
Abstract
Airborne Staphylococcus aureus causes a significant proportion of nosocomial infections. The purpose of this study was to combine real-time quantitative polymerase chain reaction with the DNA-binding agent propidium monoazide (PMA-qPCR) to assess exposures to airborne S. aureus. In this work, we generated a S. aureus aerosol and used our assay to detect viable, airborne S. aureus in a study chamber. The biological collection efficiencies of three samplers (the AGI-30 impinger, BioSampler, and Nuclepore filter sampler) were evaluated using the S. aureus aerosols. The effects of storage in collection fluid on S. aureus sampled by the AGI-30 impinger and BioSampler were evaluated. Furthermore, air samples from an intensive care unit (ICU) and a gymnasium (GYM) were subsequently used to test the performance of a BioSampler combined with our PMA-qPCR technique. The BioSampler was more effective than the AGI-30 and Nuclepore filter samplers for preserving the culturability and viability of S. aureus aerosol samples. After sampling by impingement, the loss of viable S. aureus was minimized by treating the cells with PMA prior to storage at −20°C and analyzing the samples by qPCR within 3 weeks. In field applications, we noted that traditional culture assays tended to underestimate the viable concentrations of S. aureus by approximately one order of magnitude. Overall, combining qPCR with and without PMA staining may be useful for assessing exposure to airborne S. aureus. However, a complex set of parameters that may affect the efficiency of PMA-qPCR must be taken into account before applying PMA-qPCR to bioaerosol detection.
Copyright 2014 American Association for Aerosol Research
1. INTRODUCTION
Staphylococcus aureus is an opportunistic pathogen with a wide spectrum of clinical manifestations, including scalded skin syndrome, pneumonia, food poisoning, and toxic shock syndrome (Francis et al. Citation2005). Following the introduction of methicillin into clinical use, methicillin-resistant S. aureus (MRSA) has been frequently associated with nosocomial infections (Hulten et al. Citation2010; Sacar et al. Citation2010). In Taiwan, MRSA has accounted for up to 80% of all S. aureus isolates causing nosocomial infections in hospitals since 1998 (Ho et al. Citation1999). Furthermore, the incidence of MRSA infections is increasing worldwide.
Although MRSA is known to be transmitted by close contact with infected individuals or by contaminated hands or clothing (Cooper et al. Citation2004), airborne dispersal and transmission may be involved in the progression of epidemics (Shiomori et al. Citation2002). The presence of MRSA in the nostrils of healthcare staff led Cookson et al. (Citation1989) to speculate that a possible transmission route of this bacterium is by inhalation. Indeed, a nasal carrier can disperse S. aureus into the air and was identified as the possible cause of a nosocomial outbreak (Sherertz et al. Citation1996). In addition, the presence of airborne MRSA has been found to be significantly related to the presence of MRSA-infected or -colonized patients (Wilson et al. Citation2004; Gehanno et al. Citation2009). In recent years, several studies have investigated airborne MRSA using different samplers, including the Andersen 1-STG impactor (Hsiao et al. Citation2012; Chang and Wang Citation2014), SAS 100 impactor (Agnoletti et al. Citation2014), AES air sampler (Creamer et al. Citation2014), and AGI-30 impinger (Schulz et al. Citation2012). Among these studies, active sampling has been the most common method used for surveying airborne S. aureus, and the detection of MRSA in air samples suggests that the risk of infection through inhalation may be increasing (Friese et al. Citation2012; Schulz et al. Citation2012; Creamer et al. Citation2014). However, few studies have been performed to characterize the collection efficiency and sampling stress (via impaction or impingement) on airborne S. aureus (Hsiao et al. Citation2012; Chang and Wang Citation2014).
Most previous studies have employed impactors combined with culture assays to detect airborne S. aureus or MRSA. However, these methods are slow and do not effectively detect bacteria that are in the viable but non-culturable (VBNC) stage. Considering that S. aureus in the VBNC stage is a potential cause of nosocomial infection (Pasquaroli et al. Citation2013), culture-independent assays are needed to detect viable S. aureus or MRSA. Therefore, the efficiencies of liquid (e.g., AGI-30 or BioSampler) and filter samplers, analyzed through both culture and non-culture methods, warrant further investigation. For AGI-30 or BioSampler sampling, the interpretation of results from field studies is complicated because the analysis is delayed until the samples are transported to a laboratory, and the organisms present may therefore multiply or die during transit (Thorne et al. Citation1992; Tseng and Li Citation2005). Several studies have addressed this issue by refrigerating samples until the microbes can be plated (Li and Lin Citation2001; Tseng and Li Citation2005; Chang and Wang Citation2014). However, after air sampling, the optimal conditions for storing different microbial species in collection media can vary widely and must be tested further.
To rapidly detect microorganisms, real-time quantitative polymerase chain reaction (qPCR) assays have been used to quantify target sequences with a high sensitivity and specificity. Although qPCR has been widely employed to detect S. aureus in different contexts (Fang and Hedin Citation2003; Berrada et al. Citation2006; Cattoir et al. Citation2011), it has not been adapted to monitor airborne S. aureus. Additionally, the major disadvantage to qPCR is that it cannot quantify cell viability. In recent years, viability assays combining qPCR with propidium monoazide (PMA-qPCR) have been utilized to differentiate between live and dead cells. PMA is a high-affinity, photoreactive DNA-binding agent; it can only penetrate the compromised membranes of dead cells. Upon photolysis by exposure to bright visible light, the photoreactive azido group of PMA is converted into a highly reactive nitrene radical that can form covalent cross-links with DNA (Nocker and Camper Citation2009). These cross-links prevent the subsequent qPCR amplification of the target DNA of non-viable cells. Additionally, PMA is excluded from viable cells with intact membranes and is then inactivated by exposure to visible light. Thus, only DNA from intact (i.e., viable) cells can be amplified when bacteria are treated with PMA prior to DNA extraction.
Recently, another DNA intercalating agent, ethidium bromide monoazide (EMA) was applied in an attempt to discriminate heat-inactivated S. aureus from living cells, but EMA was found to be an unsuitable indicator of the viability of Staphylococcus species (Kobayashi et al. Citation2009a). In contrast, PMA can only penetrate dead cells and has therefore been recommended as a highly selective dye to distinguish S. aureus that have been inactivated by heat or antibiotics from living bacteria (Kobayashi et al. Citation2009b, Citation2010). These results indicate that while EMA and PMA present similar mechanisms for discriminating between viable and dead bacteria, the penetration of these two dyes into bacteria could be species-dependent. Compared with EMA, PMA is a better reagent for rapidly detecting viable airborne S. aureus or MRSA (Kobayashi et al. Citation2009b).
In our previous study, we optimized PMA-qPCR to identify the viability of Acinetobacter baumannii (Tseng et al. Citation2014). However, a complex set of parameters that may affect the efficiency of PMA-qPCR must be taken into account before applying PMA-qPCR to detect different bioaerosols. The optimal conditions for PMA-qPCR may vary with microbe species (Fittipaldi et al. Citation2012). Therefore, there is a need to separately investigate the efficiency of the same technology for different microbe species. The purpose of this study was to optimize PMA-qPCR for quantifying viable S. aureus and to subsequently evaluate the effects of three bioaerosol sampling methods (the AGI-30 impinger, BioSampler, and Nuclepore filter sampler) on S. aureus recovery using culture, qPCR, and PMA-qPCR. Based on our experience, relative humidity (RH) may affect the biological efficiency of the samplers when collecting Gram-positive bacteria (Hsiao et al. Citation2012, Citation2014). Therefore, samples were taken at three levels of RH (35%, 55%, and 85%). The effects of storage on the kinetic decay curves of S. aureus sampled by the AGI-30 impinger and BioSampler in collection fluid were also evaluated. Furthermore, an environmental monitoring program using PMA-qPCR was conducted to quantify the viable concentrations of S. aureus in the air of an intensive care unit (ICU) and a gymnasium (GYM). To our knowledge, this is the first report to demonstrate the presence of viable S. aureus in field air using PMA-qPCR.
2. MATERIALS AND METHODS
2.1. Test Microorganisms
The antibiotic-sensitive S. aureus reference strain ATCC 29213 (MSSA) was purchased from the Bioresource Collection and Research Center (Hsin-Chu, Taiwan). An active MSSA culture was inoculated in lysogeny broth (LB) and incubated for 24 h at 37°C. After cultivation, the obtained microbe pellets were aseptically washed with sterile phosphate-buffered saline (PBS) and transferred to a sterile 15 ml conical centrifuge tube, which was capped and centrifuged (500 × g, 5 min). Finally, the supernatant was discarded, and the pellets were resuspended in sterile distilled water for the preparation of spray suspensions and the PMA optimization test.
2.2. Preparation of Viable and Dead S. aureus
Taking into account the DNA extraction efficiency and detection limit of our qPCR assay, cell suspensions of 105 CFU/ml were prepared and evaluated in a subsequent PMA optimization test. The prepared cell suspensions were heated at 85°C for 10 min to obtain dead cells. After heating treatment, MSSA was no longer culturable on LB agar (Difco Laboratory, Detroit, MI, USA). The unheated samples were defined as 100% controlled viable sample, whereas the heated bacteria were defined as 0% controlled viable sample. Three additional controlled viable samples (75%, 50%, and 25%) were produced from these two preparations. For each of these five samples (100%, 75%, 50%, 25%, and 0%), the culturable ratio was calculated as the average CFUs obtained from triplicate platings divided by that for the 100% controlled viable sample. In addition, the viable ratios of MSSA cells for each of the five samples were the average MSSA concentrations determined by qPCR and PMA-qPCR assays divided by that of the 100% controlled viable sample.
2.3. Optimization of the PMA Treatment
The effects of different PMA concentrations and light exposure times on the amplification of DNA from viable cells were evaluated via DNA extraction followed by qPCR analysis. PMA purchased from Biotium Inc. (Biotium, Hayward, CA, USA) was added at a final concentration of 50, 25, 10, or 2.5 μg/ml to sample tubes containing 300 μl of either viable (100% controlled viable sample) or heat-killed (0% controlled viable sample) cells (105 CFU/ml), as described in previous studies (Kobayashi et al. Citation2009b, Citation2010). For PMA staining, the tubes were placed in the dark for 5 min and then transferred to ice and exposed to a 500 W halogen light at a distance of 20 cm.
The influence of the light exposure time on both the PMA concentration and PMA-qPCR was evaluated. The optimal PMA concentration obtained from optimization tests, performed as previously described, was 10 μg/ml. Therefore, both viable and heat-treated cells were stained with 10 μg/ml PMA and then exposed to the halogen light for varying lengths of time (3, 7, or 10 min). In qPCR and PMA-qPCR assays, a positive reaction is detected by the accumulation of a fluorescent signal. The threshold cycle (Ct) is defined as the number of cycles that exceed the detection threshold during the PCR amplification. Ct levels are inversely proportional to the amount of the target gene in the sample. Therefore, the difference between the Ct values obtained for untreated and PMA-treated MSSA by qPCR was calculated for each pair of samples, according to the method described in a previous study, as follows: ΔCt = Ct with PMA − Ct without PMA (Kobayashi et al. Citation2009b). A low ΔCt indicates the presence of amplifiable DNA from viable bacteria, whereas a high ΔCt indicates that the target DNA in the sample originated from bacteria with membrane damage (Miotto et al. Citation2012).
2.4. DNA Extraction and qPCR
After exposure to light, S. aureus DNA was extracted using a QIAamp DNA mini kit (Qiagen GmbH, Hilden, Germany) according to the manufacturers’ instructions. For each sample, 50 μl of DNA extract was obtained. For the identification of S. aureus, the primers NUC1 (5′-GCG ATT GAT GGT GAT ACG GTT-3′) and NUC2 (5′-AGC CAA GCC TTG ACG AAC TAA AGC-3′) were employed to detect the S. aureus-specific nuc gene, which is a single-copy gene, as previously described (Fang and Hedin Citation2003). qPCR was conducted in a LightCycler (Roche Diagnostics GmbH, Basel, Switzerland) using a SYBR green kit (LightCycler FastStart DNA Master SYBR Green I). The reactions contained 0.25 μM of each primer (Mission Biotech Co., Taipei, Taiwan), 3 mM MgCl2, 2 μl SYBR Green I mix, 9.6 μl nuclease-free water, and 5 μl DNA extract, for a total volume of 20 μl. The qPCR program followed a previous protocol (Fang and Hedin Citation2003), with the exception that the primer annealing temperature was changed from 55°C to 60°C. For quantification, the DNA standard (nuc gene) of S. aureus was synthesized from a cloning plasmid by Mission Biotech (Taipei, Taiwan). A calibration curve was constructed by serially diluting the DNA standard, and the curve was linear over 7 orders of magnitude (0.3–1.6 × 105 copies/μl). The known quantities of plasmid DNA yielded cycle threshold (Ct) values ranging between 19 and 37 cycles, and the correlation coefficient (r) of the calibration curve was 0.99. Finally, the DNA copy number/μl of the test samples was calculated automatically by the LightCycler® software to obtain the number of viable and total S. aureus by adjusting for the sample volume in the suspension or airborne samples.
2.5. Aerosol Preparation and Test System
The test chamber was constructed of acrylic plastic and had a volume of 24.3 l (27 cm × 30 cm × 30 cm). A Collison three-jet nebulizer (BGI Collison Nebulizer, BGI Inc., Waltham, MA, USA) was used to nebulize the MSSA stock in deionized water at 20 psi with dry, filtered, compressed laboratory air. The S. aureus concentration in the nebulizer was approximately 105 CFU/ml (coefficient of variation% (CV%) = 3.0%) in each experiment. The aerosolized suspension was diluted with filtered, compressed air at 50 l/min, and this combination resulted in an aerosol residence time of 24 s. The generated S. aureus aerosol in the chamber was monitored by AGI-30, BioSampler and Nuclepore filters using culture assays every 10 min, and the generated S. aureus was stable at least for 90 min (CV% = 15%). To maintain the RH in the test chamber, a humidified gas stream was generated by passing pure compressed air through a humidity saturator. The water vapor content in the gas stream was adjusted by changing the flow rate ratio of the humidified gas stream to the dry gas stream, and the actual RH in the chamber was monitored by a hygrometer (Rotronic AG, Bassersdorf, Switzerland). To evaluate the effect of different RH values, the airborne samples were collected at three levels of RH (35%, 55%, and 85%).
2.6. Test Samplers and Sample Processing
The performance of different bioaerosol samplers in collecting airborne MSSA was evaluated using an AGI-30 impinger (Ace Glass Inc., Vineland, NJ, USA), a BioSampler (SKC Inc., Connellsville, PA, USA), and a Nuclepore filter sampler (Nuclepore®, Costar Corp., Cambridge, MA, USA). These bioaerosol devices were chosen because they are commonly used, and samples collected from the collection fluid or filters of these instruments can be analyzed using both culture and non-culture methods. The AGI-30 and BioSampler were filled with 20 ml of deionized water, while the Nuclepore filter sampler consisted of a 37-mm diameter polycarbonate membrane with 0.4-μm pores supported by cellulose pads. The AGI-30 and BioSampler were both operated at a flow rate of 12.5 l/min and for 60 min, while the Nuclepore filter was operated at 20 l/min for 60 min. In general, 30 min is usually recommended for AGI-30 and BioSampler sampling. However, to improve the detection limit for collecting S. aureus aerosol, our study increased the sampling time from 30 min to 60 min. Therefore, the remaining volumes of deionized water in the AGI-30 and BioSampler were measured to adjust the collected airborne concentration of S. aureus after sampling. The volumes of collection fluid lost to evaporation during 60 min of sampling at the three tested RH levels were 3–6 ml for the AGI-30 and 2–5 ml for the BioSampler. For the Nuclepore system, the filters were eluted after sampling by rinsing with 1 ml of sterile deionized water, and the eluate was slowly vortexed with the filter in a rotator (Vortex-2Genie, Scientific Industries Inc., NY, USA) for 30 s. The deionized water samples from the AGI-30 and BioSampler and the filter eluate from the Nuclepore system were inoculated separately onto tryptic soy agar (TSA) or analyzed via qPCR and PMA-qPCR. For bioaerosol sampling, TSA agar is most commonly used and also considered excellent general agar on which to culture a variety of bacteria. Therefore, we used TSA agar for studying the S. aureus aerosol. Finally, the culturable, viable, and total cell counts from the bioaerosol samplers were calculated based on the dilution ratio, plated volume, sampling time, and flow rate. Because the detection limit of our qPCR instrument is 0.3 copies/μl for S. aureus, the detection limit of our qPCR and PMA-qPCR methods corresponded to a minimum concentration of 2.4 × 103 cells/m3 for impingement and 100 cells/m3 for the Nuclepore filter after 60 min of sampling.
2.7. Indicators for the Evaluation of Collection Efficiency
2.7.1. Measurement of Culturability and Viability Through Culture, qPCR, and PMA-qPCR Assays
The total concentrations of S. aureus cells were determined via qPCR, and viable cell concentrations were determined via PMA-qPCR. As only DNA from viable cells is detected when bacteria are treated with PMA prior to DNA extraction, cell culturability and viability were defined as follows:
2.7.2. Calculation of the Bacterial Recovery Ratio Using CR and VR
The culturability and viability of aerosolized bacteria significantly affect the biological efficiency of a bioaerosol sampler (Lin and Li Citation1999). However, the culturability and viability of aerosolized bacteria are also affected by the culture preparation process (Lin and Li Citation1999; Tseng and Li Citation2005). To better understand the biological efficiency of the evaluated samplers for collecting airborne S. aureus, we evaluated the culturability and viability of the S. aureus in the liquid suspension used as the bioaerosol source. Consequently, the culturability ratio (CR) and viability ratio (VR) were used to adjust the initial culturability and viability to determine biological efficiency. The CR and VR were determined as the ratio of the culturability or viability obtained from different samplers to the culturability or viability obtained from suspensions according to a previous study (Chen and Li Citation2005a). These parameters are defined as follows:
where Csampler and Cnebulizer represent the culturability of MSSA from the samplers and the nebulizer, respectively, and Vsampler and Vnebulizer represent the viability of MSSA from the samplers and the nebulizer, respectively. The Cnebulizer and Vnebulizer values were obtained before air sampling.
2.8. Effect of Storage Conditions After Bioaerosol Collection
To determine the effect of storage time on the viability and culturability of MSSA, collection of airborne MSSA samples with the AGI-30 and BioSampler was conducted at 55% RH. As the BioSampler is considered to achieve better preservation of bioaerosol viability than the AGI-30, the MSSA cells collected by the AGI-30 and BioSampler may display varying degrees of cell membrane damage. Therefore, the formation of intracellular ice crystals during the storage period at freezing temperatures may cause different degrees of damage to cell membranes if damage from air sampling was already present. In contrast, the Nuclepore filter sampler was not evaluated here because the bacterial cells collected on the filters might not recover during storage due to the damage caused by impaction and dehydration stress (Li and Lin Citation2001).
After air sampling with the AGI-30 and BioSampler, each sample of collected MSSA was separated and stored for the culture assay (−20°C or 4°C), qPCR (−20°C), and PMA-qPCR (−20°C). For the culture assay, each suspension was inoculated after being stored for 0, 1, 2, 3, 4, or 5 days. The PMA-qPCR assays were subsequently divided as follows: One sample of the collection medium was directly subjected to PMA staining, DNA extraction, and freezing at −20°C, and qPCR quantification was not performed until after the indicated storage time (Pre-PMA). Another sample of the MSSA-containing collection medium was immediately stored at −20°C, and individual vials of the sample were subjected to the full PMA-qPCR assay (Post-PMA) at various time points up to 28 days. To obtain a kinetic curve of the variations in cell concentrations for up to 28 days of storage, each suspension was inoculated for the qPCR and PMA-qPCR assays after being stored for 0, 3, 7, 14, 21, or 28 days. The effect of the storage time on the variation in the viability and culturability of MSSA was determined by calculating the Ct/C0 ratio, where Ct and C0 are the cell concentrations of the simultaneously collected samples stored for t h and 0 h, respectively.
2.9. Field Sampling Locations
We collected aerosol samples from a GYM and a medical center ICU in Taiwan. These locations were chosen for sampling because S. aureus is frequently found in GYMs and ICUs and could therefore play a role in community or nosocomial transmission of S. aureus (Markley et al. Citation2012; Chen et al. Citation2014).
2.10. Field Aerosol Sampling
At the GYM and ICU, aerosol samples were collected in triplicate using a BioSampler because our study demonstrated that the BioSampler performed better than the AGI-30 and the Nuclepore filter sampler for collecting airborne MSSA. The pump and sampling apparatus were placed approximately 1 m from either a hospital bed in the ICU or fitness equipment in the GYM. In addition, the sampling locations were 3 m from square ceiling diffusers to prevent interference due to air flow. Samples were taken at a height of 1.2 m above the floor, which was within the breathing zone of people at each location. The BioSampler was operated at a flow rate of 12.5 l/min for 60 min. In the field study, we only used TSA agar to compare the concentrations of total bacterial aerosols, not to identify the S. aureus colonies that recovered on TSA agar. To rapidly identify S. aureus colonies in the field, another medium, CHROMagarTM Staph aureus (CSA) agar medium (CHROMagar Microbiology, Paris, France), was used in conjunction with the BioSampler to identify culturable S. aureus in the air according to methods we have described previously (Hsiao et al. Citation2012). The CSA agar allows for direct color-based identification and can be used to target pathogens with high specificity. Both agar media (CSA and TSA) evaluated in the field were placed in an incubator at 37°C for 24 h. To identify total and viable S. aureus cells, the collection medium in the BioSampler was analyzed via qPCR and PMA-qPCR. The sample processing procedure applied in these analyses was the same as that used in the chamber study.
2.11. Statistical Analysis
Statistically significant differences (p < 0.05) in the Ct values (ΔCt) obtained for untreated and PMA-treated MSSA between each pair of samples using various PMA concentrations and light exposure times were analyzed using the Wilcoxon signed rank test. The non-parametric Kruskal–Wallis test, followed by Scheffe's test was employed to compare (1) the CR and VR values from the culture and PMA-qPCR assays at the three different RH levels; (2) the CR and VR values from the three different bioaerosol samplers; (3) the log Ct/C0 values obtained after various storage times; (4) the numbers of total, viable, and culturable S. aureus cells from the GYM and ICU. To compare the culturable and viable ratios obtained through different assays, regression analyses were performed, and R-values were calculated. We used Spearman's rank correlation (r = Spearman's rho) to assess the association of the culturable and viable ratios determined via culture, qPCR, and PMA-qPCR. In addition, the non-parametric Wilcox rank-sum (Mann–Whitney) test was utilized to examine the differences in S. aureus concentrations between the GYM and ICU.
3. RESULTS AND DISCUSSION
3.1. Optimal PMA Concentration Needed to Bind Cellular DNA
To determine the optimal concentration of PMA, viable and dead MSSA (105 CFU/ml, corresponding to 105 copies/ml) were left untreated (0 μg/ml) or were treated with PMA (2.5, 10, 25, or 50 μg/ml) and exposed to halogen light for 7 min. shows that the ΔCt values of viable MSSA that were untreated and those that were treated with 2.5 or 10 μg/ml PMA were not significantly different (p = 0.15 and 0.20, respectively). However, treatment with 25 or 50 μg/ml PMA led to a significant different ΔCt compared with untreated samples (p = 0.03 and 0.02, respectively). Therefore, 25 and 50 μg/ml PMA reduced the amount of DNA in viable cells. For dead MSSA cells (), 2.5 μg/ml PMA was insufficient to inhibit the qPCR amplification of DNA (p = 0.22). PMA treatment at a minimum concentration of 10 μg/ml resulted in a significant increase in the Ct values derived from dead MSSA. Therefore, 10 μg/ml PMA was used in subsequent experiments because this concentration inhibited qPCR amplification from dead cells, but not viable cells.
FIG. 1. Effects of the PMA concentration on DNA binding in 105 CFU/ml of (a) viable MSSA cells and (b) dead MSSA cells. The effects of the different PMA concentrations are shown as differences in Ct values between the untreated and PMA-treated groups (2.5, 10, 25, or 50 μg/ml) following exposure to light for 7 min. *p < 0.05 compared with the respective untreated groups. The experiments were performed in triplicate, and the data are shown as the mean ± standard error of the mean.
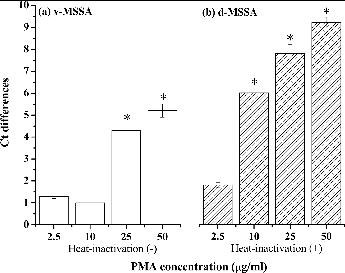
To our knowledge, there have been two previous studies that investigated the effectiveness of PMA for quantifying S. aureus (Nocker et al. Citation2006; Kobayashi et al. Citation2009b). However, no optimal PMA treatment test for S. aureus has been developed. These two studies applied 50 μM PMA (corresponding to 25 μg/ml) to distinguish between intact and dead S. aureus in suspension, but we found that this concentration of PMA may penetrate intact cell membranes. In contrast to most previous studies, which used PMA concentrations ranging from 25 to 50 μg/ml (Fittipaldi et al. Citation2012), a recent study recommend a lower PMA concentration of 10 μM (corresponding to 5 μg/ml) combined with a longer incubation time (30 min) to quantify another gram-positive species, Listeria monocytogenes (Nkuipou-Kenfack et al. Citation2013). Although making a direct comparison is difficult due to the variations in the methodology, these results indicated that the optimized PMA concentration may be affected by many parameters such as microbial species, cell concentration, the ratio between live and dead cells, and the dye incubation time (Fittipaldi et al. Citation2012). Therefore, multiple PMA concentrations may have to be assessed and optimized before PMA-qPCR is used to define the bacteria viability in different applications. EMA, another DNA-intercalating agent, has been used at a concentration of 25 μg/ml to detect Legionella pneumophila (Chang and Chou Citation2011a,Citationb) and at 100 μg/ml to detect Bacillus subtilis and Pseudomonas fluorescens in the air (Liang et al. Citation2012). However, EMA can enter both viable and dead S. aureus at a concentration of 50 μg/ml (Kobayashi et al. Citation2009a). Clearly, the optimal concentration of EMA for bioaerosol detection is species-dependent. Similar to EMA, higher concentrations of PMA (25 or 50 μg/ml) may also penetrate intact cell membranes, leading to underestimation of the viable cell concentration.
3.2. Effect of Light Exposure Times on PMA-qPCR
To test the effects of different light exposure times, viable and dead MSSA (105 CFU/ml) were left untreated or were treated with 10 μg/ml PMA and exposed to a halogen light for 3, 7, or 10 min. The difference in the obtained Ct values (ΔCt) between viable treated and untreated MSSA after 7 min was insignificant (p = 0.22; ). This result indicated that PMA did not penetrate the membrane of viable cells within 7 min of irradiation. Moreover, when dead samples were treated with PMA, a minimum light exposure time of 7 min significantly inhibited DNA amplification (p = 0.03; ). The optimal light exposure time for the PMA-qPCR assay varied from those reported in previously published studies. In general, 2–10 min of light exposure has typically been used to detect viable bacteria or viruses (Nocker et al. Citation2006, Citation2007; Kobayashi et al. Citation2009b; Parshionikar et al. Citation2010; Liu and Mustapha Citation2014). Therefore, the optimal light exposure time for DNA amplification may vary and depend on the light source and bacterial species used (Liu and Mustapha Citation2014). As with the PMA concentration, the light exposure time before PMA-qPCR has not been optimized for every species that has been tested. Both the optimal light exposure time and the optimal PMA concentration may vary among species, an idea that must be tested further.
FIG. 2. Effects of the light exposure time on the amplification of DNA from 105 CFU/ml of (a) viable MSSA cells and (b) dead MSSA cells. The effects of 3, 7, and 10 min of light exposures are shown as differences in Ct values between untreated samples and those treated with 10 μg/ml PMA. *p < 0.05 compared with the respective untreated groups. The experiments were performed in triplicate, and the data are shown as the mean ± standard error of the mean.
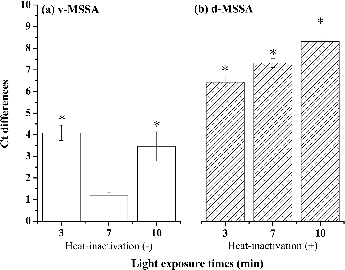
Exposure to light causes PMA to covalently bind to DNA, while unbound PMA is inactivated (Fittipaldi et al. Citation2012). Insufficient light exposure may affect the efficiency of PMA binding to the DNA or cause the residual PMA in solution to continue reacting with viable cells. In this study, when samples were exposed to light for 3 or 10 min, PMA penetrated both viable and dead MSSA. Therefore, a PMA concentration of 10 μg/ml and 7 min of exposure to light were used for subsequent experiments. The optimized PMA concentration (10 μg/ml) that we used for MSSA quantification is the same as that used to detect A. baumannii (Tseng et al. Citation2014). However, the optimal light exposure time for A. baumannii is only 3 min. We speculate that this difference may be related to different cell wall structures of gram-positive (MSSA) and gram-negative bacteria (A. baumannii). The light-absorbing properties of the bacterial samples may result in varying optimal light exposure times (Fittipaldi et al. Citation2012). To accurately report PMA-qPCR precision, the percentage of coefficient variation (CV%) was used to determine reproducibility (Hospodsky et al. Citation2010). Higher CV% represents greater variation in the sample reproducibility. Overall, the intra- and inter-sample variability values for PMA-qPCR in the current study, which refer to the degrees of agreement within a sample or between two samples, were 15% and 48%, respectively. These results were closed to those of our previous study (14% and 45%) for quantifying A. baumannii by PMA-qPCR (Tseng et al. Citation2014).
3.3. Comparison of Culturable or Viable Ratios as Measured by Culture, qPCR, and PMA-qPCR
The culturable or viable ratios of MSSA determined by three promising methods, culture, qPCR and PMA-qPCR, were compared. The culturable or viable ratio of MSSA measured by the culture, qPCR, and PMA-qPCR assays was linearly related to the controlled viable samples from 0% to 100% (). According to Spearman's rank correlation, the viable ratios measured through PMA-qPCR significantly correlated with those measured based on the culturable ratios (r = 1; p < 0.01) but not with those measured by qPCR (r = 0.6; p = 0.29). These observations show that qPCR-based viable ratios are higher than those measured by PMA-qPCR or culture assays (Liu and Mustapha Citation2014). The gradual decline in the viable ratio observed in conjunction with the decreasing controlled viable samples, as measured using qPCR, could be related to membrane protein degradation caused by heating, which in turn, could lead to DNA damage (Masters et al. Citation1998).
FIG. 3. Correlations between controlled viable samples and culturable (or viable) ratios assessed using the culture, qPCR, and PMA-qPCR assays. Three different controlled viable samples (75%, 50%, and 25%) were prepared from the 0% (heat-killed) and 100% (without heating) controlled viable samples. For each of the five controlled viable samples, the culturable (or viable) ratios of MSSA cells were defined as the average MSSA concentrations determined by culture, qPCR, and PMA-qPCR analyses divided by those of the 100% controlled viable samples. The experiments were performed in triplicate, and the data are shown as the mean ± standard error of the mean.
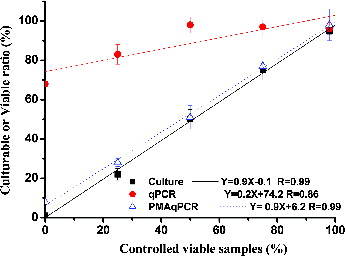
In order to apply PMA-qPCR assay for detecting airborne S. aureus, the evaluated MSSA concentration in our study (105 CFU/ml) was lower than those reported in previous studies (Nocker et al. Citation2006; Kobayashi et al. Citation2009b). When PMA-qPCR is required to quantify high cell concentrations of S. aureus, a longer PMA incubation time (>5 min) may be needed to penetrate the cell membranes of a larger number of dead cells (Nkuipou-Kenfack et al. Citation2013). Certainly, the PMA incubation time may also be affected by the PMA concentration; there is a delicate balance between many factors that has to be optimized in different environmental applications by PMA-qPCR (Fittipaldi et al. Citation2012). In comparison with different microbe species, the performance of PMA-qPCR used to identify the viable ratio of MSSA is as good as assays for identifying another important nosocomial infection pathogen, A. baumannii (Tseng et al. Citation2014). qPCR does not differentiate between dead and viable cells of A. baumannii.
Despite the fact that it involves sample heating, PMA-qPCR has also been employed to study the inactivation efficiency of different disinfectants (benzalkonium chloride, chlorine, ethanol, and isopropanol) on different bacterial species (Nocker et al. Citation2007). These compounds may also induce membrane damage. Another EMA dye combined with qPCR and heating has also been optimized for the detection of L. pneumophila, and this assay was subsequently used to monitor L. pneumophila in air and water (Chen and Chang Citation2010; Chang and Chou Citation2011b). However, PMA-qPCR was considered inadequate for identifying inactivated bacteria after a short exposure to ultraviolet radiation (Nocker et al. Citation2007). PMA-qPCR may not identify inactivated bacteria if the inactivation mechanisms do not target cell membrane integrity.
3.4. CR and VR for Different Bioaerosol Samplers
To compare the biological efficiencies of the evaluated samplers for collecting airborne MSSA, we used CR and VR to adjust the initial culturability and viability in the nebulizer. lists the CR and VR measured in MSSA using the AGI-30 sampler, BioSampler, and Nuclepore filter sampler at three RH levels (35%, 55%, and 85%). The CR values obtained for MSSA on TSA from the AGI-30 and BioSampler ranged from 0.24 to 0.68 at all three levels of RH. Although the CRs increased with the RH level, RH did not significantly affect the CR of MSSA, regardless of whether the AGI-30, BioSampler, or Nuclepore filter sampler was used (p = 0.42). Overall, the CR values measured using the BioSampler were significantly superior to those measured with the AGI-30 (p < 0.01) and the Nuclepore filter sampler (p < 0.01). Similar trends were observed for the VR values, as the VRs obtained with the BioSampler were significantly higher than those for the AGI-30 and the Nuclepore filter sampler (p < 0.01). However, the VRs were not affected by RH (p = 0.52) in either sampler type. There are no significant differences between the CRs and VRs reported for A. baumannii and MSSA under the same test conditions by a Wilcox rank-sum test (Tseng et al. Citation2014).
TABLE 1 CR and VR values obtained using the AGI-30 sampler, BioSampler, and Nuclepore filter sampler
As previously observed in studies sampling airborne Escherichia coli, Candida famata, and L. pneumophila (Chen and Li Citation2005a; Chang and Chou Citation2011b), our CR data indicated that the impingement sampling method was superior to filtration when studying the culturability of MSSA using the traditional cultivation assay. Even though the sampling volumes from 120 to 180 l (sampling flow rate × sampling time) used for Nuclepore filter sampling in previous studies were lower than ours in the current study (1,200 l), the filtration still impeded optimal culturability in comparison with the impingement sampling method (Chen and Li Citation2005a; Chang and Chou Citation2011b). For liquid impingers, particle collection by inertial impaction into the collection fluid can prevent dehydration of bioaerosols (Macher Citation1999). In the contrary, a long sampling time used with the Nuclepore filter sampler increases the yield, but viable MSSA may be lost on the filter due to dehydration during the sampling and extraction processes (Tseng and Li Citation2005).
Our results also agreed with a recent study showing that the BioSampler performed better than the AGI-30 for collecting airborne MSSA, especially when the sampling time was extended to 60 min (Chang and Wang Citation2014). The BioSampler is commonly considered to be more efficient than the AGI-30 at collecting bioaerosols because the BioSampler minimizes the re-aerosolization problem and reduces collection fluid loss (Lin et al. Citation2000). Furthermore, the relatively gentle swirling motion that occurs in the BioSampler may be more conducive to collecting culturable airborne MSSA than the violent bubbling of the AGI-30 (Lin et al. Citation2000). Compared with the sampling indicator, isolates with higher CR values correspond to species that are more resistant to the stress of air sampling. Based on the same sampling indicator, the average CR value obtained using the AGI-30 at 55% RH was 0.35, which is similar to that of fungal spores (Penicillin citrinum; 0.38), but higher than that of vegetative cell-type bioaerosols (E. coli and yeast; 0.07–0.1) and bacterial endospores (Bacillus subtilis; 0.15) (Chen and Li Citation2005b). However, using filter sampling, the CR value obtained for MSSA (0.05) was similar to that of yeast (0.038), but lower than that of P. citrinum (0.51) and B. subtilis (0.16) and higher than that of E. coli (0.005) (Chen and Li Citation2005a).
The VR parameter reflects the effect of sampling stress on the integrity of cell membranes. The VRs measured using the Nuclepore filter sampler were significantly lower than those for the AGI-30 and BioSampler, reflected in the fact that the cell membranes of MSSA collected via filtration sustained more damage than those collected via impingement. However, because CR and VR are both determined by the ratio of the concentrations obtained from the tested samplers to those obtained from suspensions, the CR and VR values obtained from samples collected by the Nuclepore filter reflect not only the biological efficiency but also the elution efficiency of the filter. The loss of MSSA during the filter elution process may therefore affect CR and VR.
In contrast, the BioSampler is recommended for achieving better preservation of S. aureus viability during aerosol sampling. The swirling motion of the liquid in the BioSampler generates fewer bubbles, which may minimize sampling stress and thereby preserve greater numbers of viable MSSA (Lin et al. Citation2000; Chang and Wang Citation2014). For BioSampler sampling, mineral oil is a non-evaporating liquid that is recommended to be used with the collection fluid for long-term bioaerosol sampling (Lin et al. Citation1999). However, it is worth noting that mineral oil may interfere with our PMA-qPCR assay. Therefore, we only used sterile water as collection fluid in the BioSampler, and the effects of mineral oil on the PMA-qPCR assay need further investigation. The present study is the first to demonstrate the performance of PMA for quantifying airborne S. aureus using VR values. Therefore, it is difficult to compare resistance levels between species due to the sampling stress based on different viability test methods (Chen and Li Citation2005b).
3.5. Effect of Storage Conditions on S. aureus Sampled with the AGI-30 and BioSampler
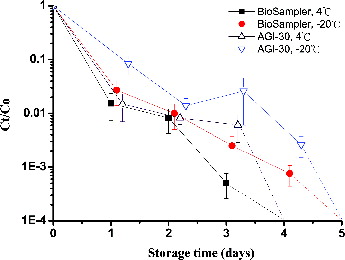
After air sampling, the effects of the storage temperature and storage time on the culturability and viability of MSSA were evaluated for the impinger and the BioSampler. Storage of MSSA in deionized water at chilled (4°C) and freezing (−20°C) temperatures markedly reduced the culturability of MSSA within a 5-day period (). We observed a 2-log decrease in MSSA titers after 2 days in the collection media from the AGI-30 and the BioSampler at both temperatures. The culturability of MSSA in the collection medium was maintained for 3 days, although a large reduction in MSSA titers was observed. To compare the stability of culturable titers and the DNA of MSSA during storage, we used deionized water as the collection medium in the AGI-30 and BioSampler. However, deionized water is not recommended for collecting airborne MSSA if these samples cannot be cultured immediately. Compared with deionized water, PBS and Tween solutions are more likely to preserve the culturability of MSSA at 4°C (Chang and Wang Citation2014). Storing MSSA in deionized water without cryoprotective agents, such as glycerol, may result in mechanical damage from ice crystals. Therefore, decreased culturability of MSSA may be related to damage from air sampling, the formation of intracellular ice crystals, and thawing (Hilton et al. Citation2001). In contrast to MSSA, the culturability of A. baumannii is more stable in the collection fluid during the storage period at 4°C (Tseng et al. Citation2014). These results agree with those from a previous study indicating that titers of A. baumannii cells in suspension are stable (<1 log decrease) for at least 2 weeks at 4°C (Rastogi et al. Citation2007).
FIG. 4. Effects of the storage time on MSSA samples collected from the AGI-30 and BioSampler that were maintained at −20°C or 4°C and evaluated using the culture assay. On the logarithmic y-axis, Ct and C0 represent the CFU concentrations of samples collected simultaneously and stored for t h and 0 h. The dotted line indicates no colony survival for the indicated storage time. The experiments were performed in triplicate, and the data are shown as the mean ± standard error of the mean.
shows the results regarding the stability of MSSA-derived DNA stored at −20°C for up to 28 days, as analyzed via qPCR or PMA-qPCR. When MSSA cells in the AGI-30 collection medium (deionized water) were stored at −20°C and then processed through qPCR or Pre-PMA, the DNA or PMA-bound DNA remained stable (p > 0.05) during the 28-day storage period (). In contrast, when MSSA cells were subjected to Post-PMA treatment and immediately stored at −20°C for durations of up to 28 days, we noted a greater variation in viability (CV% = 53%) compared with Pre-PMA samples (CV% = 15%; ). Similar results were obtained for the BioSampler (), although the log Ct obtained in the Pre-PMA treatment group after 28 days of storage was significantly lower than the log C0 (p < 0.05). The DNA of MSSA collected with the BioSampler and analyzed via qPCR or Pre-PMA at −20°C was stable (p > 0.05), with over 75% of the DNA being maintained for at least 3 weeks. However, when MSSA were processed via Post-PMA, the variation in viability was greater (CV% = 59%) than in Pre-PMA (CV% = 29%).
FIG. 5. Effects of the storage time on MSSA collected with the (a) AGI-30 and (b) BioSampler and stored at −20°C, measured via qPCR and PMA-qPCR. The PMA-qPCR data are divided into Pre-PMA and Post-PMA groups, which are described in the Methods section. On the logarithmic y-axis, Ct and C0 represent the cell concentrations in samples collected simultaneously and stored for t h and 0 h. The experiments were performed in triplicate, and the data are shown as the mean ± standard error of the mean.
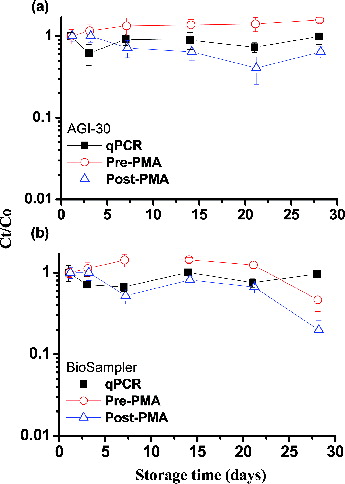
Our results indicated that both PMA-bound and unbound DNA extracted from MSSA collected with either the AGI-30 or BioSampler remained stable for up to 3 weeks when stored at −20°C. For the Post-PMA assay at −20°C, variations in DNA stability became apparent as the storage time increased (). When directly storing frozen MSSA cells without cryoprotective agents, the formation of intracellular ice crystals may cause damage to cell membranes and lead to cell death (McGann Citation1978). PMA is expected to penetrate dead cells with broken membranes when these samples are frozen. Therefore, we recommend that airborne MSSA samples collected using an impingement method with deionized water be cultured as soon as possible. If investigators cannot take samples through the full PMA-qPCR process within 24 h, airborne MSSA samples collected using impingement methods should, at a minimum, be processed immediately for PMA staining and DNA extraction. The subsequent process of qPCR quantification can then be conducted within 3 weeks. In contrast to MSSA, when the A. baumannii cells were processed by Post-PMA and frozen at −20°C, viability rapidly decreased by more than 3 logs after 7 days (Tseng et al. Citation2014). This phenomenon may be related to the fact that gram-negative bacteria are more susceptible to freezing injury than gram-positive species.
3.6. Concentrations of Airborne S. aureus in the ICU and GYM
During the sampling procedures, the mean ambient RH was 61% in the ICU and 67% in the GYM. The mean culturable concentration of total bacterial determined using the BioSampler was 365 CFU/m3 in the ICU and 916 CFU/m3 in the GYM. Overall, there was no significant difference in the total, viable, and culturable concentrations of S. aureus between the two locations (p > 0.05; ). However, the total concentrations of S. aureus were significantly higher than those of viable and culturable cells in both the ICU and GYM samples (p < 0.01). The viable concentrations of S. aureus were nearly 17 times higher than those of culturable cells, but this difference was not statistically significant in either the ICU or the GYM (p = 0.97).
FIG. 6. Total, viable, and culturable airborne S. aureus concentrations detected in samples from the ICU and GYM. *p < 0.05 compared with the respective groups. The experiments were performed in triplicate, and the data are shown as the mean ± standard error of the mean.
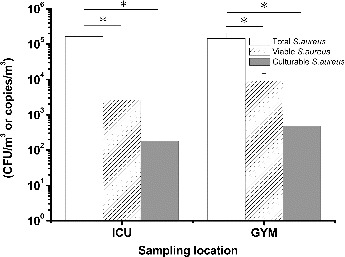
The mean concentrations of culturable S. aureus that we found in the ICU (180 CFU/m3) and the GYM (476 CFU/m3) were higher than those reported in hospital outpatient halls (10 CFU/m3; Hsiao et al. Citation2012) and residential environments in Texas (15 CFU/m3; Gandara et al. Citation2006). However, the culturable concentrations were similar to those from residential environments in Philadelphia (141 CFU/m3), as were the mean percentages of culturable S. aureus within the total culturable bacterial concentrations (51% vs. 69%) (Perez et al. Citation2011). A previous report suggested that S. aureus could be dispersed by humans (Thompson et al. Citation2014), and the concentration of this bacterium can vary depending on the type of house sampled, the geographical region, and the sampling method used. Under stressful conditions, S. aureus can enter the VBNC state and can be responsible for recurrent symptoms (Pasquaroli et al. Citation2013). The low viability of airborne S. aureus found in the ICU (1.5%) and GYM (6.2%) may be related to cleaning and disinfecting procedures implemented at these two sites. Although S. aureus has been reported be infectious at the relatively high dose of 105 organisms (Schmid-Hempel and Frank Citation2007), the viable S. aureus found in the ICU could still pose a health risk to seriously ill patients via inhalation (Shiomori et al. Citation2002).
PMA-qPCR may be useful in addressing the viability of bioaerosols; however, it still has some limitations. First, several parameters may affect the efficiency of PMA-qPCR. In addition to the parameters mentioned in the discussion, the length of the PCR amplicon, the sequence of DNA, pH, turbidity, and salt concentration may also affect the performance of PMA-qPCR (Fittipaldi et al. Citation2012). All factors require further examination in terms of the suppression of dead cell signals when PMA-qPCR is applied to detect different types of bioaerosols. Second, the detection limit of impingement combined with PMA-qPCR to detect S. aureus was relatively high in our current study. Consequently, the PMA-qPCR assay is recommended for use in GYMs or hospital environments such as ICUs or long-term health care units, where relatively high concentrations of S. aureus may be present in the air. Finally, another disadvantage of PMA-qPCR is that the method might only provide a semi-quantitative detection of the microbe species because PMA-qPCR might not detect the true fraction of viable cells. The mechanism of PMA-qPCR for identifying the microbial viability is based on membrane integrity. However, bacterial cells might already be dead before the damage to the membrane integrity can be detected (Fittipaldi et al. Citation2012). Before making definite conclusions on the viability of bioaerosols, multiple methods involving different mechanisms for identifying microbial viability should be assessed and compared.
4. CONCLUSIONS
The widespread use of antibiotics has resulted in an increased number of antibiotic-resistant bacterial strains, such as MRSA. The present study demonstrated that a PMA-qPCR assay was able to rapidly detect viable, airborne S. aureus. Because S. aureus in the VBNC state may be induced by environmental pressures and can be responsible for recurrent symptoms, there is an immediate need for air-monitoring programs to promote early awareness and prevention of viable, airborne S. aureus. However, the conditions in our chamber study might not be as complex as those found in field environments. Inhibitory effects on PMA-qPCR may occur when sampling is conducted in an environment with a variety of organic compounds. The application of PMA-qPCR to distinguish between viable and dead S. aureus cells may be performed within 90 min of sampling. However, parameters that affect the performance of the PMA-qPCR assay must be optimized when applying PMA-qPCR to detect bioaerosols.
FUNDING
This work was supported by grant MOST 103-2314-B-320-003 from the Ministry of Science and Technology, Republic of China, and grant TCMRC-P-101011-01 from Tzu Chi University, Hualien, Taiwan.
REFERENCES
- Agnoletti, F., Mazzolini, E., Bacchin, C., Bano, L., Berto, G., Rigoli, R., et al. (2014). First Reporting of Methicillin-resistant Staphylococcus aureus (MRSA) st398 in an Industrial Rabbit Holding and in Farm-Related People. Vet. Microbiol., 170:172–177.
- Berrada, H., Soriano, J. M., Manes, J., and Pico, Y. (2006). Real-Time Quantitative PCR of Staphylococcus aureus and Application in Restaurant Meals. J. Food Prot., 69:106–111.
- Cattoir, V., Merabet, L., Djibo, N., Rioux, C., Legrand, P., Girou, E., et al. (2011). Clinical Impact of a Real-Time PCR Assay for Rapid Identification of Staphylococcus aureus and Determination of Methicillin Resistance from Positive Blood Cultures. Clin. Microbiol. Infect., 17:425–431.
- Chang, C. W., and Chou, F. C. (2011a). Assessment of Bioaerosol Sampling Techniques for Viable Legionella Pneumophila by Ethidium Monoazide Quantitative PCR. Aerosol Sci. Technol., 45:343–351.
- Chang, C. W., and Chou, F. C. (2011b). Methodologies for Quantifying Culturable, Viable, and Total Legionella pneumophila in Indoor Air. Indoor Air, 21:291–299.
- Chang, C. W., and Wang, L. J. (2014). Methods for Quantifying Staphylococcus aureus in Indoor Air. Indoor Air. doi:10.1111/ina.12124.
- Chen, N. T., and Chang, C. W. (2010). Rapid Quantification of Viable Legionellae in Water and Biofilm Using Ethidium Monoazide Coupled with Real-Time Quantitative PCR. J. Appl. Microbiol., 109:623–634.
- Chen, P. S., and Li, C. S. (2005a). Sampling Performance for Bioaerosols by Flow Cytometry with Fluorochrome. Aerosol Sci. Technol., 39:231–237.
- Chen, P. S., and Li, C. S. (2005b). Bioaerosol Characterization by Flow Cytometry with Fluorochrome. J. Environ. Monit., 7:950–959.
- Chen, Y., Zhao, W., Liu, H., Song, Q., Wang, Y., Zhao, J., et al. (2014). Occurrence of Nosocomial Methicillin-Resistant Staphylococcus aureus as a Marker for Transmission in a Surgical Intensive Care Unit in China. Am. J. Infect. Control, 42:436–438.
- Cookson, B., Peters, B., Webster, M., Phillips, I., Rahman, M., and Noble, W. (1989). Staff Carriage of Epidemic Methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol., 27:1471–1476.
- Cooper, B. S., Stone, S. P., Kibbler, C. C., Cookson, B. D., Roberts, J. A., Medley, G. F., et al. (2004). Isolation Measures in the Hospital Management of Methicillin Resistant Staphylococcus aureus (MRSA): Systematic Review of the Literature. BMJ, 329:533–541.
- Creamer, E., Shore, A. C., Deasy, E. C., Galvin, S., Dolan, A., Walley, N., et al. (2014). Air and Surface Contamination Patterns of Meticillin-resistant Staphylococcus aureus on Eight Acute Hospital Wards. J. Hosp. Infect., 86:201–208.
- Fang, H., and Hedin, G. (2003). Rapid Screening and Identification of Methicillin-Resistant Staphylococcus aureus from Clinical Samples by Selective-Broth and Real-Time PCR Assay. J. Clin. Microbiol., 41:2894–2899.
- Fittipaldi, M., Nocker, A., and Codony, F. (2012). Progress in Understanding Preferential Detection of Live Cells Using Viability Dyes in Combination with DNA Amplification. J. Microbiol. Methods, 91:276–289.
- Francis, J. S., Doherty, M. C., Lopatin, U., Johnston, C. P., Sinha, G., Ross, T., et al. (2005). Severe Community-onset Pneumonia in Healthy Adults Caused by Methicillin-resistant Staphylococcus aureus Carrying the Panton-Valentine Leukocidin Genes. Clin. Infect. Dis., 40:100–107.
- Friese, A., Schulz, J., Hoehle, L., Fetsch, A., Tenhagen, B. A., Hartung, J., et al. (2012). Occurrence of Mrsa in Air and Housing Environment of Pig Barns. Vet. Microbiol., 158:129–135.
- Gandara, A., Mota, L. C., Flores, C., Perez, H. R., Green, C. F., and Gibbs, S. G. (2006). Isolation of Staphylococcus aureus and Antibiotic-Resistant Staphylococcus aureus from Residential Indoor Bioaerosols. Environ. Health Perspect., 114:1859–1864.
- Gehanno, J. F., Louvel, A., Nouvellon, M., Caillard, J. F., and Pestel-Caron, M. (2009). Aerial Dispersal of Methicillin-Resistant Staphylococcus aureus in Hospital Rooms by Infected or Colonised Patients. J. Hosp. Infect., 71:256–262.
- Hilton, C. L., Mackey, B. M., Hargreaves, A. J., and Forsythe, S. J. (2001). The Recovery of Arcobacter butzleri nctc 12481 from Various Temperature Treatments. J. Appl. Microbiol., 91:929–932.
- Ho, M., McDonald, L. C., Lauderdale, T. L., Yeh, L. L., Chen, P. C., and Shiau, Y. R. (1999). Surveillance of Antibiotic Resistance in Taiwan, 1998. J. Microbiol. Immunol. Infect., 32:239–249.
- Hospodsky, D., Yamamoto, N., and Peccia, J. (2010). Accuracy, Precision, and Method Detection Limits of Quantitative PCR for Airborne Bacteria and Fungi. Appl. Environ. Microbiol., 76:7004–7012.
- Hsiao, P. K., Chen, W. T., Chang, K. C., Ke, Y. J., Kuo, C. L., and Tseng, C. C. (2012). Performance of CHROMagar Staph aureus and CHROMagar MRSA for Detection of Airborne Methicillin-resistant and Methicillin-sensitive Staphylococcus aureus. Aerosol Sci. Technol., 46:297–308.
- Hsiao, P. K., Cheng, C. C., Chang, K. C., Yiin, L. M., Hsieh, C. J., and Tseng, C. C. (2014). Performance of CHROMagar VRE Medium for the Detection of Airborne Vancomycin-resistant/sensitive Enterococcus Species. Aerosol Sci. Technol., 48:173–183.
- Hulten, K. G., Kaplan, S. L., Lamberth, L. B., Slimp, K., Hammerman, W. A., Carrillo-Marquez, M., et al. (2010). Hospital-Acquired Staphylococcus aureus Infections at Texas Children's Hospital, 2001–2007. Infect. Control Hosp. Epidemiol., 31:183–190.
- Kobayashi, H., Oethinger, M., Tuohy, M. J., Hall, G. S., and Bauer, T. W. (2009a). Unsuitable Distinction between Viable and Dead Staphylococcus aureus and Staphylococcus epidermidis by Ethidium Bromide Monoazide. Lett. Appl. Microbiol., 48:633–638.
- Kobayashi, H., Oethinger, M., Tuohy, M. J., Hall, G. S., and Bauer, T. W. (2009b). Improving Clinical Significance of PCR: Use of Propidium Monoazide to Distinguish Viable from Dead Staphylococcus aureus and Staphylococcus epidermidis. J. Orthop. Res., 27:1243–1247.
- Kobayashi, H., Oethinger, M., Tuohy, M. J., Hall, G. S., and Bauer, T. W. (2010). Distinction between Intact and Antibiotic-inactivated Bacteria by Real-Time PCR after Treatment with Propidium Monoazide. J. Orthop. Res., 28:1245–1251.
- Li, C. S., and Lin, Y. C. (2001). Storage Effects on Bacterial Concentration: Determination of Impinger and Filter Samples. Sci. Total Environ., 278:231–237.
- Liang, Y., Wu, Y., Sun, K., Chen, Q., Shen, F., Zhang, J., et al. (2012). Rapid Inactivation of Biological Species in the Air Using Atmospheric Pressure Nonthermal Plasma. Environ. Sci. Technol., 46:3360–3368.
- Lin, W. H., and Li, C. S. (1999). Collection Efficiency and Culturability of Impingement into a Liquid for Bioaerosols of Fungal Spores and Yeast Cells. Aerosol Sci. Technol., 30:109–118.
- Lin, X., Reponen, T. A., Willeke, K., Grinshpun, S. A., Foarde, K. K., and Ensor, D. S. (1999). Long-term Sampling of Airborne Bacteria and Fungi into a Non-Evaporating Liquid - Measurement of Effective Airflow Rate and Collection Efficiency. Atmos. Environ., 33:4291–4298.
- Lin, X. J., Reponen, T., Willeke, K., Wang, Z., Grinshpun, S. A., and Trunov, M. (2000). Survival of Airborne Microorganisms During Swirling Aerosol Collection. Aerosol Sci. Technol., 32:184–196.
- Liu, Y., and Mustapha, A. (2014). Detection of Viable Escherichia coli O157:H7 in Ground Beef by Propidium Monoazide Real-Time PCR. Int. J. Food Microbiol., 170:48–54.
- Macher, J. (1999). Bioaerosols, Assessment and Control, American Conference of Governmental Industrial Hygienists, Cincinnati, OH.
- Markley, J. D., Edmond, M. B., Major, Y., Bearman, G., and Stevens, M. P. (2012). Are GYM Surfaces Reservoirs for Staphylococcus aureus? A Point Prevalence Survey. Am. J. Infect. Control, 40:1008–1009.
- Masters, C. I., Miles, C. A., and Mackey, B. M. (1998). Survival and Biological Activity of Heat Damaged DNA. Lett. Appl. Microbiol., 27:279–282.
- McGann, L. E. (1978). Differing Actions of Penetrating and Nonpenetrating Cryoprotective Agents. Cryobiology, 15:382–390.
- Miotto, P., Bigoni, S., Migliori, G. B., Matteelli, A., and Cirillo, D. M. (2012). Early Tuberculosis Treatment Monitoring by Xpert(R) Mtb/Rif. Eur. Respir. J., 39:1269–1271.
- Nkuipou-Kenfack, E., Engel, H., Fakih, S., and Nocker, A. (2013). Improving Efficiency of Viability-PCR for Selective Detection of Live Cells. J. Microbiol. Methods, 93:20–24.
- Nocker, A., and Camper, A. K. (2009). Novel Approaches toward Preferential Detection of Viable Cells Using Nucleic Acid Amplification Techniques. FEMS Microbiol. Lett., 291:137–142.
- Nocker, A., Cheung, C. Y., and Camper, A. K. (2006). Comparison of Propidium Monoazide with Ethidium Monoazide for Differentiation of Live Vs. Dead Bacteria by Selective Removal of DNA from Dead Cells. J. Microbiol. Methods, 67:310–320.
- Nocker, A., Sossa, K. E., and Camper, A. K. (2007). Molecular Monitoring of Disinfection Efficiency Using Propidium Monoazide in Combination with Quantitative PCR. J. Microbiol. Methods, 70:252–260.
- Parshionikar, S., Laseke, I., and Fout, G. S. (2010). Use of Propidium Monoazide in Reverse Transcriptase PCR to Distinguish between Infectious and Noninfectious Enteric Viruses in Water Samples. Appl. Environ. Microbiol., 76:4318–4326.
- Pasquaroli, S., Zandri, G., Vignaroli, C., Vuotto, C., Donelli, G., and Biavasco, F. (2013). Antibiotic Pressure Can Induce the Viable but Non-Culturable State in Staphylococcus aureus Growing in Biofilms. J. Antimicrob. Chemother., 68:1812–1817.
- Perez, H. R., Johnson, R., Gurian, P. L., Gibbs, S. G., Taylor, J., and Burstyn, I. (2011). Isolation of Airborne Oxacillin-Resistant Staphylococcus aureus from Culturable Air Samples of Urban Residences. J. Occup. Environ. Hyg., 8:80–85.
- Rastogi, V. K., Wallace, L., and Smith, L. S. (2007). Disinfection of Acinetobacter baumannii-contaminated Surfaces Relevant to Medical Treatment Facilities with Ultraviolet C Light. Mil. Med., 172:1166–1169.
- Sacar, S., Sayin Kutlu, S., Turgut, H., Cevahir, N., Hircin Cenger, D., and Tekin, K. (2010). Epidemiology and Associated Factors for Nosocomial Methicillin-Resistant Staphylococcus aureus Infection in a Tertiary-Care Hospital. Epidemiol. Infect., 138:697–701.
- Schmid-Hempel, P., and Frank, S. A. (2007). Pathogenesis, Virulence, and Infective Dose. PLoS Pathog., 3:1372–1373.
- Schulz, J., Friese, A., Klees, S., Tenhagen, B. A., Fetsch, A., Rosler, U., et al. (2012). Longitudinal Study of the Contamination of Air and of Soil Surfaces in the Vicinity of Pig Barns by Livestock-Associated Methicillin-resistant Staphylococcus aureus. Appl. Environ. Microbiol., 78:5666–5671.
- Sherertz, R. J., Reagan, D. R., Hampton, K. D., Roberston, K. L., Thomas, R., and Gwaltney, J. M. (1996). A Cloud Adult: The Staphylococcus aureus-Virus Interaction Revisted. Ann. Intern. Med., 124:539–547.
- Shiomori, T., Miyamoto, H., Makishima, K., Yoshida, M., Fujiyoshi, T., Udaka, T., et al. (2002). Evaluation of Bedmaking-Related Airborne and Surface Methicillin-Resistant Staphylococcus aureus Contamination. J. Hosp. Infect., 50:30–35.
- Thompson, K. A., Copley, V. R., Parks, S., Walker, J. T., and Bennett, A. M. (2014). Staphylococcus aureus Dispersal from Healthy Volunteers. Am. J. Infect. Control, 42:260–264.
- Thorne, P. S., Kiekhaefer, M. S., Whitten, P., and Donham, K. J. (1992). Comparison of Bioaerosol Sampling Methods in Barns Housing Swine. Appl. Environ. Microbiol., 58:2543–2551.
- Tseng, C. C., Hsiao, P. K., Chang, K. C., Cheng, C. C., Yiin, L. M., and Hsieh, C. J. (2014). Detection of Viable Antibiotic-Resistant/Sensitive Acinetobacter baumannii in Indoor Air by Propidium Monoazide Quantitative Polymerase Chain Reaction. Indoor Air. doi: 10.1111/ina.12165.
- Tseng, C. C., and Li, C. S. (2005). Collection Efficiencies of Aerosol Samplers for Virus-containing Aerosols. J. Aerosol Sci., 36:593–607.
- Wilson, R. D., Huang, S. J., and Mclean, A. S. (2004). The Correlation between Airborne Methicillin-Resistant Staphylococcus aureus with the Presence of MRSA Colonized Patients in a General Intensive Care Unit. Anaesth. Intensive Care, 32:202–209.