
Abstract
In aerosol research, a common approach for the collection of particulate matter (PM) is the use of filters in order to obtain sufficient material to undertake analysis. For subsequent chemical and toxicological analyses, in most cases the PM needs to be extracted from the filters. Sonication is commonly used to most efficiently extract the PM from the filters. Extraction protocols generally involve 10–60 min of sonication. The energy of ultrasonic waves causes the formation and collapse of cavitation bubbles in the solution. Inside the collapsing cavities the localized temperatures and pressures can reach extraordinary values. Although fleeting, such conditions can lead to pyrolysis of the molecules present inside the cavitation bubbles (gases dissolved in the liquid and solvent vapors), which results in the production of free radicals and the generation of new compounds formed by reactions with these free radicals. For example, simple sonication of pure water will result in the formation of detectable levels of hydroxyl radicals. As hydroxyl radicals are recognized as playing key roles as oxidants in the atmosphere the extraction of PM from filters using sonication is therefore problematic. Sonication can result in significant chemical and physical changes to PM through thermal degradation and other reactions. In this article, an overview of sonication technique as used in aerosol research is provided, the capacity for radical generation under these conditions is described and an analysis is given of the impact of sonication-derived free radicals on three molecular probes commonly used by researchers in this field to detect reactive oxygen species (ROS) in PM.
Copyright 2014 American Association for Aerosol Research
INTRODUCTION
Ultrasound is a sound wave with a frequency greater than 20 kHz and exposure to ultrasonic sound waves can be responsible for a number of physical and chemical changes. The effect of ultrasound in a liquid medium was first recognized almost 100 years ago, however the most significant interest in sonochemistry started in 1960s, when ultrasonic cleaning baths became popular for cleaning laboratory glassware, forming dispersions and cell disruption. Since then, sonochemistry has been mainly used in synthesis (organic and inorganic) and polymer chemistry (initiation of polymerization and degradation) (Thompson and Doraiswamy Citation1999). In the last 30 years there has been an increased interest in the use of ultrasound to destroy organic pollutants present in water and/or wastewater, as this approach is safe, clean and does not cause secondary pollution (Adewuyi Citation2001).
In this article, a short review of sonication techniques and their radical-generating capacities will be given, as well as an overview of the implications for organic PM research. It will also provide recent evidence demonstrating radical generation upon sonication.
Fundamentals of Ultrasonic Irradiation
When ultrasound propagates through a liquid it produces alternating compressions and rarefactions of the liquid being irradiated, resulting in positive and negative pressures being exerted on the liquid. During rarefaction, when a large negative pressure is applied to the liquid, the average distance between the molecules increases and the physical structure of the liquid breaks down, resulting in the generation of voids or cavities. These cavitation bubbles may contain gases, solvent vapor and any volatile substance present in the liquid.
Acoustic cavitation can be divided into two types, stable and transient. Stable cavities oscillate radially about some equilibrium size for many acoustic cycles, whereas transient cavities grow with each acoustic cycle until they reach an unstable size and then violently collapse during the compression part of the wave. It has been determined that the size at which these cavities collapse depends on the liquid and the frequency of sound; for example, at 20 kHz this critical size is ∼170 μm (Suslick Citation1990). During the collapse the localized temperature and pressure within the cavity become extremely high, around 5000 K and 500 atm, respectively (Suslick et al. Citation1986). Several factors can affect acoustic cavitation, including reaction temperature, hydrostatic pressure, ultrasonic frequency, acoustic power, the nature of dissolved gases and the physicochemical properties of the solvent.
The Chemistry of Ultrasonic Degradation
High local temperatures and pressures within collapsing cavities (both stable and transient) are considered to be responsible for the chemical effects of ultrasonic irradiation. Sonochemical reactions can happen in three different regions: the interior of gas bubbles, at the thin liquid layer between the collapsing gas bubble and the bulk solvent and throughout the bulk of the solution. As a result of extremely high temperatures and pressures, the interior of the gas bubbles become like a micro combustion chamber where pyrolysis occurs for the molecules present inside the cavitation bubbles. These molecules can include gases, solvent vapor and any volatile species present in the solution. When water is used as a solvent, such conditions will lead to the formation of highly reactive species such as hydrogen atoms and hydroxyl radicals. If molecular oxygen is present, even atomic oxygen can be formed (Adewuyi Citation2001). Evidence for the formation of H atoms and OH radicals in the sonolysis of water was first provided through electron spin resonance (ESR) and spin-trapping studies (Makino et al. Citation1983). They are generated via the following reactions:
These radicals can attack solute molecules or combine to form H2, H2O2 or water. If there are no hydroxyl radical scavengers, the most common reaction upon sonolysis of water is dimerization of the hydroxyl radical or disproportion of hydro-peroxyl radicals to produce hydrogen peroxide (Petrier et al. Citation1992; Mark et al. Citation1998).
The second important region for free radical generation is the thin liquid layer immediately surrounding the collapsing cavity, which has been estimated to extend around 200 nm from the cavity surface and to generate temperatures of around 2000 K over lifetimes of <2 μs (Suslick et al. Citation1986). This layer typically contains less volatile species and surfactants. Thermolysis and radical abstraction reactions readily occur in this region, and it is the region where the radicals produced can react with chemical species present in the bulk liquid. In this layer, oxidative degradation by hydroxyl radicals may take place if solutes are present in sufficiently high concentrations. However if the solute is not volatile enough to enter this region, the process may not be enhanced by the use of ultrasound.
The third important region is the bulk liquid. Although no primary sonochemical reactions occur in the bulk liquid, a small portion of free radicals produced in the cavities or at the interfacial region may escape into the bulk liquid and react with the compounds present there.
The sonolytic production of free radicals in aqueous mixtures of solutes with vapor pressures higher than water (e.g., acetone, acetonitrile, methanol, ethanol) has also been demonstrated previously using the ESR in combination with spin trapping (Krishna et al. Citation1989; Riesz et al. Citation1993), where in addition to hydrogen atoms and hydroxyl radicals, carbon-centered radicals were also found. Cavitation can occur in organic solvents as well. The sonolysis of organic liquids (e.g., dimethylformamide, methylformamide, dimethylacetamide, toluene, n-alcohols, n-alkanes, cyclohexane, dioxane, and tetrahydrofuran) has also been investigated previously using ESR and spin trapping (MiöÌk and Riesz Citation1996a,Citationb). Various carbon nitrogen centered radicals (depending on the liquid) have been identified during the exposure of argon saturated organic liquids to 50 kHz ultrasound (MiöÌk and Riesz Citation1996a,Citationb).
At very high frequencies (i.e., several hundred kHz), the cavitational effect is reduced, so in the past, most sonochemical reactions were carried out at frequencies between 20 and 50 kHz. Lower frequency ultrasound produces more violent cavitation (i.e., higher localized temperatures and pressures at the cavitation site), but higher frequency ultrasound can actually increase the number of free radicals in the system. Despite less violent cavitation with higher frequency ultrasound (due to shorter cavity lifetime and, thus, smaller maximum cavity size), this frequency results in more cavitational events and, therefore, more opportunities for free radicals to be generated (Crum Citation1995).
Ultrasonic Reactors
Ultrasound is mainly generated by placing the reactor in an ultrasonic bath or by introducing the ultrasonic emission source directly into the solution. Ultrasonic cleaning baths are most widely used as they provide a convenient and non-expensive source of ultrasound. However, the ultrasonic power supplied to the reaction mixture can be relatively low due to the poor energy transfer. In order to maintain a consistent ultrasonic intensity, the liquid level in the bath, the solvent level in the ultrasonic reactor and the position of the reaction vessel (e.g., the flask containing the solution and the filters) must be carefully controlled (Thompson and Doraiswamy Citation1999). On the other hand, an ultrasonic probe (horn) can be directly immersed in the reaction solution and it is preferred over an ultrasonic bath, as it is more energy efficient (i.e it delivers ultrasonic power directly into the liquid). However both types of ultrasonic reactors can give uneven distribution of cavitational activity, with ultrasonic intensity decreasing with the increasing distance from the horn or transducer in the case of ultrasonic bath (Gogate et al. Citation2002).
Degradation of Organic Compounds by Ultrasonic Irradiation
Over the last three decades ultrasonic irradiation has been studied extensively in the context of removing organic compounds from wastewaters. The compounds studied include aromatic compounds, such as phenol, chlorophenols, benzene, toluene, xylene, polycyclic aromatic hydrocarbons (PAHs), as well as chlorinated aliphatic hydrocarbons, alcohols, explosives, herbicides, pesticides, organic dyes and other organic compounds (reviewed in Adewuyi Citation2001). The controlling mechanism for the destruction of these materials by ultrasound is usually dependent on the compound in question as well as the cavitational intensity, which depends on the operating conditions of the ultrasonic reactor. Pyrolysis within collapsing cavities plays the dominant role in the degradation of hydrophobic and volatile compounds, while the polar and less volatile compounds (e.g., phenol, chlorophenol, etc.) degrade mainly by reaction with OH radicals in the bulk phase or in the interfacial region between the collapsing cavity and the bulk solution, depending on their hydrophobicity. The degradation rate of non-volatile compounds is generally lower than for volatile compounds, as volatile compounds are more easily transferred into the interior of a cavity (Goel et al. Citation2004). The timescales of treatment reported in the literature are generally in the range of minutes to several hours for complete degradation (Adewuyi Citation2001). Even though ultrasonic irradiation is effective in degrading organic pollutants, total mineralization (degradation to CO2 and H2O) is difficult to achieve with ultrasound alone, even with extended periods of ultrasonic irradiation.
Consequences of Ultrasonic Irradiation in Aerosol Research
When PM is extracted from filters using ultrasound, the extreme temperatures and pressures created in the interior of the collapsing bubbles can induce changes in the chemical composition of material that has been collected on the filters in a manner similar to the process used to degrade organic pollutants in wastewaters. Recently Mutzel et al. found that even as little as 15 min of ultrasonic agitation of secondary organic aerosol (SOA) filter samples resulted in detectable chemical changes to the collected PM. Notably sonication caused an increase in PM-bound peroxides and the degradation of some SOA compounds (Mutzel et al. Citation2013). Hasson and Paulson have found that that after 15 min of sonication of deionized water, H2O2 concentrations were an order of magnitude higher than the levels expected in the PM samples collected on filters (Hasson and Paulson Citation2003). Similarly, Khurshid et al. have shown that 10 min sonication of blank filters in dichlorofluorescein/horseradish peroxidase (DCFH/HRP) solution results in increase of fluorescence intensity of the solution (Khurshid et al. Citation2014). Huang and Wang have used 10 min sonication to extract PM from filters and have also reported that field blanks resulted in a background fluorescence increase of DCFH/HRP solution which was 25–75% of the fluorescence of PM samples (Hung and Wang Citation2001). In a similar study (Venkatachari et al. Citation2007) it was found that the fluorescence intensity of the blank filters was 28–60% of the fluorescence of the PM samples. However, these studies have not reported a connection between these surprisingly high background H2O2 concentrations/fluorescence intensities and the effects of sonochemistry (i.e., radical/ROS formation). A very recent study (Fuller et al. Citation2014) has reported that 15 min sonication gave results where ROS concentrations of blank filter samples were higher than the values obtained for sample filters on which PM was collected. This was explained (and experimentally confirmed) by generation of H2O2 from two hydroxyl radicals created upon sonication. Despite this result and the broad understanding of the propensity for ultrasound to induce chemical changes, sonication remains the most commonly used method for the extraction of PM collected onto filters for subsequent chemical and toxicological analyses. The aim of this study was to further emphasize the chemical effects of sonication in the context of aerosol research. Herein we examined the ability of three forms of diagnostic probe commonly used in aerosol research to detect and quantify levels of reactive oxygen species (ROS) in PM. Without any detailed analysis of products resulting from sonication, we clearly demonstrate that each probe showcases that substantial chemical reaction is induced when the probe solution is exposed to ultrasonic irradiation in an ultrasonic bath.
METHODOLOGY
Chemicals
Three molecular probes commonly used for detection of ROS in PM samples were used to investigate the generation of radicals and ROS upon sonication of laboratory grade deionized water, ethanol/deionized water mixture and dimethyl sulfoxide (DMSO). The three probes were dithiotreitol (DTT), 2,7-dichlorofluorescein diacetate (DCFH-DA) and 9,10-bis(phenylethynyl)anthracene-nitroxide (BPEAnit).
Dithiotheritol (DTT) is a strong reducing agent and it has been used extensively to measure ROS formation in presence of redox active species which are able to oxidise DTT to its disulfide form (Kumagai et al. Citation2002). The DTT probe was dissolved in water to achieve a concentration of ∼1 mM.
DCFH-DA is a fluorescence based probe which has been commonly used for detecting ROS in PM samples. DCFH-DA first gets activated by sodium hydroxide to form non-fluorescent DCFH, which turns into its fluorescent derivative in the presence of ROS (such as H2O2, organic peroxides and OH radicals) and a catalytic enzyme HRP. DCFH-DA was dissolved in 50 mL of 10/90 ethanol/water (v/v) mixture to produce a concentration of ∼10 μM. 30 min prior to sonication the DCFH-DA solution was treated with 5 mL of 0.01 M NaOH under dark conditions in order to cleave the acetate groups (i.e., to generate the reactive entity, dichlorofluorescein [DCFH]). Immediately before sonication, horseradish peroxidase enzyme (HRP; ∼0.5 units/mL) was added to the DCFH solution to catalyze the reaction between DCFH and ROS species
BPEAnit has also been used previously to detect PM-related ROS. It is a weakly fluorescent compound that becomes strongly fluorescent upon radical trapping or redox activity. BPEAnit has strong ability to trap carbon- and sulphur-centred radicals and when dissolved in DMSO it can also detect peroxyl and hydroxyl radicals (Stevanovic et al. Citation2012). While DTT and DCFH-DA are both commercially available, BPEAnit was synthesized in our laboratory (Fairfull-Smith and Bottle Citation2008). The BPEAnit probe was dissolved in DMSO to produce a concentration ∼10 μM.
Sonication Experiments
Sonication was performed by placing a reaction flask with the probe solution (10 mL) in an ultrasonic bath (Elmasonic S30(H)) with a frequency of 37 kHz and power of 80 W. The sonication times were 15, 30 and 60 min, except for DTT where maximum sonication time was 30 min. These experiments were repeated 3–4 times. Considering that ultrasonic baths are known to have uneven energy transfer, an experiment was performed to test the uniformity of energy transfer within the ultrasonic bath. For this purpose, three reaction flasks, each containing the same volume (10 mL) of the same solution (either BPEAnit or DCFH) were placed next to each other in the ultrasonic bath and exposed to ultrasonic irradiation for 15, 30, and 60 min. In order to examine the formation of ROS/radicals even after turning the ultrasonic bath off, experiments with the BPEAnit and DCFH were performed where the probe was added to 30 min sonicated solvent and to a non-sonicated solvent.
Spectroscopy
Fluorescence emission of BPEAnit and DCFH was measured using a Varian Cary eclipse spectrofluorimeter. In the case of BPEAnit the excitation wavelength was set to 430 nm, whereas in the case of DCFH, the excitation wavelength was set to 485 nm. Absorbance spectra of DTT were measured using a Varian Cary 50 UV-VIS Spectrophotometer. Fluorescence and absorbance spectra were recorded at different sonication times, ranging from t = 0 min to t = 60 min. The spectra were recorded around 30 min upon completion of sonication. For each probe a spectrum was recorded for a blank (i.e., non-sonicated) sample at the same times as the times of sonication.
Calibration Assays
In the case of DCFH/HRP system, the fluorescent intensities of sonicated samples at 524 nm were converted into equivalent H2O2 concentrations using a calibration curve obtained by adding standard solutions of H2O2 to the DCFH/HRP mixture to obtain 0–400 nM H2O2 in the final solutions. Blank sample was prepared by mixing DCFH/HRP with laboratory grade deionized water. Spectra were recorded after 30 min of incubation.
In the case of BPEAnit, a calibration curve was obtained by plotting known concentrations of fluorescent derivative of BPEAnit (BPEAnit-Me) against fluorescence intensity at 485 nm. This calibration curve was used to calculate the amount of BPEAnit that was converted to a fluorescent product upon sonication, which corresponds to the amount of ROS/radicals trapped by BPEAnit upon sonication.
RESULTS AND DISCUSSION
BPEAnit is a weakly fluorescent compound but upon reaction with carbon- or sulfur-centred radicals and/or ROS, a strongly fluorescent product is formed (a) (Fairfull-Smith and Bottle Citation2008). b shows that sonication of BPEAnit in DMSO results in an increase of fluorescence emission of BPEAnit that is correlated with increased sonication. Therefore, the increase of fluorescence indicates that sonication of BPEAnit solution in DMSO results in the formation of radicals and/or ROS. Previous studies have demonstrated radical formation upon sonication – sulfur trioxide radical anion (SO3·−) was identified as the main product of the sonication of the argon saturated 75/25 DMSO/water mixture (Kondo et al. Citation1993). In addition, our recent study has shown that sonication of DMSO in the presence of BPEAnit results in formation of methoxyamine, methanesulfinamide and methanesulfonamide, which are products of BPEAnit and methyl (CH3·), methanesulfoxyl (CH3SO·) and methanesulfonyl (CH3S(O)2·) radicals, respectively (Stevanovic et al. Citation2012).
FIG. 1. (a) Formation of a fluorescent product upon reaction of BPEAnit with a radical; (b) an example of fluorescence spectra of BPEAnit upon sonication in DMSO; (c) average ROS/radical concentrations for different sonication times. The experiment was repeated 3 times and the error bars present one standard deviation.
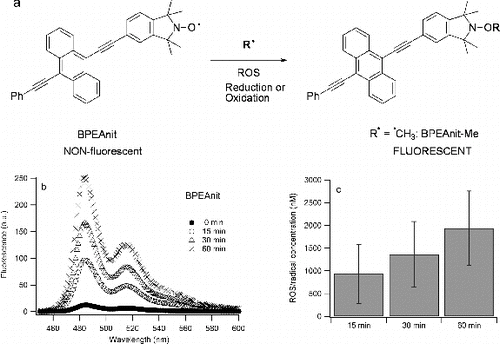
Based on the calibration curve obtained by plotting known concentrations of BPEAnit-Me against their fluorescence intensity, the ROS/radical concentrations for different sonication times were calculated. The values obtained for the amount of BPEAnit converted into a fluorescent product either by radical trapping or redox activity (i.e., ROS/radical concentrations) upon 15, 30, and 60 min were 0.93 ± 0.65 μM, 1.36 ± 0.72 μM, and 1.94 ± 0.81 μM, respectively. The amounts of radicals/ROS generated upon sonication are around two orders of magnitude higher than the values obtained for PM samples using the same probe (Miljevic et al. Citation2010; Crilley et al. Citation2012), which emphasises the extent of radical generation upon sonication of DMSO. However, if the probe was added to the 30 min sonicated DMSO it was found that ROS/radicals trapped by the probe were ∼50 nM.
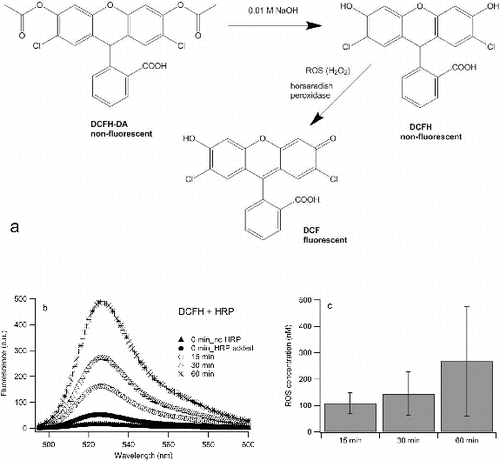
The scheme of ROS-induced oxidation of DCFH in the presence of HRP is shown in a and b shows that sonication of DCFH in 10/90 ethanol/water mixture in the presence of HRP also results in an increase of fluorescence with increased sonication. Of further significance also shows (black circles) that the fluorescence of DCFH increases after HRP is added in the solution (prior to sonication). The role of HRP is to catalyse the reaction between H2O2 and DCFH, but it has been demonstrated previously that DCFH reacts directly with HRP even in the absence of H2O2 to give fluorescent products (DCF) (Rota et al. Citation1999). This is a clear demonstration of the questionable value of DCF as an ROS probe. However, despite DCF's limitations it is notable that further significant increases in fluorescence occur upon sonication. This is likely to involve the generation of hydrogen peroxide, which, in turn, reacts with HRP to initiate another oxidative cycle of DCFH. As mentioned above, a common reaction of the radicals formed during the sonication of water is the dimerization of hydroxyl radicals or the disproportionation of hydrogen-peroxyl radicals to give hydrogen peroxide (Petrier et al. Citation1992, Mark et al. Citation1998). Fluorescence intensities from four sonication experiments were converted into ROS concentrations using the calibration curve. It is important to note that fluorescence of DCFH/HRP mixture increases with time and therefore, the increase in fluorescence intensity of this blank (i.e., non-sonicated) sample was subtracted from the fluorescence intensities of sonicated samples. The average ROS concentrations are shown in c. The values obtained for concentrations of ROS generated upon 15, 30 and 60 min were 108 ± 39 nM, 144 ± 82 nM, and 268 ± 208 nM, respectively. This is comparable to the results of Fuller et al. (Citation2014), who reported ROS concentrations of 80 nM generated during 15 min of sonication of pure HPLC water. ROS concentrations were also calculated when adding DCFH/HRP to 30 min sonicated deionized water and it was found that they were ∼55 nM, which is three times less than the ROS concentrations obtained for 30 min sonication of water with DCFH/HRP present in the solution during the sonication step. The total ROS concentrations generated upon sonication were found to be of the same order of magnitude to the ROS concentrations measured in PM samples using the same probe (See et al. Citation2007; Venkatachari et al. Citation2007; Wang et al. Citation2011). Without HRP there is still an increase of fluorescence with sonication (not shown), however the increase is ∼25 times smaller than in the presence of HRP. Similar fluorescence responses were observed when pure ethanol or water were used as solvents.
FIG. 2. (a) Hydrolysis of DCFH-DA and ROS-induced oxidation of DCFH; (b) an example of fluorescence spectra of DCFH upon sonication in 10% ethanol/90% water in the presence of HRP; (c) average ROS concentrations for different sonication times. The experiment is repeated 3 times and error bars present one standard deviation.
There is a significant difference between ROS/radicals measured by BPEAnit and DCFH. This difference can be explained by a different solvent that was used for each of these two probes. Sonication of different solvents will result in formation of different radicals and possibly different amounts of radicals being formed. In addition, these two probes are not equally specific for the same type of radicals/ROS.
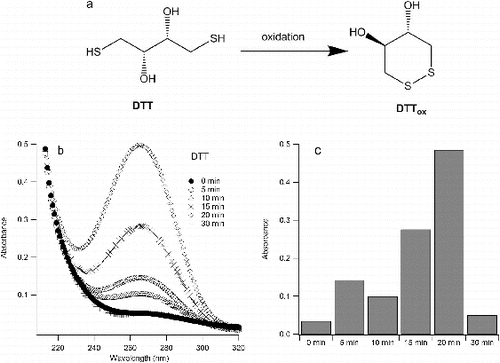
Sonication of aqueous solutions of DTT resulted in its oxidation, which is evident from its absorbance spectrum (). Considering that DTT and its oxidized form have different UV-VIS spectra, the oxidation of DTT can be monitored by changes in the UV-VIS spectrum. Because of its cyclic structure (a), oxidized DTT has an absorption peak at ∼270 nm, while reduced DTT starts to absorb only below 240 nm (Cleland Citation1964; Klitgaard et al. Citation2006). It can be seen from that the absorbance at 270 nm increases with increased sonication (from t = 0 min to t = 20 min) and then at t = 30 min drops down to its initial value (i.e., at t = 0 min). This suggests that the amount of oxidized DTT increases during the first 20 min of sonication. DTT has been shown to react with ROS, such as OH radical and hydrogen peroxide to give oxidized DTT (Redpath Citation1973; Netto and Stadtman Citation1996). After this time the oxidized (cyclic) DTT can be further oxidized to sulfinic or sulfonic acid (Capozzi and Modena Citation1974). The molar extinction coefficient of oxidized DTT was found to be 285 at ∼270 nm (λmax) (Klitgaard et al. Citation2006) and based on that value it was calculated that after 20 min of sonication approximately all of DTT has been converted into a cyclic product (i.e., oxidized DTT). The reduction of absorbance at ∼270 nm after 20 min indicates the destruction of cyclic DTT.
FIG. 3. (a) Oxidation of DTT; (b) absorbance spectra of oxidized DTT upon sonication in water; (c) absorption of oxidized DTT at 270 nm (λmax).
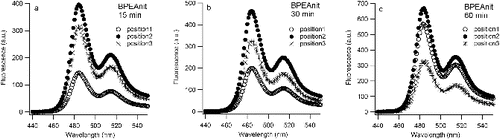
Sonication experiments performed in this study (at least three with each probe) were found to give reproducible trends (i.e., each probe displayed an increased response with sonication time). Notably, the actual reproducibility between experiments was poor, even with careful attention paid to repeat each the experiment with the same solution and conditions. Moreover, a plot of the fluorescence emission arising from sonication of the BPEAnit probe in DMSO reveals that fluorescence emission does not increase linearly with sonication time (). This reflects the fact that cavitational activity in ultrasonic reactors is uneven (Gogate et al. Citation2002 and references therein) and is very dependent on the exact position of the vessel when placed in the ultrasonic bath. With the experiment where a sonicated BPEAnit solution was monitored over time, the position of the reaction flask within the ultrasonic bath varied over the course of the experiment. For each fluorescence measurement, the flask was removed from the sonication bath and then replaced for further sonication, without paying attention to the exact position within the bath. This is an important issue that impacts on reproducibility and reliability for aerosol research where sonication is used to treat filters used to accumulate particulates. To determine the potential significance of this aspect for aerosol research we tested the influence of the placement of the vessel in the ultrasonic bath on the level of radicals/ROS detected. For that purpose, three reaction flasks containing the same solution of BPEAnit in DMSO were placed next to each other in the ultrasonic bath (position 1 was left, position 2 was middle and position 3 was right () and simultaneously sonicated for 15, 30, and 60 min. As shown in Figure 5, the fluorescence intensity of the same BPEAnit solutions at different positions in the ultrasonic bath was different. For all three sonication times the flask with the highest fluorescence response was in the middle (position 2) of the ultrasonic bath. In some cases the fluorescence response from position 2 was more than 50% higher than fluorescence intensity at the position 1 or 3. This suggests that position 2 is the closest to the transducer system of the ultrasonic bath, i.e., that the ultrasonic intensity was the strongest at position 2. Different fluorescence intensities and greater fluorescence response from position 2 (middle of the bath) were also obtained for simultaneously sonicated DCFH + HRP solutions. This experiment explains large error bars obtained for the average ROS/radical concentrations (c and c).
FIG. 4. Fluorescence intensity at 485 nm of BPEAnit upon sonication in DMSO.
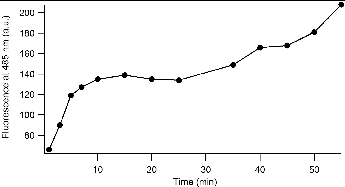
FIG. 5. Fluorescence spectra of BPEAnit (a, b, and c) at three different positions (position 1: left, position 2: middle, and position 3: right) in the ultrasonic bath upon simultaneous sonication for 15, 30, and 60 min. Fluorescence spectra at t = 0 min was the same for all three solutions, with intensity of 10 a.u. at 485 nm (fluorescence maximum).
CONCLUSION
This study demonstrates that sonication of dimethyl sulfoxide, water and ethanol/water mixtures leads to the generation of radical species, which is in accordance with previous literature. In the context of aerosol research this means that the formation of these species can alter the character of PM collected onto filters. Therefore, the standard use of sonication for extraction of organic material from filters should be avoided, if possible. Instead, alternative methods such as vortexing, magnetic stirrer or orbital shaking should be explored in more detail and validated in terms of extraction efficiency. In addition, if sonication is applied in the presence of the molecular probes for detection of ROS and radicals, such as the ones used in this study, sampling artifacts (e.g., probe responses arising from ultrasound-induced radicals) should be taken into consideration when interpreting results. The amounts of ROS trapped by the probes used in this study were found to be comparable or higher to the previously reported ROS measured in PM samples using the same probe. Furthermore, if sonication is applied to remove PM from filters the position within the ultrasonic bath of the vessel containing the filters should be carefully controlled as this can lead to variations in ROS generation of over 50%.
FUNDING
This work was supported by the Australian Research Council Discovery Grant (DP120100126).
REFERENCES
- Adewuyi, Y. G. (2001). Sonochemistry: Environmental Science and Engineering Applications. Ind. Eng. Chem. Res., 40:4681–4715.
- Capozzi, G., and Modena, G. (1974). The Chemistry of the Thiol Group. The Chemistry of Functional Groups. S. Patai, John Wiley & Sons.
- Cleland, W. W. (1964). Dithiothreitol, a New Protective Reagent for SH Groups*. Biochemistry, 3:480–482.
- Crilley, L. R., Knibbs, L. D., Miljevic, B., Cong, X., Fairfull-Smith, K. E., et al.(2012). Concentration and Oxidative Potential of On-Road Particle Emissions and Their Relationship with Traffic Composition: Relevance to Exposure Assessment. Atmos. Environ., 59:533–539.
- Crum, L. A. (1995). Comments on the Evolving Field of Sonochemistry by a Cavitation Physicist. Ultrason. Sonochem., 2:S147–S152.
- Fairfull-Smith, K. E., and Bottle, S. E. (2008). The Synthesis and Physical Properties of Novel Polyaromatic Profluorescent Isoindoline Nitroxide Probes. Eur. J. Organic Chem., 32:5391–5400.
- Fuller, S. J., Wragg, F. P. H., Nutter, J., and Kalberer, M. (2014). Comparison of On-Line and Off-Line Methods to Quantify Reactive Oxygen Species (ROS) in Atmospheric Aerosols. Atmos. Environ., 92:97–103.
- Goel, M., Hongqiang, H., Mujumdar, A. S., and Ray, M. B. (2004). Sonochemical Decomposition of Volatile and Non-Volatile Organic Compounds—A Comparative Study. Water Res., 38:4247–4261.
- Gogate, P. R., Tatake, P. A., Kanthale, P. M., and Pandit, A. B. (2002). Mapping of Sonochemical Reactors: Review, Analysis, and Experimental Verification. AIChE J., 48:1542–1560.
- Hasson, A. S., and Paulson, S. E. (2003). An Investigation of the Relationship Between Gas-Phase and Aerosol-Borne Hydroperoxides in Urban Air. J. Aerosol Sci., 34:459–468.
- Hung, H. F., and Wang, C. S. (2001). Experimental Determination of Reactive Oxygen Species in Taipei Aerosols. J. Aerosol Sci., 32:1201–1211.
- Khurshid, S. S., Siegel, J. A., and Kinney, K. A. (2014). Indoor Particulate Reactive Oxygen Species Concentrations. Environ. Res., 132:46–53.
- Klitgaard, S., Neves-Petersen, M. T., and Petersen, S. B. (2006). Quenchers Induce Wavelength Dependence on Protein Fluorescence Lifetimes. J. Fluoresc., 16:595–609.
- Kondo, T., Kirschenbaum, L. J., Kim, H., and Riesz, P. (1993). Sonolysis of Dimethyl-Sulfoxide Water Mixtures: A Spin-Trapping Study. J. Phys. Chem., 97:522–527.
- Krishna, C. M., Kondo, T., and Riesz, P. (1989). Sonochemistry of Alcohol-Water Mixtures. Spin-Trapping Evidence for Thermal Decomposition and Isotope-Exchange Reactions. J. Phys. Chem., 93:5166–5172.
- Kumagai, Y., Koide, S., Taguchi, K., Endo, A., Nakai, Y., Yoshikawa, T., and Shimojo, N. (2002). Oxidation of Proximal Protein Sulfhydryls by Phenanthraquinone, a Component of Diesel Exhaust Particles. Chem. Res. Toxicol., 15:483–489.
- Makino, K., Mossoba, M. M., and Riesz, P. (1983). Chemical Effects of Ultrasound on Aqueous Solutions. Formation of Hydroxyl Radicals and Hydrogen Atoms. J. Phys. Chem., 87:1369–1377.
- Mark, G., Tauber, A., Laupert, R., Schuchmann, H.-P., Schulz, D., Mues, A., and von Sonntag, C. (1998). OH-Radical Formation by Ultrasound in Aqueous Solution – Part II: Terephthalate and Fricke Dosimetry and the Influence of Various Conditions on the Sonolytic Yield. Ultrason. Sonochem., 5:41–52.
- Miljevic, B., Heringa, M. F., Keller, A., Meyer, N. K., Good, J., et al. (2010). Oxidative Potential of Logwood and Pellet Burning Particles Assessed by a Novel Profluorescent Nitroxide Probe. Environ. Sci. Technol., 44:6601–6607.
- Mišík, V., and Riesz, P. (1996a). EPR Study of Free Radicals Induced by Ultrasound in Organic Liquids II. Probing the Temperatures of Cavitation Regions. Ultrason. Sonochem., 3:25–37.
- Mišík, V., and Riesz, P. (1996b). Recent Applications of EPR and Spin Trapping to Sonochemical Studies of Organic Liquids and Aqueous Solutions. Ultrason. Sonochem., 3:S173–S186.
- Mutzel, A., Rodigast, M., Iinuma, Y., Böge, O., and Herrmann, H. (2013). An Improved Method for the Quantification of SOA Bound Peroxides. Atmos. Environ., 67:365–369.
- Netto, L. E. S., and Stadtman, E. R. (1996). The Iron-Catalyzed Oxidation of Dithiothreitol Is a Biphasic Process: Hydrogen Peroxide Is Involved in the Initiation of a Free Radical Chain of Reactions. Arch. Biochem. Biophys., 333:233–242.
- Petrier, C., Jeunet, A., Luche, J. L., and Reverdy, G. (1992). Unexpected Frequency Effects on the Rate of Oxidative Processes Induced by Ultrasound. J. Am. Chem. Soc., 114:3148–3150.
- Redpath, J. L. (1973). Pulse Radiolysis of Dithiothreitol. Radiat. Res., 54:364–374.
- Riesz, P., Kondo, T., and Carmichael, A. J. (1993). Sonochemistry of Acetone and Acetonitrile in Aqueous Solutions. A Spin Trapping Study. Free Radical Res., 19: s45–s53.
- Rota, C., Chignell, C. F., and Mason, R. P. (1999). Evidence for Free Radical Formation During the Oxidation of 2′-7′-Dichlorofluorescin to the Fluorescent Dye 2′-7′-Dichlorofluorescein by Horseradish Peroxidase: Possible Implications for Oxidative Stress Measurements. Free Rad. Biol. Med., 27:873–881.
- See, S. W., Wang, Y. H., and Balasubramanian, R. (2007). Contrasting Reactive Oxygen Species and Transition Metal Concentrations in Combustion Aerosols. Environ. Res., 103:317–324.
- Stevanovic, S., Miljevic, B., Eaglesham, G. K., Bottle, S. E., Ristovski, Z. D., and Fairfull-Smith, K. E. (2012). The Use of a Nitroxide Probe in DMSO to Capture Free Radicals in Particulate Pollution. Eur. J. Org. Chem., 2012:5908–5912.
- Suslick, K. S. (1990). Sonochemistry. Science, 247:1439–1445.
- Suslick, K. S., Hammerton, D. A., and Cline, R. E. (1986). Sonochemical hot spot. J. Am. Chem. Soc., 108:5641–5642.
- Thompson, L. H., and Doraiswamy, L. K. (1999). Sonochemistry: Science and Engineering. Ind. Eng. Chem. Res., 38:1215–1249.
- Venkatachari, P., Hopke, P. K., Brune, W. H., Ren, X. R., Lesher, R., Mao, J. Q., and Mitchel, M. (2007). Characterization of Wintertime Reactive Oxygen Species Concentrations in Flushing, New York. Aerosol Sci. Technol., 41:97–111.
- Wang, Y., Hopke, P. K., Sun, L., Chalupa, D. C., and Utell, M. J. (2011). Laboratory and Field Testing of an Automated Atmospheric Particle-Bound Reactive Oxygen Species Sampling-Analysis System. J. Toxicol., 2011:419476, 9 pages.