HuffmanJ.A., UlbrichI.M., CanagaratnaM.R., WorsnopD.R., et al. 200842636–657
- NemitzE., SuttonM.A. 200441025–1045
- NemitzE., SuttonM.A., WyersG.P., OtjesR.P., MennenM.G., van PuttenE.M., et al. 200441007–1024
- NgN.L., CanagaratnaM.R., JimenezJ.L., ZhangQ., UlbrichI.M., WorsnopD.R. 201145910–916
- NgN.L., ChhabraP.S., ChanA.W. H., SurrattJ.D., KrollJ.H., KwanA.J., et al. 200775159–5174Doi: 5110.5194/acp-5157-5159-2007
- OliveiraP.H. F., ArtaxoP., PiresC. Jr, de LuccaS., ProcópioA., HolbenB., et al. 200759B338–349Doi: 310.1111/j.1600-0889.2007.00270.x
- PöschlU., MartinS.T., SinhaB., ChenQ., GuntheS.S., HuffmanJ.A., et al. 20103291513–1516Doi: 1510.1126/science.1191056
- RizzoL.V., ArtaxoP., KarlT., GuentherA.B., GreenbergJ. 201044503–511
- RobinsonN.H., HamiltonJ.F., AllanJ.D., LangfordB., OramD.E., ChenQ., et al. 2011111039–1050Doi: 1010.5194/acp-1011-1039-2011
- RuijgrokW., TiebenH., EisingaP. 199731399–415
- RummelU., AmmannC., GutA., MeixnerF.X., AndreaeM.O. 20021078050Doi: 8010.1029/2001JD000520
- SchneiderJ., FreutelF., ZornS.R., ChenQ., FarmerD.K., JimenezJ.L., et al. 20111111415–11429Doi: 11410.15194/acp-11411-11415-12011
- SeinfeldJ.H., PankowJ.F. 200354121–140
- ShillingJ.E., ChenQ., KingS.M., RosenoernT., KrollJ.H., WorsnopD.R., et al. 20099771–782
- SpracklenD.V., JimenezJ.L., CarslawK.S., WorsnopD.R., EvansM.J., MannG.W., et al. 20111112109–12136Doi: 12110.15194/acp-12111-12109-12011
- TrebsI., LaraL.L., ZeriL.M. M., GattiL.V., ArtaxoP., DlugiR., et al. 20066447–469
- VongR.J., VongI.J., VickersD., CovertD.S. 2010105749–5758
- WeselyM.L., HicksB.B. 2000342261–2282
- WolfeG.M., ThorntonJ.A., MckayM., GoldsteinA.H. 2011117875–7891Doi: 7810.5194/acp-7811-7875-2011
- WolfeG.M., ThorntonJ.A., YatavelliR.L. N., MckayM., GoldsteinA.H., LaFranchiB., et al. 20099615–634
- WortonD.R., GoldsteinA.H., FarmerD.K., DochertyK.S., JimenezJ.L., GilmanJ.B., et al. 20111110219–10241Doi: 10210.15194/acp-10211-10219-12011