Abstract
A particle into liquid sampler (PILS) has been directly coupled to an accurate mass atmospheric ionization time-of-flight mass spectrometer (ToF) for use in the speciation of secondary organic aerosol (SOA) formed in the University of California—Riverside, College of Engineering–Center for Environmental Research and Technology (CE-CERT) atmospheric chambers. To this end, the PILS has been optimized for direct continuous injection into the ToF and the PILS-ToF system has been used to obtain real-time mass spectral traces of the particle phase products of atmospheric chamber reactions. The PILS-ToF system has been initially applied to SOA formed from α-pinene dark ozonolysis and isoprene photooxidation. The characterization of the PILS-ToF system includes experiments on the well understood α-pinene/O3 system, which verifies the performance of the tool. The PILS-ToF tool is then used to provide new insight into the chemical composition of the SOA formed from isoprene photooxidation. For the first time, time resolved traces of oligomer formation during isoprene photooxidation are observed using the PILS-ToF. The unique subunits of oligomer series are reported. The PILS-ToF system is demonstrated to be a powerful tool for obtaining real-time electro spray ionization and chemical ionization mass spectral speciation of aerosols.
Copyright 2013 American Association for Aerosol Research
1. INTRODUCTION
The chemical characterization of secondary organic aerosols (SOA) formed is required for complete understanding of SOA reaction mechanisms. Toward this end, work on sampling and instrumentation for the mass spectral chemical characterization of aerosols has been prolific in recent years, yielding aerosol mass spectral techniques that have varying aerosol sampling and sample ionization methods (Eygluenent et al. Citation2008; Cross et al. Citation2009; Faulhaber et al. Citation2009). Currently two mass spectral-based methods appear to have gained popularity in the field of aerosol chemical characterization.
The first technique is in situ aerosol mass spectrometry using a focused particle beam with electron impact ionization (Jimenez et al. Citation2009), hereafter referred to as Aerosol Mass Spectrometry (AMS). Data from the AMS can provide information on the oxidation state of the aerosol through analysis of the mass fragmentation. This method has led to a greater understanding of oxidation processes, both in the ambient and in environmental chamber studies (Jimenez et al. Citation2009). Nevertheless, AMS mass spectral analysis leads to many small ion fragments requiring intense data analysis and giving little direct aerosol chemical speciation.
The second technique is filter sampling followed by ex-situ liquid chromatography, coupled to an atmospheric pressure ionization mass spectrometer (Surratt et al. Citation2006; Szmigielski et al. Citation2007; Surratt et al. Citation2010; Lin et al. Citation2013). This technique has led to the direct identification of large molecular weight chemical species in secondary organic aerosols (i.e., oligomers). Liquid chromatography allows for increased detection signal while generally producing a simple mass spectrum upon separation. With atmospheric pressure ionization techniques, such as matrix assisted laser desorption ionization (MALDI), chemical ionization, and electrospray ionization (ESI), ions are generally observed as the parent molecule with an extra hydrogen (in the case of positive ionization) or absent a hydrogen (in the case of negative ionization). However, filter techniques do not provide for the necessary time resolution to readily observe the evolution of transients in particle phase reaction intermediates.
Here, a new mass spectral technique is developed that directly couples the particle-into-liquid sampler (PILS), as developed and refined by Orsini et al. (Citation2003), to the atmospheric ionization inlet of an accurate mass time-of-flight mass spectrometer; this system is termed here the PILS-ToF. The PILS-ToF is to used to chemically speciate the particle-phase products of SOA formed in the University of California, Riverside, College of Engineering-Center for Environmental Research and Technology (CE-CERT) atmospheric chamber using “soft” ionization that leads to fewer ion fragments while providing high time resolution. The ability to chemically speciate SOA generated in real-time allows the observation of transient particle-phase intermediates in real-time.
To date, the PILS has been directly coupled either to ion chromatographs (IC) or total organic carbon (TOC) analyzers (Orsini et al. Citation2003; Peltier et al. Citation2007). Offline PILS-ToF measurements were published by Bateman et al. on limonene ozonolysis (Bateman et al. Citation2010). However, we report here the direct injection of a PILS sample into the ionization source of the mass spectrometer that allows for real-time chemical speciation of aerosol particles. This method is particularly effective for characterizing the chemical mechanisms of particle formation as intermediates can be observed.
Here the PILS-ToF system is validated against other particle measurement methods, both chemical and physical. The PILS-ToF is further applied to the characterization of particle phase reactions occurring during the dark ozonolysis of α-pinene and the photooxidation of isoprene with NOx. These two hydrocarbon oxidant systems were chosen for study as they are reported to form particle phase oligomers and are atmospherically relevant (Hall and Johnston Citation2010; Surratt et al. Citation2010.
2 MATERIALS AND METHODS
2.1 PILS-ToF
The PILS head (Brechtel Manufacturing, Inc.) is described in detail by Orsini et al. and is only briefly outlined here (Orsini et al. Citation2003). The PILS collects particles from the chamber through a ½′′ sample line, to reduce pressure drop in sample collection, into a Venturi. The Venturi expands from ½′′ at its inlet to 2′′ at its outlet. Steam is injected at the entrance of the Venturi through a 1/16′′ stainless steal tube located in the annular region of the Venturi. Steam is produced from in-house purified high pressure liquid chromatography (HPLC) grade water with a measured resistivity of greater than 18.2 MΩ and less than 5 ppb total organic carbon. The steam condenses around the aerosol particles in the 2′′ inner diameter by 5′′-long finned tube. The water soluble organic material dissolves, or is encapsulated, and the entrained droplets are focused by a second nozzle onto a quartz impaction plate. At the quartz impaction plate, a wash flow of HPLC water flushes the impacted liquid droplets to the HPLC pump responsible for injecting the sample into the ionization source of the Agilent Accurate Mass 6210 ToF. A flow diagram of the PILS-ToF is shown in with corresponding stream flows provided in .
Table 1 PILS instrument flow-rates
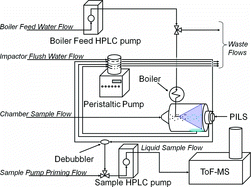
It should be noted that for small chamber volumes the PILS sample flow rate of 15 L/min limits the experiment duration. A gas denuder was not used in application of the PILS, thus observed chemical species include both soluble oxidation products and particle products. Future use of a gas denuder would prevent collection of sorbed water soluble gases.
The ToF has a multimode ionization source, allowing for both positive and negative atmospheric pressure chemical ionization or electrospray ionization. The mass spectrometer is tuned weekly and calibrated daily and has an average mass accuracy at 200 Da of ±0.0020 m/z units (20 ppm). The mass spectrometer was tuned and calibrated using solution of mixed molecular weight fluorocarbons, Agilent part number G1946-85004, by methods laid out in Agilent procedures (Agilent Citation2007). PILS-ToF data collected 5 min prior to the start of the chamber reaction (start of reaction coincides with the addition of ozone for dark reactions or lights on for photooxidation reactions) is used for background spectra and subtracted from all subsequent mass spectral signals.
2.2 Chamber Experiments
Experiments were conducted in the large and mezzanine chambers at UCR/CE-CERT, described in detail elsewhere (Carter et al. Citation2005; Nakao et al. Citation2011). Briefly, the large chamber facility consists of two 90 m3 FEP Teflon® film reactors located in a temperature-controlled room, which is continuously flushed with purified air. The reactors are attached to a rigid collapsible frame to minimize diffusion of contaminants into the reactors by maintaining a positive differential pressure with respect to the chamber's surroundings. The chamber has four banks of 112 W, 350-nm black lights (272 lights in total) providing a k1 of 0.51 min−1 (Carter et al. Citation2005).
The mezzanine chamber is in a 2.5 m × 3 m × 7.8 m enclosure covered with reflective aluminum sheets and is illuminated with 170 of 40 W black lights with peak intensity at 350 nm with a measured k1 of 0.6 min−1. Within this enclosure is a 12 m3 FEP Teflon chamber. A minimum of 1-m space is maintained between the chamber surface and black lights to avoid excessive heating at the surface of the film. Additionally, six fans are used to mix the air inside the enclosure with conditioned room air to minimize heating within the enclosure. The enclosure temperature is typically within 25°C–28°C.
Connected to the chambers are in-house built scanning mobility particle sizers (SMPS), similar to those described by Cocker et al. (Citation2001), along with the PILS-ToF described above. Hydrocarbon concentration is tracked by gas valve sampling gas chromatography (GC) using an Agilent 6890 N GC equipped with a flame ionization detector (FID). NO and NOx concentration is measured during reactions by Thermo Environmental Instruments Inc. model 42C trace level NO-NO2-NOy analyzer. O3 concentration is measured by a Dasibi Environmental Corporation model 1003-AH. CO concentration is measured by a Thermo Environmental Instruments Inc. model 48C trace level CO analyzer. All quantitative gas-phase instrumentation is calibrated to a gas standard prior to each chamber experiment.
Liquid hydrocarbon is introduced to the chamber by placing a known aliquot of hydrocarbon injected into a glass bulb, where a flowing particle-free purified air stream evaporates the hydrocarbon into the chamber. CO injections into the chamber are performed by flowing gas from a UHP CO cylinder at a specific flow-rate over a set amount of time (CO is used as a hydroxyl radical scavenger). O3 is injected by flowing particle-free purified air through a laboratory built O3 generator. NO injection is performed using UHP NO, a vacuum rack, and known volume glass bulb. All experiments reported here are performed under dry low relative humidity (RH < 1%) with no aerosol seed.
3 RESULTS AND DISCUSSIONS
PILS-ToF measurements were recorded for two reaction systems, the dark ozonolysis of α-pinene and the NO photooxidation of isoprene. During SOA formation and aging, both reaction systems have been reported to form second generation and higher large molecular weight oligomer products (Gao et al. Citation2010; Surratt et al. Citation2010; Sato et al. Citation2011). These oligomers have been identified by aerosol filter sampling followed by atmospheric pressure ionization based mass spectrometry: either MALDI, chemical ionization, or ESI. These oligomers serve as an optimum test bed for evaluation of the PILS-ToF system's real-time aerosol chemical characterization capabilities. It has been confirmed by others that oligomer products are not products of the ionization source, but are indeed formed during the SOA chamber reactions (Camredon et al. Citation2010; Gao et al. Citation2010; Sato et al. 2011); the data presented here support these findings. Experimental parameters and the current SOA chamber experiments are summarized in . It should be noted that the as built PILS-ToF system cannot distinguish a difference between isomers of an identified molecular formula.
Table 2 Experimental conditions and results
As part of the PILS-ToF characterization, PILS-ToF chamber background characterization experiments were performed. These experiments included: pure-air only runs (with and without black lights), photooxidation of 250 ppb NO in clean air, photooxidation 250 ppb of NO2 in clean air, and photooxidation of 3 ppm H2O2 in clean air. Background experiments were performed in the absence of any measurable hydrocarbon concentration, as measured by GC-FID. Results from the PILS-ToF during these background experiments showed no significant signal change in the total ion counts throughout these experiments consistent with no significant particle formation observed by the SMPS. For the experimental work here, the chamber generated aerosol concentrations are an order of magnitude higher than ambient. Further characterization of the instruments signal-to-noise and lower detection limits are necessary before application of the PILS-ToF to ambient.
3.1 α-Pinene Ozonolysis
α-Pinene dark ozonolysis experiments were conducted as outlined above (Section 2). The initial CO, ozone, and α-pinene concentrations were 30 ppm, 125 ppb, and 150 ppb, respectively (; experiment EPA 1311A). PILS-ToF mass spectra taken during SOA formation from α-pinene ozonolysis used positive mode ESI with data recorded from m/z 60 to 1000.
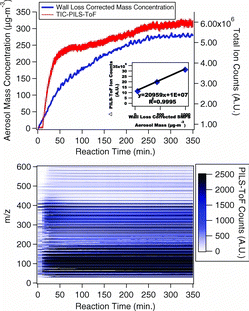
The SMPS and PILS-ToF total ion count (TIC) traces for SOA formation during EPA1311 are presented in . shows that α-pinene particle formation measured by the PILS-ToF follows the particle formation measured by the SMPS. The inset graph in Figure 2a shows the change in ion-counts measured by the PILS-ToF during α-pinene SOA particle formation versus SMPS maximum particle mass concentration for MEZ102309, MEZ102709, and MEZ110509 experiments. The increasing initial α-pinene concentrations form maximum particle mass concentration from 50–993 μg/m3. Where the mass concentration is calculated from the SMPS measured volume concentration and a density of 1 kg/L is assumed. The PILS-ToF signal has a strong linear correlation (R2 = 0.9995) with the maximum SMPS measured particle mass concentration (assuming a density of 1 g-cm−3). This result suggests that the majority of the α-pinene SOA formed is water-soluble (Engelhart et al. Citation2008) and suggests that >99% of the soluble organic aerosol mass can be speciated with the PILS-ToF analytical technique. It should be noted that the PILS-ToF data reported in the inset to , MEZ102309, MEZ102709, and MEZ110509, all used the APCI ionization source in the ToF. Experiments in the mezzanine chamber had a maximum duration of 4 h due to deflation of the small volume chamber due to the large PILS-ToF flow rate of 15 L/min. The PILS-ToF flow-rate was observed to have no appreciable effect on the length of an experiment for the large chambers. Furthermore, inert tracer concentration, as measured by GC-FID, showed that no dilution occurred during large chamber experiments.
The PILS-TOF TIC signal varies as particles age. Thus one can observe the changes in TIC signal for the range of explored m/z ions as a function of time (). In each mass spectral scan presented is an average of 5 min of mass spectra. For computational efficiency in the production of a PILS-ToF contour plot, the mass spectral resolution is reduced to unit mass. The PILS-ToF signal in shows that a clear and coherent mass spectral pattern emerging at 10 min into the reaction of α-pinene and ozone as particle nucleation occurs.
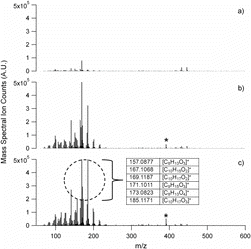
gives the detailed PILS-ToF mass spectra of EPA1311A at 10 min, 50 min, and 380 min into the reaction. At 50 min, once particle nucleation is complete, the PILS-ToF mass spectrum can be categorized into two mass spectral regimes above and below m/z 250 (.). Table S1, found in the online supplemental information (SI), gives the PILS-ToF molecular formula assignments for all mass spectral peaks of the peaks with intensity 1000 ion-counts or greater observed in , the final state mass spectrum for experiment EPA1311A, and these ions corresponding mass defect. These assigned ions account for 82% of the total ion counts observed in . Furthermore, the matches in Table S1, where applicable are matched to mass spectral data found in the literature (Docherty et al. Citation2005; Yu et al. Citation1999; Yasmeen et al. Citation2010; Yasmeen et al. Citation2011; Kristensen et al. Citation2013).
Masses below m/z 250 broadly correspond to the compounds observed by Yu et al. (Yu et al. Citation1999). This mass spectral regime below m/z 250 is dominated by positive ions 139.1224, 141.1136, 155.1331, 157.1293, 169.1484, 171.1294, 173.1104, 185.1466, 199.1278, and 201.1645 corresponding to the parent particle phase compounds norpinone, 2,2-dimethyl-cyclobutyl-1,3-dicarboxaldehyde, norpinonaldehyde, 2,2-dimethyl-3-formyl-cyclobutyl-methanoic acid, pinaldehyde, norpinonic acid isomers, norpinic acid, pinonic acid, C10H14O3 (which has an unknown structure), and 10-hydroxyl pinonic acid. These ions alone, without their secondary fragments, contribute 44% to the PILS-ToF TIC and are widely reported in the literature (Yu et al. Citation1999). Analysis of the PILS-ToF mass spectra indicates that 80% of the total PILS-ToF ion counts observed after nucleation can be attributed to the ten ions corresponding to the ten molecules listed directly above.
Although it appears that carboxylic acids and aldehydes with pinene-based backbones dominate the mass spectra, there are significant peaks observed with m/z above 300. These high m/z features have been broadly attributed to oligomers (Gao et al. Citation2004, 2010; Camredon et al. Citation2010). When looking at , the onset of nucleation is observed immediately, corresponding to the initial introduction of ozone. Small oligomer formation seems to occur concurrently with particle nucleation. The predominant ion observed by the PILS-ToF throughout SOA formation is at m/z 391.3839 ( and ).
Camredon et al. (Citation2010) assign m/z 391 as a sodium ion adduct of a molecule with molecular weight of 368.4212 Da. Gao et al. (Citation2010) assigns a negative ion of molecular weight m/z 389.1655 (which would have an equivalent positive ion at m/z 391.1655) to C18H30O9. Camredon et al. (Citation2010) further support their assignment through use of an MCM v.3.1 model result showing a molecule of molecular weight 368.4212, and molecular formula C19H28O7, as a probable product of the ester formation reaction of pinic acid (M = 186.2060 Da) and 10-hydroxy-pinonic acid (M = 200.2327 Da) in which a water molecule is lost. The ion mass defect of m/z 391.3839, observed here by the PILS-ToF, supports the findings of Camredon et al. (Citation2010).
A molecule of the positive ion at m/z 369.2837 is also observed, at low intensity. The mass difference between this dimer molecule observed here and that of the molecule observed and modeled by Camredon et al. (Citation2010) is 0.0271 Da. Although this mass difference is inside the accuracy of 0.002 of the calibrated mass spectrometer used here, it is an order of magnitude less than the difference between the observation reported here and the measurement by Gao et al. (Citation2010) of 0.2184. The observation here supports the work of Camredon et al. in their dimer assignment, who assign and model this single dimer species of molecular weight 368 (Camredon et al. Citation2010).
Particle products at an m/z above 550 continue to form during particle ageing (). These high mass species appear 20 min after the initial addition of ozone. PILS-ToF spectra from SOA formed in α-pinene dark ozonolysis supports the findings in the literature while validating the applicability and performance of the PILS-ToF in the characterization of water-soluble SOA products. The technique can also be applied to elucidate the chemical properties of lesser-studied SOA systems.
3.2 Isoprene NO Photooxidation
Isoprene NO photooxidation was performed in the presence of H2O2 in the UCR/CE-CERT chamber. The initial concentration of NO, H2O2, and isoprene, used in the isoprene photooxidation experiment were 500 ppb, 3 ppm, and 250 ppb, respectively (, EPA 1353A). PILS-ToF spectra taken during SOA formation from isoprene photooxidation used negative mode ESI with data recorded from m/z 60 to 1000. Negative mode ESI was chosen to allow for comparison to offline liquid mass spectral data collected and reported by Surratt et al. (Citation2006) and Sato et al. (Citation2008).
shows the change in the PILS-ToF TIC signal for unit resolution m/z as a function of time. The isoprene dataset is processed as in Section 3.1 for α-pinene SOA. a–f gives detailed PILS-ToF mass spectra at chosen discrete times during the isoprene photooxidation reaction.
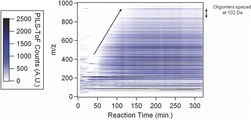
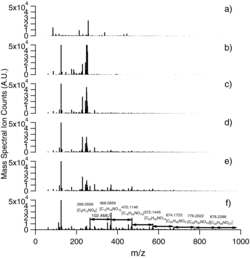
The temporal evolution of chemical species in isoprene SOA () is different than α-pinene SOA (). In , large molecular weight compounds (>m/z 500) are observed to form over time. High molecular weight products are observed to grow in between 50 and 125 min after lights are turned on. The observed mass spectra change very little after 125 min, indicating little chemical ageing of the isoprene chamber SOA.
shows the PILS-ToF background spectra, taken prior to the beginning of photolysis. The spectrum in is subtracted from all subsequent spectra. – gives intermediate spectra taken directly prior, during, and directly after SOA particle nucleation (particle nucleation occurred at 30 min into the reaction) at 35 min, 60 min, and 75 min into the reaction, respectively. The spectra in is taken at 100 min, during particle mass growth, and is taken at 300 min, the final reaction state PILS-ToF spectra.
In , five ions account for more than 50% of the background spectra. These ions are m/z: 112.9861, 213.1139, 226.9793, 231.1243, and 257.1767. Mass-to-charge 112.9861, the largest intensity peak in the background spectra accounting for 33% of the background spectra and 226.9793 can be attributed to sodium trifluoroacetate, used to tune the ToF prior to PILS-ToF data collection (Moini et al. Citation1998). The other background ions, m/z 213.1139, 231.1243, and 257.1986, can be attributed to hydrocarbon contaminants from a rubber liquid chromatography sample septa.
A close examination of the PILS-ToF TIC trace () reveals a peak of transient ions begins forming at 25 min into the reaction, which has significantly less intensity by 50 min into the reaction. , the PILS-ToF mass spectrum corresponding to reaction time of 35 min, shows the bulk of these transient ions to be between 200 and 300 m/z ion. The major transient ions present are m/z: 248.8974, 249.8969, 250.8960, 251.8944, 253.8930, and 243.9032 making up 23%, 17%, 10%, 9%, 6%, and 3% of the mass spectrum, respectively. These ions account for 65% of the total mass spectral abundance at 35 min into the reaction. These ions could not be identified due to their extraordinary negative mass defect.
shows that by the end of the reaction, these transient ions are barely visible in the overall mass spectrum, accounting for less than 1% of the total mass spectrum. shows a major oligomerization mass spectral pattern consistent with the formation of nitro-oligoesters, as reported by Surratt et al. (Citation2010) and Sato et al. (Citation2011). The dimer nitro-oligoester C8H13NO9 has a calculated molecular weight of 267.0590, the expected ion weight [M-H−] of 266.0512, and an observed ion m/z of 266.0556. The trimer is observed at m/z 368.0856 a difference or 102 Da. Each further oligomer is observed at the interval of 102 Da, corresponding to the addition of the monomer. The highest nitro-oligoester observed is the nine-unit oligomer observed above the background is at an m/z 980.2569.
In addition to the nitro-oligoester, many other oligoestsers were observed during the NO photooxidation of isoprene. Some of these oligoesters seen in have been observed previously, but some others are previously unreported and are given a molecular formula assignment here. Table S2 (SI) gives the PILS-ToF molecular formula assignments for all mass spectral peaks of the peaks with intensity 1000 ion-counts or greater observed in , the final state mass spectrum for experiment EPA1353A, and these ions corresponding mass defect. These assigned ions account for 78% of the total ion counts observed in .
The final state PILS-ToF mass spectrum is further presented and analyzed by use of the Van Krevelen Method which yields a Van Krevelen Diagram (Van Krevelen Citation1950), shown in a, b. The Van Krevelen plot is made by simply calculating the H:C and O:C ratios for mass spectral molecular matches of peaks observed in the final state mass spectrum shown in , whose ion matches are given in detail in Table S2 (Surratt et al. Citation2010; Sato et al. Citation2011; Nguyen et al. 2011; Lin et al. 2012). In , the molecular formulas are broken up into two categories, those previously observed in the literature and those matched in this work. The molecules previously observed account for only 14% of the total ion counts shown in , while the molecules matched in this work make up the remaining 64% of the total ion counts. All PILS-ToF mass spectral ions, which have been previously reported by Surratt et al. (Citation2006) or Sato et al. (Citation2008) appear on one of three lines of , where these lines are captioned a, b, and c.
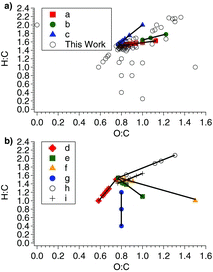
shows six new lines indicating six additional particle phase chemical mechanisms, labeled lines d, e, f, g, h, and i. Those chemical mechanistic lines shown in make up 57% of the observed final state PILS-ToF mass spectrum shown in . The lines in and are highly correlated, with R2 equal to or approaching 1. The linear regressed formulas and correlation coefficients of lines a–i in and are provided in . Table S2 references those ions observed here and observed previously with notations correlating the peak match to the corresponding line on the Van Krevelen plots.
Table 3 Parameters for Van Krevelen lines
Line a of contains oligoesters and nitro-oligoestrers observed by both Surratt et al. (Citation2010) and Sato et al. (Citation2011). The oligoesters on line a begin with the monomer C5H8O5 and the monomer unit [-C4H6O3-] up to C29H43O23. Line a also contains the most intense nitro-oligoesters with repeating subunit 102 Da seen clearly in observed by both Surratt et al. (Citation2010) and Sato et al. (Citation2011). Line b corresponds to those oligomers having two nitro-ester groups where these molecules are introduced by Sato et al. (Citation2011). Molecules on line c are also all oligoesters, but begin with monomer C4H8O4. It should be noted that common analysis of a Van Krevelen Diagram would assign line a of to oxidation or reduction reactions and lines b and c of to hydration or condensation reactions (Van Krevelen Citation1950; Kim et al. Citation2003).
In , most molecules falling on line d can be attributed to oligoesters with an initial building block of m/z 267.0596 corresponding to an assigned molecular match of C12H12O7. These unpublished four oligoesters lie on line d each with a 102 Da spacing. This oligomeric spacing of 102 Da is the same as oligoesters reported by Surratt et al. The observed m/z for the PILS-ToF mass spectral peaks lying on line d are 369.0887, 471.1178, 573.1476, and 675.1766. These four observed PILS-ToF mass spectral peaks match the molecular formulas of C16H18O10, C20H24O13, C24H30O16, and C28H36O19, respectively. In addition to the five oligoesters, separated by a repeating subunit of m/z 102, there is an additional mass spectral molecular match lying on line e having an observed m/z of 407.1186, which is matched to a molecular formula of C16H24O12. The ion counts on line d account for only 1% of the total ion counts observed in .
Line e of again corresponds to a series of oligoesters separated by both a repeating subunit of m/z 102 and 58. The molecular matches on line e of having a separation of m/z 102 are 189.0441, 291.0765, 393.1063, 495.1360, and 597.1638 having molecular matches of C7H10O6, C11H16O9, C15H22O12, C19H28O16, and C23H34O18, respectively. Each of these oligoesters on line f separated by m/z 102, starting at m/z 291.0765, has an intermediate oligoester offset by 58 Da. These offset esters are found at m/z 349.0812, 451.1133, and 553.1430, corresponding to molecular matches of C13H18O11, C17H24O14, and C21H30O17, respectively. Two other PILS-ToF molecular matches fall on line e having m/z 304.0260 and 335.0973; these have been assigned molecular matches of C10H11NO10 and C13H20O10, respectively. The molecular matches on line e account for 2% of the PILS-ToF ions observed in .
Line f of shows two apparent series of oligoesters separated by m/z 102. The first set of ion molecular matches separated by m/z 102 are: 307.0718, 409.1024, 511.1320, and 613.1606, who have molecular matches of: C11H16O10, C15H22O13, C19H28O16, and C23H33O19, respectively. The second set of molecular matches lying on line f of separated by m/z 102 are: 321.0861, 423.1150, 525.1438, and 627.1725, whose molecular matches are: C12H18O10, C16H24O13, C20H30O16, and C24H36O20, respectively. The molecular match for m/z 146.9714 of C4H3O6 also lies on line f of with no clear correlation to an oligomeric series. This ion match has an extraordinary mass error of −150.3 ppm. The ion molecular matches on line f of account for 2% of the PILS-ToF total ion counts observed in .
Observed ions that have been matched to lie on line g of make up 50% of the total ion counts observed in . One ion alone, at m/z 123.9869, constitutes 49% of the total ion counts observed. The ion m/z 124.9891 is matched to the molecule C5H2O4. The other two ions on line g, m/z 126.9938 and 129.0243, match to molecules C5H4O4 and C5H6O4, respectively (these ions have high mass errors of –77.7 and 38.7 ppm, respectively). These ions on line g appear to be simply highly oxygenated forms of the five carbon backbone of isoprene.
On line h, a series of apparent nitro-oligoesters is observed. These nitro-oligoesters are again separated by m/z 102, indicative of the addition the monomer observed at m/z 119.0391 (this ion has a high mass error of 34.8 ppm) matched to C4H8O4 as proposed by Surratt et al. (Citation2010). The nitro-oligoesters lying on line h of are m/z 366.1010, 468.1215, and 570.1480 that match to the molecules C9H21NO14, C13H27NO17, and C17H33NO20. In addition to the three nitro-oligoesters, two ions that have no apparent reaction series at 539.1519 and 569.1668 are observed matched to the molecules C17H32O19 and C22H24O17, respectively. The ions observed to fall on line h of make up 1% of the total ion counts of .
In on line i of , four of six ions match to a 17 carbon backbone. These ions are m/z 435.1182, 469.1214, 500.1232, and 531.1283, matched to the molecules C17H24O13, C17H26O15, C17H27NO16, and C17H28N2O17, respectively. The two ions that are not matched to a molecule with 17 carbons are 583.1538 and 614.1557, matched to the molecules with 22 carbons, C22H32O18 and C22H33NO19, respectively. The ions shown to lie on line i of account for 1% of the total ion counts in .
4 CONCLUSIONS
A new time-resolved mass spectral method is presented by coupling the PILS to a “soft” ionization ToF-MS. Data presented here use reactions of two different hydrocarbon systems known to form SOA in chamber studies to validate the PILS-ToF. The PILS-ToF total ion counts are validated to the physical measurement of aerosol mass by SMPS, where the correlation is shown to be linear with an offset. The PILS-ToF is then validated chemically by comparing the chemical characterization results to those previously reported for dark α-pinene ozonolysis and the photooxidation of isoprene and NO using other mass spectral methods. Additionally, for NO photooxidation of isoprene, the PILS-ToF elucidated five new potential oligomeric paths while showing that a single ion matching a highly oxygenated five carbon structure, the same number of carbons of isoprene, accounted for 49% of the total ion counts observed by the PILS-ToF. Furthermore, the mass spectral ions previously observed for SOA produced by isoprene photooxidation only account for 14% of the PILS-ToF ions-counts, while new ion matches in this work make up 64%.
As for the PILS-ToF chemical characterization capabilities, it is shown here that the PILS-ToF agrees with previously reported filter sampling results “soft-ionization” mass spectral results for both reaction systems.
The PILS-ToF shows utility due to its time-resolution and ionization method, electrospray or chemical ionization opposed to electron impact ionization used by most aerosol mass spectrometers. The time resolution allows for aerosol transients or intermediates to be observed, while the “soft” ionization makes molecular identification much easier as complex software packages are not necessary for data analysis, as is the case for high-resolution AMS data.
PILS_ToF_Supplementary_091013.zip
Download Zip (32.1 KB)REFERENCES
- Agilent Technologies Inc. (2007). Agilent 6200 Series ToF and 6500 Series Q-ToF LC/MS System. Agilent Technologies Inc., Santa Clara, CA.
- Bateman, A.P., Nizkorodov, S.A., Laskin, J., and Laskin, A. (2010). High-Resolution Electro-Spray Ionization Mass Spectrometry Analysis of Water-Soluble Organic Aerosols Collected with a Particle into Liquid Sampler. Anal. Chem., 82:8010–8016.
- Camredon, M., Hamilton, J.F., Alam, M.S., Wyche, K.P., Carr, T., White, I.R., et al. (2010). Distribution of Gaseous and Particulate Organic Composition During Dark α-Pinene Ozonolysis. Atmos. Chem. Phys., 10:2893–2917.
- Carter, W.P. L., Cocker, D.R. I., Fitz, D.R., Malkina, I.L., Bumiller, K., Sauer, C.G., et al. (2005). A New Environmental Chamber for the Evaluation of Gas-Phase Chemical Mechanisms and Secondary Aerosol Formation. Atmos. Environ., 39:7768–7788.
- Cocker, D.R., Flagan, R.C., and Seinfeld, J.H. (2001). State-of-the-Art Chamber Facility for Studying Atmospheric Chemistry. Environ. Sci. Technol., 35:2594–2601.
- Cross, E.S., Onasch, T.B., Canagaratna, M., Jayne, J.T., Kimmel, J., Yu, X.-Y., et al. (2009). Single Particle Characterization Using a Light Scattering Coupled to a Time-of-Flight Aerosol Mass Spectrometer. Atmos. Chem. Phys., 9:7769–7793.
- Docherty, K.S., Wu, W., and Ziemann, P.J. (2005). Contributions of Organic Peroxides to Secondary Aerosol Formed from Reactions of Monterpenes with O3. Environ. Sci. Technol., 39:4049–4059.
- Engelhart, G.J., Asa-Awuku, A., Nenes, A. and Pandis, S.N. (2008). CCN Activity and Droplet Growth Kinetics of Fresh and Aged Monoterpene Secondary Organic Aerosol. Atmos. Chem. Phys., 8:3937–3949.
- Eygluenent, G., Le Person, A., Dron, J., Monod, A., Voisin, D., Mellouki, A., et al. (2008). Simple and Reversible Transformation of an APCI/MS/MS Into an Aerosol Mass Spectrometer: Development and Characterization of a New Inlet. Aerosol Sci. Technol., 42:182–193.
- Faulhaber, A.E., Thomas, B.M., Jimenez, J.L., Jayne, J.T., Worsnop, D.R., and Ziemann, P.J. (2009). Characterization of a Thermodenuder-Particle Beam Mass Spectrometer System for the Study of Organic Aerosol Volatility and Composition. Atmos. Meas. Techniques, 2:15–31.
- Gao, Y., Hall, W.A. I. V., and Johnston, M.V. (2010). Molecular Composition of Monoterpene Secondary Organic Aerosol at Low Mass Loading. Environ. Sci. Technol., 44:7897–7902.
- Gao, S., Keywood, M., Ng, N.L., Surratt, J., Varutbangkul, V., Bahreini, R., et al. (2004). Low-Molecular-Weight and Oligomeric Components in Secondary Organic Aerosol from the Ozonolysis of Cycloalkenes and a-Pinene. J. Phys. Chem. A, 108:10147–10164.
- Hall, W.A. I. V., and Johnston, M.V. (2010). Oligomer Content of a-Pinene Secondary Organic Aerosol. Aerosol Sci. Technol., 45:37–45.
- Jimenez, J.L., Canagaratna, M.R., Donahue, N.M., Prevot, A.S. H., Zhang, Q., Kroll, J.H., et al. (2009). Evolution of Organic Aerosols in the Atmosphere. Science, 326:1525–1529.
- Kim, S., Kramer, R.W., and Hatcher, P.G. (2003). Graphical Method for Analysis of Ultrahigh-Resolution Broadband Mass Spectra of Natural Organic Matter, the Van Krevelen Diagram. Anal. Chem., 75:5336–5344.
- Kristensen, K., Enggrob, K.L., King, S.M., Worton, D.R., Platt, S.M., Mortensen, R., et al. (2013). Formation and Occurrence of Dimer Esters of Pinene Oxidation Products in Atmospheric Aerosols. Atmos. Chem. Phys., 13:3763–3776.
- Lin, Y.-H., Zhang, Z., Docherty, K.S., Zhang, H., Budisulistiorini, S.H., Rubitschun, C.L., et al. (2012). Isoprene Epoxydiols as Precursors to Secondary Organic Aerosol Formation: Acid-Catalyzed Reactive Uptake Studies with Authentic Compounds. Environ. Sci. Technol., 46:250–258.
- Lin, Y.-H., Zhang, H., Pye, H.O. T., Zhang, Z., Marth, W.J., Park, S., et al. (2013). Epoxide as a Precursor to Secondary Organic Aerosol Formation from Isoprene Photooxidation in the Presence of Nitrogen Oxides. Proc. Natl. Acad. Sci. U.S.A., 110:6718–6723, S6718/6711-S6718/6716.
- Moini, M., Jones, B.L., Rogers, R.M., and Longfei, J. (1998). Sodium Trifluoroacetate as a Tune/Calibration Compound for Positive- and Negative-Ion Electrospray Ionization Mass Spectrometry in the Mass Range 100–4000 Da. J. Am. Soc. Mass Spectrom., 9:977–980.
- Nakao, S., Shrivastava, M., Nguyen, A., Jung, H., and Cocker, D., III (2011). Interpretation of Secondary Organic Aerosol Formation from Diesel Exhaust Photooxidation in an Environmental Chamber. Aerosol Sci. Technol., 45:964–972.
- Nguyen, T.B., Laskin, J., Laskin, A., and Nizkorodov, S.A. (2011). Nitrogen-Containing Organic Compounds and Oligomers in Secondary Organic Aerosol Formed by Photooxidation of Isoprene. Environ. Sci. Technol., 45:6908–6918.
- Orsini, D.A., Ma, Y., Sullivan, A., Sierau, B., Baumann, K. and Weber, R.J. (2003). Refinements to the Particle-into-Liquid Sampler (PILS) for Ground and Airborne Measurements of Water Soluble Aerosol Composition. Atmos. Environ., 37:1243–1259.
- Peltier, R.E., Weber, R.J., and Sullivan, A.P. (2007). Investigating a Liquid-Based Method for Online Organic Carbon Detection in Atmospheric Particles. Aerosol Sci. Technol., 41:1117–1127.
- Sato, K. (2008). Detection of Nitrooxypolyols in Secondary Organic Aerosol Formed from the Photooxidation of Conjugated Dienes under High-NOx Conditions. Atmos. Environ., 42:6851–6861.
- Sato, K., Nakao, S., Clark, C.H., Qi, L., and Cocker Iii, D.R. (2011). Secondary Organic Aerosol Formation from the Photooxidation of Isoprene, 1,3-Butadiene, and 2,3-dimethyl-1,3-butadiene Under High NOx Conditions. Atmos. Chem. Phys., 11:7301–7317.
- Surratt, J.D., Chan, A.W. H., Eddingsaas, N.C., Chan, M., Loza, C.L., Kwan, A.J., et al. (2010). Reactive Intermediates Revealed in Secondary Organic Aerosol Formation from Isoprene. Proc. Nat. Acad. Sci., 107:6640–6645.
- Surratt, J.D., Murphy, S.M., Kroll, J.H., Ng, N.L., Hildebrandt, L., Sorooshian, A., et al. (2006). Chemical Composition of Secondary Organic Aerosol Formed from the Photooxidation of Isoprene. J. Phys. Chem. A, 110:9665–9690.
- Szmigielski, R., Surratt, J.D., Vermeylen, R., Szmigielska, K., Kroll, J.H., Ng, N.L., et al. (2007). Characterization of 2-methylglyceric Acid Oligomers in Secondary Organic Aerosol Formed from the Photooxidation of Isoprene Using Trimethylsilylation and Gas Chromatography/Ion Trap Mass Spectrometry. J. J. Mass Spectrom., 42:101–116.
- Van Krevelen, D.W. (1950). Graphical-Statistical Method for the Study of Structure and Reaction Processes of Coal. Fuel, 29:269–284.
- Yasmeen, F., Szmigielski, R., Vermeylen, R., Gomez-Gonzalez, Y., Surratt, J.D., Chan, A.W. H., et al. (2011). Mass Spectrometric Characterization of Isomeric Terpenoic Acids from the Oxidation of α-pinene, Î2-pinene, d-limonene, and Δ3-carene in Fine Forest Aerosol. J. Mass Spectrom., 46:425–442.
- Yasmeen, F., Vermeylen, R., Szmigielski, R., Iinuma, Y., Boge, O., Herrmann, H., et al. (2010). Terpenylic Acid and Related Compounds: Precursors for Dimers in Secondary Organic Aerosol from the Ozonolysis of α- and Î2-pinene. Atmos. Chem. Phys., 10:9383–9392.
- Yu, J., Cocker, D.R. , III, Griffin, R.J., Flagan, R.C., and Seinfeld, J.H. (1999). Gas-Phase Ozone Oxidation of Monoterpenes: Gaseous and Particulate Products. J. Atmos. Chem., 34:207–258.