
Abstract
A particle-into-liquid sampler coupled with ion chromatograph (PILS-IC) for the on-line measurement of inorganic ions has been modified by the insertion of two ion-exchange pre-concentration cartridges that enrich the sample during the period of the IC analysis. The limits of detection of the modified instrument were 10-15 times lower and the time coverage 24 times higher (from 2 to 48 min per hour) than those of the original PILS-IC setup. The instrumental performance in terms of recovery and break-through volume from the cartridges was satisfactory. The modified PILS-IC was operated in comparison with a diffusion denuder line and with a high-resolution time-of-flight aerosol mass spectrometer (HR-TOF-AMS) during a short intensive measurement period organized in the framework of the European Monitoring and Evaluation Programme (EMEP), a co-operative program for monitoring and evaluation of the long-range transmission of the air pollutants in Europe. The instrument showed a quantitative response in agreement with the results of the diffusion lines, and an ability to trace fine concentration variations not so different from the performance of the much more complex HR-TOF-AMS. From the time patterns of the ion concentrations measured by the modified PILS-IC, it was possible to obtain useful information about the variations in the air quality and in the strength of the particulate matter sources.
Copyright 2015 American Association for Aerosol Research
1. INTRODUCTION
Measuring the chemical composition of atmospheric particulate matter (PM) at high time resolution is an exciting challenge in atmospheric pollution research. Time changes in PM concentration and composition depend on the variations in the pollutant mass amount released in the atmosphere as well as in the air volume available for its dispersion (mixing properties of the atmosphere). These variations are generally fast (minutes/hours) and cannot be caught by the traditional PM measurements, carried out on a 24-h sampling basis. Particularly in the case of sporadic events, such as long-range transport from neighboring countries or local occasional emissions, time-integrated measurements may lead to a loss of information about the behavior of individual PM sources. The identification of these events, in fact, requires a synchronization of PM composition data with meteorological data (mainly wind direction and velocity), that are easily available at high time frequency (Wittig et al. Citation2004; Wu and Wang Citation2006; Lee et al. Citation2008; Gao et al. Citation2011).
For a time-resolved determination of PM components, it is generally possible to sample on filter media; by using this technique, the time resolution can reach 2–3 h in moderately polluted atmospheres (Perrino et al. Citation2010). However, this method is time-consuming and expensive; in addition, significant analytical uncertainties due to filter blanks may be introduced, particularly in the case of low concentrations.
As regards the determination of soluble inorganic ions, an essential issue in studies about secondary pollution, the use of filters also favors the occurrence of sampling artifacts due to the interaction between gaseous and particulate species (e.g., nitric acid and sodium chloride) and to the volatilization of semi-volatile species (ammonium nitrate and ammonium chloride; Tsai and Perng Citation1998; Chow et al. Citation2005; Yu et al. Citation2006; Vecchi et al. Citation2009). These sampling artifacts can be efficiently reduced by using diffusion lines, that is, a number of diffusion denuders and a filter pack set in series, but the use of this technique increases laboratory work and analytical costs even more (Allegrini et al. Citation1994; Perrino, Ramirez et al. Citation2001).
During the last decade, several devices aimed to continuously collect and analyze atmospheric particles on a short time-scale (from seconds to few minutes) have been developed. These techniques do not involve sampling on filters and are thus free from the above sampling artifacts. To name some, the steam-jet aerosol collector (SJAC; Khlystov et al. Citation1995), the aerosol mass spectrometer (AMS; Jayne et al. Citation2000; Allan et al. Citation2003), the flash volatilization nitrate monitor (R&P8400N; Stolzenburg and Hering Citation2000), the ambient ion monitor-ion chromatograph (AIM-IC; Markovic et al. Citation2012), and the particle-into-liquid sampler coupled with ion chromatograph (PILS-IC; Weber et al. Citation2001).
PILS-IC is one of the most used devices for online measurement of water-soluble inorganic ions in PM. Briefly, the air flow is denuded of gaseous species and aerosol particles are grown in a saturated water vapor chamber to form droplets that are collected by inertial impact on a collection plate. The plate is then washed by deionized water and the resulting solution is directly analyzed for anions and cations by two ion chromatographs, allowing the simultaneous analysis of Na+,NH4+, K+, Mg2+, Ca2+, Cl−,NO2−, NO3− and SO42−, in about 15 min (Weber et al. Citation2001; Orsini et al. Citation2003).
The analytical performances of the PILS-IC in its original setup are affected by two main limitations. First, the dilution of the sample by the washing solution leads to high detection limits, which make the system unsuitable for low-polluted areas (Wang et al. Citation2013). Most important, the time coverage is low (about 2–5 min per hour), as only the sample amount contained inside the loop at the time of the injection is analyzed, while the sample amount collected during the time of the analysis is discarded. Many attempts have been made to improve the performance of the original PILS setup in terms of time coverage and sensitivity, by acting on the PILS flow rates, the time duration of the chromatographic analysis (length of the column and flow rate of the mobile phase), and the capacity of the IC sampling loop (Timonen et al. Citation2010 and cited therein). However, all these possibilities have limitations and influence each other (e.g., an increase in the sample loop size would result in an increase in sensitivity but in a broadening of the chromatographic peaks, a reduction in the chromatogram length would increase the time coverage but it would decrease the separation).
A different possibility is constituted by the use of sample pre-concentration systems, which has been up to now applied to different devices (Simon and Dasgupta Citation1995; Markovic et al. Citation2012).
We report here the optimization and the validation of a PILS-IC integrated with an automated pre-concentration system (pcPILS-IC) for the semi-continuous analysis of the inorganic ions in PM. In order to decrease the detection limits and increase the temporal coverage of the acquired data, the original instrument has been modified by adding two ion-exchange cartridges enriching the sample during the period of the IC analysis. The optimized system has been then used in the field during a short intensive measurement period organized in the framework of the European Monitoring and Evaluation Programme (EMEP; a co-operative program for monitoring and evaluation of the long-range transmission of the air pollutants in Europe) and conducted at a peri-urban site near Rome, Italy (Montelibretti, EMEP site IT01). In order to evaluate the analytical reliability of the modified PILS-IC, the results were compared with those obtained by two additional methods: a high-resolution time-of-flight aerosol mass spectrometer (HR-TOF-AMS) and a denuder-filter pack assembly (diffusion line), working at 5-min and 3-h time resolution, respectively.
2. EXPERIMENTAL SECTION
2.1. PILS-IC System
The schematic assembly of the PILS-IC used in this study is reported in the online supplemental information (SI; Figure S1). Ambient air was sampled through a polyethylene cyclone with 2.5 μm cut-off at the flow rate of 15 L min−1. In order to avoid the interference of gaseous species (HCl, HNO2, HNO3, SO2, NH3), two annular diffusion denuders were inserted in the sampling line, downstream of the cyclone. The denuders, made of pyrex glass, were 200 mm in length, and 33 and 30 mm in outside and inside diameter, respectively. The first denuder, for the removal of acid gases, was coated with 1% Na2CO3+ 2% glycerine in ethanol-water solution (50:50); the second one, for the removal of ammonia, was coated with 1% H3PO4 in the same solution. At the operative flow rate, the removal efficiency of these annular denuder is better than 95% (Allegrini et al. Citation1987; Perrino et al. Citation1990; Allegrini et al. Citation1994). Loading capacity of each denuder is above 1 mg, so they could be substituted every three days (Perrino and Gherardi Citation1999). Downstream of the denuder, the air sample entered the PILS (Metrohm AG, Herisau, Switzerland). In a condensation growth chamber, atmospheric particles were mixed with a supersaturated steam and grew into droplets large enough to be collected by an impactor. A water solution was introduced at the top of the impactor plate and transported the droplets down to its base, then through a system able to remove the air bubbles from the liquid stream and finally to the sampling loop of the IC system. The water solution was spiked with 50 μg L−1 of LiBr (© Merck KGaA, Darmstadt, Germany) used as internal standard. All the solutions entering and exiting the PILS were managed by a single eight-channel peristaltic pump (model 205S, Watson-Marlow Inc., Wilmington, MA, USA) run at a speed of 24 RPM; the operative flow rates are reported in the SI (Table S1).
Cations were analyzed by a Dionex DX-120 equipped with an ION-PAC CS12A column; anions were determined by a Dionex ICS-1000 equipped with an ION-PAC AS12A column (Thermo Fisher Scientific Inc. Waltham, MA, USA). The eluent phases were methanesulfonic acid 0.1 M for cations and Na2CO3/NaHCO3 0.5 M/0.5 M for anions.
2.2. Pre-Concentration System
The connection between PILS and ICs is usually performed by two six-way valves complemented with two capillaries (sample injection loops). In this work (), the loops were replaced with two cartridges packed with ion-exchange resins (ION-PAC CG12A for cations and ION-PAC AG12A for anions, Thermo Fisher Scientific Inc., Waltham, MA, USA). During the load phase (configuration as in a), the ions collected by the PILS were retained in the cartridges; during the inject phase (configuration as in b), the ions were eluted, in back-flush, and directly transported to the chromatographic column. The solutions used to elute the cartridges were the same used for the IC analysis. The automatic positioning of the valves in the load and inject configurations was managed by the Chromeleon 7.2 Chromatography Software (Thermo Scientific™ Dionex™ Chromeleon™ 7.2 Chromatography Data System).
FIG. 1. Schematic diagram of the six-way valve and of the cartridge used for sample pre-concentration and injection.
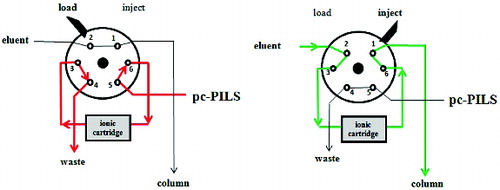
The introduction of the pre-concentration cartridges made it possible to extend the load phase to include also the time duration of the analysis, so as to collect a significant sample amount that in the original setup of the PILS-IC would have been lost. In this configuration the sample is lost only during the inject phase, when the solution from the PILS is directed to the IC drains.
The load time was set to 12 min, which was the minimum time needed to acquire a complete chromatogram for both anions and cations. The inject time was evaluated by connecting the loaded cartridges directly to the IC detector and by measuring the elution time of the resulting single broad peak. A quantitative (≥98%) elution of the ionic species from the cartridge was obtained after 3 min. In summary, the addition of the described pre-concentration system led to a significant gain in the measurement time coverage, which increased from 2 min h–1 of the traditional PILS-IC (considering a sampling loop of 150 μL and a loading flow rate of 0.3 mL min−1) to 48 min h–1 of the pcPILS-IC.
In order to obtain a comparable time coverage without using a pre-concentration cartridge, it would be possible to increase the loop volume, and/or to decrease the inlet flow rate to the IC, and/or to reduce the duration of the chromatographic run. In all these cases, however, a small improvement in the sensitivity would be obtained. A further improvement of the time coverage and sensitivity of the system could be achieved by decreasing the total flow rate of the PILS, but this might be responsible for a decrease in the overall sampling efficiency of the system.
2.3. Sampling Site and Additional Instrumentations
The field performance of the pcPILS-IC was evaluated during one of the intensive measurement periods organized by the EMEP Task Force on Measurement and Modelling (http://www.nilu.no/projects/ccc/tfmm/index.html). Measurements were carried out from 30 January to 10 February 2013 at the facilities of C.N.R. Institute of Atmospheric Pollution Research in Montelibretti, a peri-urban area about 25 km from Rome (EMEP site IT01, 42°06′13.2′′N, 12°37′48.0′′E, 48 m a.s.l.).
Ancillary measurements included the main meteorological parameters (wind speed and direction, temperature, atmospheric pressure, relative humidity, rain amount) and the natural radioactivity due to the short-life beta-decay products of Radon, determined by means of an automated monitor operating over a 1-h time basis (PBL Mixing Monitor, FAI Instruments, Fonte Nuova, Rome, Italy). Natural radioactivity has been proved to be a reliable good proxy of the mixing properties of the lower boundary layer and a valuable tool for the interpretation of pollution events (Perrino, Pietrodangelo et al. Citation2001; Vecchi et al. Citation2004; Perrino et al. Citation2009; Canepari et al. Citation2014).
In addition to pcPILS-IC, the measurement of inorganic ions was carried out by two other systems working at high temporal resolution: a high-resolution time-of-flight aerosol mass spectrometer (HR-TOF-AMS; De Carlo et al. Citation2006) and a diffusion line (diffusion denuders-filter pack assembly; Perrino et al. Citation1990; Perrino and Gherardi 1999; Perrino, Ramirez et al. Citation2001).
The HR-TOF-AMS (Aerodyne Research Inc., Billerica, MA, USA) provides size-resolved chemical analysis of the non-refractory species (at 600°C, operating temperature of the vaporizer) in submicron particles (sulfate, nitrate, ammonium, non-sea-salt chloride and organics). The concentrations reported in this work were acquired by the HR-TOF-AMS, operating in “V” ion path modes (higher sensitivity, lower mass resolution), every 5 min. The characterization of the chemical speciation and of the size distribution were coupled by analyzing the particle time-of-flight (Allan et al. Citation2003). The AMS has a default collection efficiency of about 50% due to the transmission characteristics of the standard aerodynamic lens (Liu et al. Citation2007). The experimental collection efficiency was estimated using the denuder results and applying the procedure described by Middlebrook et al. (Citation2012). All data were analyzed using the standard AMS software SQUIRREL v1.53 within Igor Pro 6.2 (WaveMetrics Inc., Lake Oswego, OR, USA). The concentrations obtained by the AMS were corrected for the experimental collection efficiency and averaged over the pcPILS-IC sampling periods.
Diffusion lines have been used at the EMEP station of Montelibretti since 1994 (http://www.nilu.no/projects/ccc/index.html). They are comprised of five annular denuders set in series: two coated with NaF (0.05% in ethanol–water solution 80:20), two with Na2CO3 and glycerol (1% + 1% in ethanol–water solution 50:50), and one with H3PO3 (1% in ethanol–water solution 80:20). The denuders are followed by a cyclone (cut size of 2.5 μm at the operative flow rate of 15 L min−1) and by a filter pack comprised of one Teflon filter, one Nylon filter, and one paper filter impregnated with the same H3PO3 solution used to coat the fifth denuder. This sampling line allows the determination of hydrochloric acid, nitric acid, nitrous acid, sulfur dioxide, and ammonia in the gaseous phase, and chloride, nitrite, nitrate, sulfate, ammonium, sodium, calcium, potassium, and magnesium in the particulate phase (coarse and fine fractions). In particular, the configuration of the filter-pack is devised in order to take into account nitric acid, hydrochloric acid, and ammonia possibly evolved from ammonium nitrate and ammonium chloride particles retained on the Teflon filter (HCl and HNO3 are retained by the nylon filter, NH3 is trapped by impregnated filter). The removal of the gaseous species on the denuders allows a correct discrimination between the gaseous and the particulate phases. Details about the rationale for the choice of the denuder coatings are reported in Perrino et al. (Citation1990). Diffusion lines are generally operated for 24 h; for this inter-comparison with pcPILS-IC and HR-TOF-AMS during the first three days (30 January–1 February) the sampling duration was decreased to 3 h, while the original 24-h sampling duration was kept for the rest of the period.
3. RESULTS AND DISCUSSION
3.1. Optimization of the pcPILS-IC
The analytical performances of the pre-concentration cartridges were evaluated by performing cartridge breakthrough volume tests, cartridge recovery tests, and calibration of the whole system.
3.1.1. Breakthrough Volumes and Recoveries
To calculate the breakthrough volumes of the pre-concentration cartridges (i.e., the volume of solution above which the ion is no longer retained because of the saturation of the ion-exchange resin) standard solutions of anions (Cl−, NO2−, NO3−, Br−, SO42−, 100 μg L−1) and of cations (Li+, Na+, NH4+,K+, Mg2+, Ca2+, 100 μg L−1) were introduced by means of a peristaltic pump into the anionic and cationic cartridge, respectively. The outgoing solutions were collected in aliquots of 5 mL and analyzed by IC.
The results are reported in . As the cartridges are functionally similar to the columns used for IC analyses, the species are released from the cartridges in the same order as the chromatographic elution. The breakthrough volumes varied from 30 mL (chloride) to 125 mL (bromide); Mg2+, Ca2+, NO3−, and SO42− were still retained in the cartridges after a volume of 250 mL. The loading capacity of the cartridges is satisfactory: considering the operative conditions of the pcPILS-IC and the ion concentration usually found in the atmosphere, saturation phenomena resulting in analyte loss can be excluded for all ions (the loading capacity for Cl−, the less retained species, is 3 μg, corresponding to an atmospheric concentration of 17 μg m–3).
TABLE 1 Analytical performances of the pre-concentration cartridges (break-through volume, capacity, and recovery percentage): limit of detection (LOD) and limit of quantification (LOQ) of the pc-PILS-IC and the PILS-IC (N = 6)
Recovery tests were performed by using the same standard solutions. For each ion, the recovery was calculated as the ratio between the amount loaded into the cartridge and the amount eluted by the cartridge after 3 min, at the flow rate of 1.2 mL min−1 (operative conditions of the system). Percent recoveries are reported in . Values above 95% were obtained for all the considered species, with a repeatability ranging from 2% to 12%.
3.1.2. Calibration of the pcPILS-IC and Calculation of the Atmospheric Concentration
The calibration of the system was carried out by introducing the standard solutions of anions and of cations (see 3.1.1) directly into the six-way valves equipped with the cartridges, by using a peristaltic pump. The amount (μg) of each ion retained by the cartridge (load phase) and then injected into the chromatographic system (inject phase) was calculated by measuring the solution volume eluted from the cartridge. The correlation between the injected amounts and the measured areas was linear for all the considered ions up to a concentration of 100 μg L–1. In all cases, Pearson coefficients were higher than 0.95.
In the field application of the pcPILS-IC, the sample collected by washing the impactor plate is partitioned into five lines: two of them are connected to the injection systems of the ICs while three are directed to the waste (see Figure S1 in the SI). As the partitioning among these five lines cannot be accurately controlled, the sample volumes introduced into the two cartridges during the pre-concentration phase cannot be exactly determined. For this reason, to correctly calculate its air concentrations (Ci), the amount of each ion (Qi) obtained by the chromatogram has to be corrected by using an internal standard (LiBr). The correction factor was obtained as the ratio between the theoretical amount of the internal standard (Li+ for cations, Br− for anions) entering the impact plate during the pre-concentration time (QIS-in; calculated by considering Li+ and Br− concentrations in the washing solution and its flow rate) and the amount of the same standard measured in each chromatographic run (QIS-out). This correction factor also takes into account the sample dilution caused by the water condensing on particles in the PILS growth chamber.
The atmospheric concentration (Ci, μg m–3) was then calculated as:
where V is the air volume (m3) sampled during the pre-concentration time.
It is worth noting that this quantitative procedure introduces some uncertainty due to the progressive deterioration of the peristaltic tube supplying the LiBr solution to the impactor plate. This deterioration originates slight variations in the flow rate and then small differences between the theoretical value of QIS-in (calculated by considering the initial flow rate) and its actual value. In this study, this problem was managed through a close control and frequent substitution of the peristaltic tube. Anyway, it could be more efficiently solved by replacing the peristaltic pump with a more stable and reliable system, such as a syringe pump or a piston pump.
3.1.3. LODs and LOQs
Limit of Detection (LOD) and limit of quantification (LOQ) of the pcPILS-IC were calculated by evaluating the blank values, according to the IUPAC definitions, or the background noise in case the blank values were not detectable (nitrite and nitrate). Operatively, the blank values were obtained by placing a clean filter-pack (see Section 2.3) at the inlet of the system and carrying out the analysis with and without the pre-concentration unit.
LODs and LOQs are reported in . In spite of the broadening of the peaks due to the presence of the pre-concentration cartridge (see Figure S2 in the SI), the values obtained by using the pcPILS-IC were 10–15 times lower than those obtained with the original setup of the PILS-IC. For Na+ and Cl−, the improvement was lower because of the high blank values in the deionized water. It is worth noting that the sensitivity of the pcPILS-IC may be easily tuned by varying the loading time. Considering the breakthrough volumes of the cartridge, the loading time can be increased up to at least 1 h without any significant further broadening of the chromatographic peaks.
3.2. Comparison with Denuder-Filter Pack and HR-TOF-AMS
The results obtained by the pcPILS-IC during the intensive measurement period were compared to the data obtained by a diffusion line and by a HR-TOF-AMS. The good performances of these two techniques have been well documented by comparison with side-by-side online instruments or with off-line filter methods (Lee et al. Citation2008; Markovic et al. Citation2012). To date, however, none of these systems by itself can be considered as a reliable reference method for the field validation of the pcPILS-IC. Diffusion lines are an effective method for the quantitative determination of inorganic ions without the artifacts due to the solid-vapor equilibrium of ammonium salts, but their typical time coverage (24 h) cannot be decreased below 2–3 h because of the long analytical work they require and the non-negligible contribution of the blanks. On the other side, the HR-TOF-AMS allows high time-resolution measurements but its results have to be corrected for the collection efficiency.
In this work, we chose to compare the pcPILS-IC results averaged over 3 and 24 h with the data yielded by the diffusion lines, in order to evaluate the quantitative response of the pcPILS-IC. To evaluate its behavior at high time-frequency (15 min) we used the data yielded by the HR-TOF-AMS, averaged on the same time duration.
The linear regressions between the results obtained by the diffusion lines (24 h and 3 h) and the pcPILS-IC are reported in the SI (Figure S3). A good correlation was obtained for all ions (R2 > 0.8). In all cases the slope was close to 1 and the intercept close to zero, showing a good quantitative agreement between the concentration values measured by the two techniques. Slightly worse results were obtained for chloride and sodium, probably due to the higher blank values of the diffusion denuders, typical of this species, and to a possible release of NaCl used as coating layer in the first two denuders of the diffusion line.
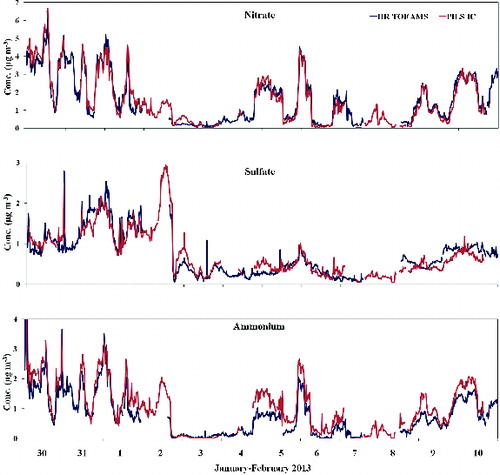
shows the temporal pattern of NH4+, NO3−, and SO4 obtained during the whole intensive measurement period by the pcPILS-IC and the HR-TOF-AMS. For all three ions, the time variability of the measured concentrations were in very good agreement along the whole period (R2 > 0.8), confirming the satisfactory performances of the pcPILS-IC in tracing even fine time variations in the concentration of ions. These results were obtained in spite of the fact that the HR-TOF-AMS detects only non-refractory species, as in the area and period of the study nitrate and sulfate were mainly in the form of ammonium salts.
FIG. 2. Comparison of nitrate, sulfate, and ammonium concentration as measured by the pcPILS-IC and by the HR-TOF-AMS.
3.3. Results of the Monitoring Campaign
The ability of the pcPILS-IC to provide reliable data for the interpretation of rapidly changing atmospheric pollution events is discussed on the basis of the results obtained during the intensive field study (29 January–10 February). This period was characterized by climatic conditions typical of the wintertime in Central Italy, with moderately low temperatures and very variable wind speed and direction.
For an overall characterization of the period, we report the time pattern of natural radioactivity (). This variable allows an overall evaluation of the mixing properties of the lower atmosphere: high values of natural radioactivity indicate a low air volume available for pollutant dispersion, low values indicates efficient atmospheric mixing (by advection or thermal convection) (Farao et al. Citation2014; Perrino et al. Citation2014). Briefly, the typical day-night pattern of natural radioactivity during atmospheric stability periods consists in increasing values from sunset to early morning (night-time stability) and decreasing values starting from sunrise, with a minimum during the warm hours, due to thermal convection. Atmospheric stability lasting for a number of consecutive days is generally responsible for a remarkable increase in the concentration of atmospheric pollutants, particularly of secondary species. During advection phenomena, instead, natural radioactivity remains at constant low values. The occurrence of advection is generally responsible for a fast change in the air quality, which may improve or worsen according to origin of the incoming air masses.
FIG. 3. Time pattern of natural radioactivity during the intensive measurement period.
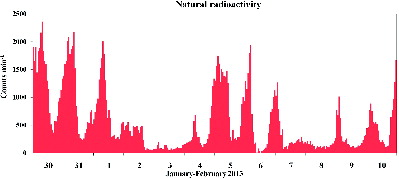
The study period can be ideally divided into three sub-periods. The first one, from the beginning of the study to 1 February, was characterized by fog and very stagnant conditions during the night, and the early and the late morning. During these three days, especially during the first one, unusually high values of natural radioactivity were measured during the late morning (until about noon); as a possible explanation, the presence of fog might have delayed the atmospheric mixing produced by thermal convection. The following sub-period, from the afternoon of 1 February to the afternoon of 4 February, was characterized by advection (constantly low values of natural radioactivity), with wind gust up to 15 m s–1 and daily mean intensity up to 5.8 m s−1 on 2 February. The rest of the study period was generally characterized by night time stability (with the exception of the night between 7 and 8 February, when the wind increased again) and moderate atmospheric mixing during the day hours.
The time patterns of the ion concentrations determined by the pcPILS-IC are reported in and . During the first sub-period, the most noticeable feature was the simultaneous increase of ammonium and nitrate (R2 > 0.86). The concentration of ammonium nitrate followed the pattern of natural radioactivity during the night and was particularly sensitive to the re-stabilization of the atmosphere during the morning hours. During the same days, the results of the pcPILS-IC showed an unusual increase in the concentration of particulate nitrite (up to 0.8 μg m–3), possibly due to the dissolution of nitrous acid, generated during the night, into the fog droplets.
FIG. 4. Time patterns of chloride, nitrite, sodium, magnesium, potassium, and calcium determined by the pcPILS-IC.
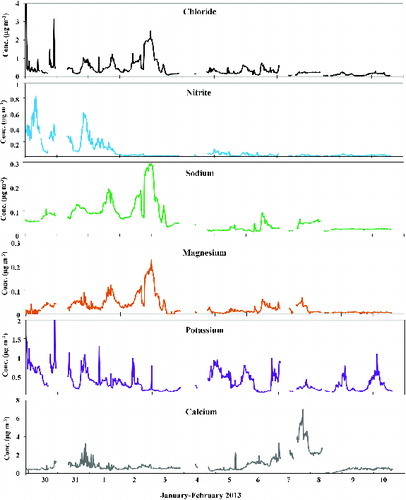
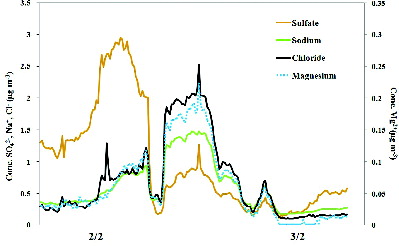
The second period was characterized by the simultaneous increase of sodium, chloride, and magnesium, all tracers of sea-salt. As reported in , which refers to 2 and 3 February, the concentration of the three sea-salt tracers increased between 9 AM and 4 PM of the first day, then abruptly decreased and finally increased again around 6 PM, resulting in high values throughout the night. It is worth noting that the first of the two peaks was also associated with peaks in other ions, particularly by sulfate, while during the second event the concentration of all ions was very low except for the sea-salt tracers. The back trajectories of the air masses, calculated by the HYbrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT) model (accessed via NOAA ARL READY website [http://www.arl.noaa.gov/HYSPLIT.php], and reported in the SI [Figure S4]), show that the air masses followed two different trajectories: the first one, quite constant in direction until about 5 PM, was originated from North Africa and traveled over the southern part of the Tyrrhenian Sea; the second one was originated from the area of France. The different origin of the air masses had a clear impact on the air quality at the receptor, as the air masses coming from North Africa were characterized by a poorer quality than those coming from the North, as already observed during a different field study by Perrino et al. (Citation2010).
FIG. 5. Time pattern of SO42−, Na+, Cl−, and Mg2+ concentration during 2 and 3 February.
During the last part of the period, the time variations in the concentration of ions were driven by the mixing properties of the lower atmosphere. This is particularly true for nitrate, ammonium, and potassium, which closely follows the time pattern of natural radioactivity. In the case of potassium, the agreement with the natural radioactivity pattern indicates that the influence of the local sources was negligible and that diffuse sources were mainly responsible for the time variation of its concentration. As the main winter source of fine potassium is biomass burning, it is reasonable that the residential buildings in the area surrounding the sampling site were responsible for the concentration of this species at the receptor.
4. CONCLUSIONS
The addition of a pre-concentration step to a PILS-IC allowed a remarkable increase of the time coverage of this online technique (from 2 to 48 min per hour). At the operating conditions, the pre-concentration cartridges showed recoveries above 95% for all the ions and values of the breakthrough volumes high enough to exclude saturation phenomena. Detection limits were 10–15 times lower than those obtained by using the original PILS-IC setup.
The modification of the PILS-IC into pc-PILC-IC is very cheap, as it only consists in the substitution of the injection loop of the IC with a commercial pre-concentration cartridge. Further advantages of the system are its flexibility, as its time resolution and sensitivity can be easily tuned by varying the cartridge loading time.
A field inter-comparison with a diffusion line showed that the quantitative response of the pcPILS-IC is satisfactory; the very good agreement with the time pattern of sulfate, nitrate, and ammonium measured by a HR-TOF-AMS demonstrated that the pcPILS-IC is able to trace fine time variations in the concentration of ions.
The use of the modified instrument during a field campaign yielded very reliable results and allowed an accurate interpretation of the air quality variations during the study period.
supplemental_file_for_pubblication.docx
Download MS Word (827.1 KB)ACKNOWLEDGMENTS
S. Dalla Torre, E. Rantica, and T. Sargolini of CNR-IIA are gratefully acknowledged for their valuable experimental work.
REFERENCES
- Allan, J. D., Jimenez, J. L., Williams, P. I., Alfarra, M. R., Bower, K. N., Jayne, J. T., Coe, H., and Worsnop, D. R. (2003). Quantitative Sampling Using an Aerodyne Aerosol Mass Spectrometer 1. Techniques of Data Interpretation and Error Analysis. J. Geophys. Res., 108(D3):4090, doi:10.1029/2002JD002358.
- Allegrini, I., De Santis, F., Di Palo, V., Febo, A., Perrino, C., Possanzini, M., and Liberti, A. (1987). Annular Denuder Method for Sampling Reactive Gases and Aerosols in the Atmosphere. Sci. Total Environ., 67:1–16.
- Allegrini, I., Febo, A., Perrino, C., and Masia, P. (1994). Measurement of Atmospheric Nitric-Acid in Gas-Phase and Nitrate in Particulate Matter by Means of Annular Denuders. Int. J. Environ. Anal. Chem., 54:183–201.
- Canepari, S., Astolfi, M. L., Farao, C., Maretto, M., Frasca, D., Marcoccia, M., and Perrino, C. (2014). Seasonal Variations in the Chemical Composition of Particulate Matter: A Case Study in the Po Valley. Part II: Concentration and Solubility of Micro- and Trace-Elements. Environ. Sci. Pollut. Res., 21:4010–4022.
- Chow, J. C., Watson, J. G., Lowenthal, D. H., and Magliano, K. L. (2005). Loss of PM2.5 Nitrate from Filter Samples in Central California. J. Air Waste Manage., 55:1158–1168.
- De Carlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L. (2006). Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer. Anal. Chem., 78:8281–8289.
- Farao, C., Canepari, S., Perrino, C., and Harrison, R. M. (2014). Sources of PM in an Industrial Area: Comparison between Receptor Model Results and Semiempirical Calculations of Source Contributions. Aerosol Air Qual. Res., 14:1558–1572.
- Gao, X., Yang, L., Cheng, S., Gao, R., Zhou, Y., Xue, L., Shou, Y., Wang, J., Wang, X., Nie, W., Xu, P., and Wang, W. (2011). Semi-Continuous Measurement of Water-Soluble Ions in PM2.5 in Jinan, China: Temporal Variations and Source Apportionments. Atmos. Environ., 45:6048–6056.
- Jayne, J. T., Leard, D. C., Zhang, X., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R. (2000). Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles. Aerosol Sci. Technol., 33:49–70.
- Khlystov, A., Wyers, G. P., and Slanina, J. (1995). The Steam-Jet Aerosol Collector. Atmos. Environ., 29:2229–2234.
- Lee, T., Yu, X-Y., Kreidenweis, S. M., Malm, W. C., and Collett, J. L. (2008). Semi-Continuous Measurement of PM2.5 Ionic Composition at Several Rural Locations in the United States. Atmos. Environ., 42:6655–6669.
- Liu, P. S. K., Deng, R., Smith, K. A., Williams, L. R., Jayne, J. T., Canagaratna, M. R., Moore, K., Onasch, T. B., Worsnop, D. R., and Deshler, T. (2007). Transmission Efficiency of an Aerodynamic Focusing Lens System: Comparison of Model Calculations and Laboratory Measurements for the Aerodyne Aerosol Mass Spectrometer. Aerosol Sci. Technol., 41:721–733.
- Markovic, M. Z., VandenBoer, T. C., and Murphy, J. G. (2012). Characterization and Optimization of an Online System for the Simultaneous Measurement of Atmospheric Water-Soluble Constituents in the Gas and Particle Phases. J. Environ. Monitor., 14:1872–1884.
- Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R. (2012). Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer Using Field Data. Aerosol Sci. Technol., 46:258–271.
- Orsini, D. A., Ma, Y., Sullivan, A., Sierau, B., Baumann, K., and Weber, R. J. (2003). Refinements to the Particle-Into-Liquid Sampler for Ground and Airborne Measurements of Water Soluble Aerosol Compositions. Atmos. Environ., 37:1243–1259.
- Perrino, C., Canepari S., Catrambone, M., Dalla Torre, S., Rantica, E., and Sargolini, T. (2009). Influence of Natural Events on the Concentration and Composition of Atmospheric Particulate Matter. Atmos. Environ., 43:4766–4779.
- Perrino, C., Canepari, S., Pappalardo, S., and Marconi, E. (2010). Time-Resolved Measurements of Water-Soluble Ions and Elements in Atmospheric Particulate Matter for the Characterization of Local and Long-Range Transport Events. Chemosphere, 80:1291–1300.
- Perrino, C., Catrambone, M., Dalla Torre, S., Rantica, E., Sargolini, T., and Canepari, S. (2014). Seasonal Variations in the Chemical Composition of Particulate Matter: A Case Study in the Po Valley. Part I: Macro-Components and Mass Closure. Environ. Sci. Pollut. Res., 21:3999–4009.
- Perrino, C., De Santis, F., and Febo, A. (1990). Criteria for the Choice of a Denuder Sampling Technique Devoted to the Measurement of Atmospheric Nitrous and Nitric Acids. Atmos. Environ., 24:617–626.
- Perrino, C., and Gherardi, M. (1999). Optimization of the Coating Layer for the Measurement of Ammonia by Diffusion Denuders. Atmos. Environ., 33:4579–4587.
- Perrino, C., Pietrodangelo, A., and Febo, A. (2001). An Atmospheric Stability Index Based on Radon Progeny Measurements for the Evaluation of Primary Urban Pollution. Atmos. Environ., 35:5235–5244.
- Perrino, C., Ramirez, D., and Allegrini, I. (2001). Monitoring Acidic Air Pollutants Near Rome by Means of Diffusion Lines: Development of a Specific Quality Control Procedure. Atmos. Environ., 35:331–341.
- Simon, P. K., and Dasgupta, P. K. (1995). Continuous Automated Measurement of the Soluble Fraction of Atmospheric Particulate Matter. Anal. Chem., 67:71–78.
- Stolzenburg, M. R., and Hering, S. V. (2000). Method for the Automated Measurement of Fine Particle Nitrate in the Atmosphere. Environ. Sci. Technol., 34:907–914.
- Timonen, H., Aurela, M., Carbone, S., Saarnio, K., Saarikoski, S., Makela, T., Kulmala, M., Kerminen, V.-M., Worsnop, D. R., and Hillamo, R. (2010). High Time-Resolution Chemical Characterization of the Water-Soluble Fraction of Ambient Aerosols with PILS-TOC-IC and AMS. Aerosol Sci. Technol., 3:1063–1074.
- Tsai, C.-J., and Perng, S.-N. (1998). Artifacts of Ionic Species for Hi-Vol PM10 and PM10 Dichotomous Samplers. Atmos. Environ., 32:1605–1613.
- Vecchi, R., Marcazzan, G., Valli, G., Ceriani, M., and Antoniazzi, C. (2004). The Role of Atmospheric Dispersion in the Seasonal Variation of PM1 and PM2.5 Concentration and Composition in the Urban Area of Milan (Italy). Atmos. Environ., 38:4437–4446.
- Vecchi, R., Valli, G., Fermo, P., D’Alessandro, A., Piazzalunga, A., and Bernardoni, V. (2009). Organic and Inorganic Sampling Artefacts Assessment. Atmos. Environ., 43:1713–1720.
- Wang, D., Pakbin, P., Saffari, A., Shafer, M. M., Schauer, J. J., and Sioutas, C. (2013). Development and Evaluation of a High-Volume Aerosol-Into-Liquid Collector for Fine and Ultrafine Particulate Matter. Aerosol Sci. Technol., 47:1226–1238.
- Weber, R. J., Orsini, D., Daun, Y., Lee, Y. N., Klotz, P. J., and Brechtel, F. (2001). A Particle-Into-Liquid Collector for Rapid Measurement of Aerosol Bulk Chemical Composition. Aerosol Sci. Technol., 35:718–727.
- Wittig, A. E., Takahama, S., Khlystov, A. Y., Pandis, S. N., Hering, S., Kirby, B., and Davidson, C. (2004). Semicontinuous PM2.5 Inorganic Composition Measurements during the Pittsburgh Air Quality Study. Atmos. Environ., 38:3201–3213.
- Wu, W. S., and Wang, T. (2006). On the Performance of a Semi-Continuous PM2.5 Sulphate and Nitrate Instrument under High Loadings of Particulate and Sulphur Dioxide. Atmos. Environ., 41:5442–5451.
- Yu, X.-Y., Lee, T., Ayres, B. R., Kreidenweis, S. M., and Collett, J. L. (2006). Loss of Fine Particle Ammonium from Denuded Nylon Filters. Atmos. Environ., 40:4797–4807.