
Abstract
A particle trap laser desorption mass spectrometer (PT-LDMS) has been developed for the online measurements of the chemical composition of submicron aerosol particles. The PT-LDMS was evaluated by both laboratory and ambient measurements, with the focus being the quantification of sulfate aerosols. Ammonium sulfate ((NH4)2SO4) is generally the predominant form of sulfate aerosols in urban air; hence, it is used as a material for laboratory experiments and calibration. Major fragments of (NH4)2SO4 were observed at mass-to-charge ratios (m/z) of 48 (SO+) and 64 (SO2+). The dependence of sensitivity (expressed as the ratio of m/z 48 signal to sulfate mass) on laser power and cell temperature was investigated. An intercomparison of PT-LDMS with a commercial sulfate particle analyzer (SPA) and filter sampling was performed in Tokyo. Good agreement was observed between SPA and filter analysis (slope = 0.98, r2 = 0.99). Although the mass concentration of sulfate measured by PT-LDMS exhibited a tight correlation with that measured by SPA, the mass concentration measured by PT-LDMS tended to underestimate that measured by SPA (slope = 0.70, r2 = 0.96). While the discrepancy can be mainly attributed to the difference in size cut between PT-LDMS (approximately PM1) and SPA (PM2.5), differences in vaporization efficiency were also found to be important.
Copyright © 2016 American Association for Aerosol Research
EDITOR:
1. Introduction
Online measurements of aerosols are important for improving our understanding of the formation and loss processes of ambient aerosols. In particular, quantification of mass concentration of sulfate aerosols is of high priority because sulfate aerosols are typically the major components of fine particulate mass in the troposphere (Altshuller Citation1973; Drewnick et al. Citation2003; Hogrefe et al. Citation2004). Ammonium sulfate ((NH4)2SO4) is a predominant form of sulfate aerosols found in urban areas and in the regions significantly affected by anthropogenic emissions (Saltzman et al. Citation1983; Pilinis and Seinfeld Citation1987; Schwab et al. Citation2004).
The instruments currently employed for online measurements of sulfate aerosols are roughly categorized into dry and wet methods. Dry methods are often based on thermal desorption techniques and include the Aerodyne aerosol mass spectrometer (AMS; Aerodyne Research Inc., USA) (Jayne et al. Citation2000), thermal desorption chemical ionization mass spectrometer (TDCIMS) (Voisin et al. Citation2003), and sulfate particulate analyzer (SPA; Thermo-Scientific, USA) (Schwab et al. Citation2006). Wet methods are based on online water extraction techniques followed by chemical analysis; these methods include a steam jet aerosol collector (SJAC) (Khlystov et al. Citation1995) and particle-into-liquid sampler (PILS) (Weber et al. Citation2001). These are often coupled with ion chromatography for the quantification of bulk compositions of aerosols.
Among these instruments, the Aerodyne AMS has been utilized by a number of researchers, and useful insights into the formation of sulfate aerosols have been reported (Zhang et al. Citation2005, Citation2007). Generally, the routine calibration of an AMS is performed using pure ammonium nitrate (NH4NO3) particles because of the high particle collection efficiency (CE) for NH4NO3. The quantification of sulfate aerosols in ambient air is performed by assuming representative values of collection efficiency and relative ionization efficiency (RIE) (Allan et al. Citation2003). As discussed below, the thermal decomposition products of (NH4)2SO4 are known to significantly vary depending on the temperature of vaporizer. In fact, earlier AMS studies indicated that the detection efficiency of (NH4)2SO4 exhibited significant dependence on vaporizer temperature (Alfarra Citation2004). For these reasons, there is a need to revisit the quantification of sulfate aerosols by thermal decomposition techniques and to reconsider the choice of appropriate materials for calibration.
We have developed a particle trap laser desorption mass spectrometer (PT-LDMS), and demonstrated the proof of concept (Takegawa et al. Citation2012). The PT-LDMS developed has been categorized as a dry method, as mentioned previously. This is the first report of quantification of mass concentrations of ambient sulfate aerosols by PT-LDMS. Laboratory experiments for determining the operating conditions of PT-LDMS and intercomparisons with other instruments, including an SPA and a filter sampling method, are presented. Possible uncertainties in quantification by PT-LDMS and SPA are discussed in detail.
2. Experimental
2.1. PT-LDMS
The hardware and data acquisition software have been used according to the previous version reported by Takegawa et al. (Citation2012) with some modifications. Herein, we summarize the important specifications. Aerosol particles are introduced into a vacuum chamber through an aerodynamic lens (Liu et al. Citation1995). The majority of gas molecules contained in sample air can be removed by a skimmer cone, which results in a concentration of aerosol particles along the centerline of the lens (Allan et al. Citation2003). Aerosol particles are collected on a particle trap comprising layers of micro-structured mesh, and are subsequently vaporized by a carbon dioxide (CO2) laser (ULR-10, Universal Laser System, Inc., USA). The vaporization mechanism included both direct heating of aerosol particles due to light absorption and thermal conduction from the heated surface of particle trap, as described in Takegawa et al. (Citation2012). The particle trap is enclosed by a gold (Au)-coated aluminum cell (a quartz cell was used in the previous version), which is connected to a quadrupole mass spectrometer (QMS; gas-tight ionizer type, PrismaPlus, Pfeiffer Vacuum, GmbH, Germany). The vaporized gas from collected aerosol particles is ionized by electron impact ionization (EI) with an electron energy of 60 eV. Ion currents at some selected m/z values (Im/z) are detected by a secondary electron multiplier (SEM) using the multiple ion detection mode of QMS.
Compared with the previous version presented by Takegawa et al. (Citation2012), the coating material of the trap was changed from pure platinum (Pt) to a mixture of Pt and tungsten (W) to increase melting point. The separation distance between the mesh layers was changed from 200 to 250 μm to facilitate machining. A 20-μm thick Au film was inserted between the particle trap and holder to improve thermal conductivity. The trap holder was cooled by circulating water at 15°C. A small gap was introduced between the holder and the cell for improving thermal insulation. These modifications enable heating of cell and cooling of trap. The former is necessary to reduce the adsorption of the evolved gas, while the latter is needed to reduce the loss of particle compounds by evaporation during collection.
The laser power (Plaser) was continuously monitored using a beam splitter and thermopile sensor. A beam homogenizer (Sumitomo Electric, Japan) was installed in optical path for generating a nearly top-hat-shaped laser beam, although the shape of the actual beam at the position of the particle trap was not precisely measured. This is beneficial in terms of both homogeneous heating of the particle trap and avoiding penetration of trap by laser beam. A radiation thermometer (IGA140, LumaSense Technologies Inc., USA) was placed outside the chamber for monitoring the temperature of particle trap. The thermometer observed near-infrared radiation emitted from the trap heated by the laser passing through a quartz window of vacuum chamber. Note that the temperature data should be treated as a lower limit because both the emissivity of the trap and the transmission efficiency of the quartz window were assumed to be unity.
The total measurement cycle was of 15 min, including the time for particle collection, laser desorption analysis, cooling of the trap, and buffer time for stabilization. After aerosol particles in sample air were collected on the particle trap for 7 min, the inlet valve was closed (SS-43GF4 with MS-142ACX, Swagelok, Inc., USA). The laser was switched on for 1.5 min to detect the gas evolved from collected particles 1 min after the inlet valve had been closed. The inlet valve was kept closed for 5.5 min after the laser was switched off for cooling the trap.
The inlet flow rate of PT-LDMS was calibrated using a standard flow meter (DryCal DC-Lite Air Flow Calibrator, Brandt Instruments, Inc., USA). The mass concentration is expressed as μg m−3 at 25°C and 1 atm.
2.2. Other instruments
The SPA quantifies the mass concentration of sulfate aerosols by a thermal decomposition/reduction method coupled with a sulfur dioxide (SO2) monitor (Schwab et al. Citation2006). Sulfate aerosol particles are reduced to SO2 in a converter tube heated to 1000°C and subsequently detected by a pulsed UV fluorescence method. The detected signals include signals from sulfate aerosols and those from gas-phase SO2. We did not use denuder to remove gas-phase interference. The mass concentration of sulfate was calculated by subtracting the data obtained for particle-free air (zero air) from the data obtained for particle-loaded air.
Filter sampling and analysis were performed by Murata Corp (http://www.murata-s.co.jp/). PM2.5 samples were collected using a PM2.5 aerosol sampler (MCAS-SJ, Murata Keisokuki Service Co. Ltd., Japan) and subsequently analyzed by ion chromatography in the laboratory. Miyakawa et al. (Citation2015) have described the collection and analysis procedure in detail.
2.3. Laboratory experiment
The dependence of sensitivity on Plaser and cell temperature (Tcell) was investigated in the laboratory prior to ambient measurements. The particle generation and evaluation system was practically the same as that described by Takegawa et al. (Citation2005). It comprised collision atomizer (Model 3076, TSI, Inc., USA), diffusion dryer, differential mobility analyzer (DMA; Model 3081, TSI, Inc., USA), and condensation particle counter (CPC, Model 3022, TSI, Inc., USA). Monodisperse aerosols with known chemical compositions were generated and introduced into PT-LDMS.
We used (NH4)2SO4 particles for the laboratory experiment because they are typically the predominant form of sulfate aerosols in urban air. As described by Takegawa et al. (Citation2012), major fragments of (NH4)2SO4 were observed at m/z 48 (SO+) and 64 (SO2+). A mobility diameter (dm) of 250 nm was chosen for most of the experiments because of the high particle transmission efficiency through aerodynamic lens at this diameter (Miyakawa et al. Citation2014). The effect of multiple charged particles was estimated using the same method as that described by Takegawa and Sakurai (Citation2011) to calculate the mass of sulfate introduced into PT-LDMS.
In addition to (NH4)2SO4, potassium nitrate (KNO3) was used as the reference because of its simple molecular structure and fragmentation pattern (Takegawa et al. Citation2012). Details of the experimental results for KNO3 particles are given in the online supplementary information (SI). The quantification of nitrate aerosols is also an important issue, but it is beyond the scope of this article. A detailed investigation of nitrate aerosols, including KNO3 and NH4NO3, is currently underway and will be presented elsewhere.
2.4. Ambient measurements
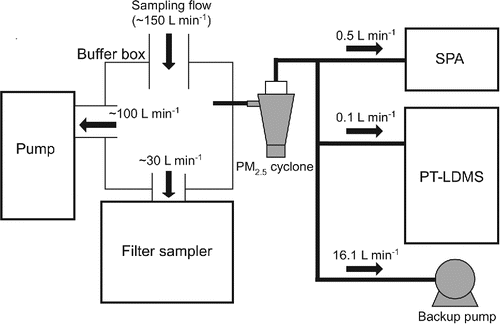
Ambient measurements were performed in November 2013 at a building on the campus of the Research Center for Advanced Science and Technology, the University of Tokyo. Although the intercomparison was also performed in March 2013, as qualitatively discussed by Miyakawa et al. (Citation2014), their data were not utilized in this study. shows the experimental setup for ambient measurements. Ambient air aspirated from a window was introduced into a buffer box so that all instruments could take samples from the same air volume. The inner surface of the buffer box was coated with aluminum foils and grounded to avoid electrostatic charges. The filter samples were collected every 12 h. The SPA and PT-LDMS were equipped with a PM2.5 cyclone (URG-2000-30EH, URG, Inc., USA), while the size cut of PT-LDMS was approximated as PM1 (limited by the transmission efficiency of aerodynamic lens).
Figure 1. Schematic diagram of the experimental set up for ambient measurements.
The zero levels of the PT-LDMS signals were measured every hour by replacing the sample air with zero air generated using a high-efficiency particulate air (HEPA) filter. The PT-LDMS was routinely calibrated using monodisperse (NH4)2SO4 particles during the measurement period by the particle generation system described in Section 2.3.
The zero levels of SPA signals were measured every hour using a three-way valve and a HEPA filter placed upstream of the converter. The valve was switched between particle-loaded and zero air. Although the SO2 monitor of SPA was calibrated using SO2 standard gas prior to the measurement period, the overall calibration of SPA was performed using monodisperse (NH4)2SO4 particles after the period. The three-way valve was not used for the correction of gas-phase SO2 during calibration because substantial amount of SO2 outgas from DMA was present, presumably originating from HEPA filters for generating sheath air. Instead, the contribution of gas-phase SO2 was estimated by turning off the high voltage of DMA.
2.5. Data reduction
2.5.1. PT-LDMS
As described in Section 2.1, ion currents at some selected m/z values were detected (m/z 14, 16, 28, 30, 44, 48, 55, and 64). The m/z 14 signal was dominated by N+ originating from the nitrogen present in ambient air with the inlet valve open, and was used to correct the temporal drift of ion detection efficiency. A similar correction process was also carried out by Takegawa et al. (Citation2012), who used m/z 32 (O2+) instead of m/z 14. Although this section focuses on the quantification of sulfate aerosols, the equations presented here are also applicable to other compounds such as nitrate.
summarizes the key parameters used in this study and their abbreviations. The ion current above the background level was accumulated over the laser irradiation time to obtain an integrated ion signal at each m/z . Note that the superscript X (= “amb” or “cal”) represents ambient or calibration conditions, respectively. The mass concentration of sulfate
was calculated using the difference in the integrated ion signal between particle-loaded and zero air
:
[1]
Table 1. Key factors affecting quantification
These equations are also used to estimate possible uncertainties in quantification (Section 4.3).
2.5.2. SPA
The mass concentration of sulfate aerosols measured by SPA was calculated using the difference of the mixing ratio of SO2 between particle-loaded and zero air (net SO2 mixing ratio, ΔχX):
[5]
ΔχX is expressed as follows:[6] where
is the mixing ratio of SO2 for particle-loaded air, and
is the mixing ratio of SO2 for zero air. FSPA is the correction factor determined by calibration:
[7]
Notably, both and
were scaled to the same standard, which is critical for the interpretation of intercomparison results.
3. Results
3.1. Laboratory experiment
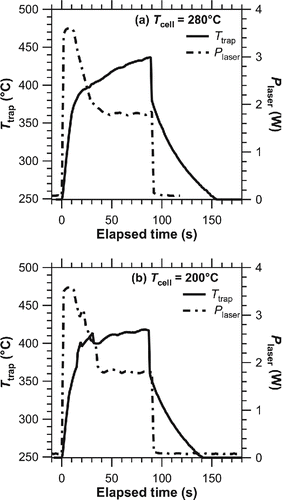
The radiation temperature of the particle trap was primarily controlled by the Plaser density and thermal properties of the trap (i.e., absorbance and thermal conductance). shows an example of Plaser and radiation temperature profiles at a certain laser power setting. The Plaser was modulated (overshoot at the beginning) to achieve a rapid evolution of radiation temperature. The Plaser and trap temperature (Ttrap) were estimated using data points between 30 s and 90 s after the laser was switched on (i.e., “plateau time period”).
Figure 2. Example of Ttrap (solid line, left axis) and Plaser (dashed line, right axis) profiles at Tcell of (a) 280°C and (b) 200°C.
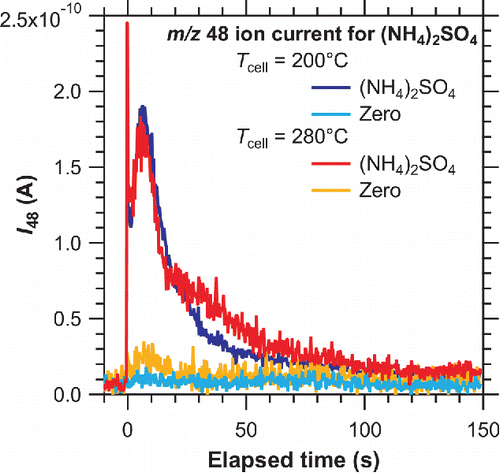
We measured sensitivity under various Plaser (Ttrap) and Tcell conditions. shows the evolution of the m/z 48 ion currents for (NH4)2SO4 particles obtained at a Plaser of ∼4 W (Ttrap of ∼550°C) and Tcell values of 200°C and 280°C. The accumulated mass of sulfate was 18 and 8 ng for Tcell = 200°C and 280°C conditions, respectively. The ion current for Tcell = 200°C was scaled down by a factor of 0.44 (= 8/18) to clarify the comparison of two signals. The ion current signal was characterized by a sharp peak immediately after the laser was switched on and a broad peak of 10 s after the laser was switched on. This result suggests that the particle vaporization process comprised at least two thermal desorption mechanisms, as mentioned earlier. Notable changes were observed for the ion current at m/z 48 for (NH4)2SO4 in the latter period of laser irradiation (30–90 s) with increasing Tcell.
Figure 3. Evolution of mass normalized m/z 48 ion current for (NH4)2SO4 particles obtained at a Ttrap of ∼550°C and Tcell of 200°C (blue and turquoise lines) and 280°C (red and orange lines). The accumulated mass of sulfate was 18 and 8 ng for Tcell = 200°C and 280°C conditions, respectively. The ion current for Tcell = 200°C was scaled down by a factor of 0.44 (= 8/18) to clarify the comparison of two signals.
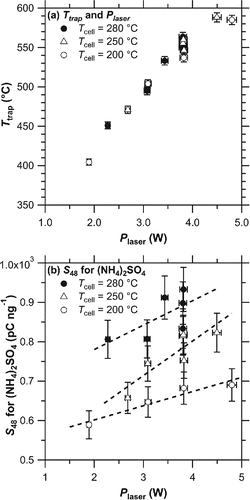
shows the relationship between Ttrap and Plaser under three Tcell conditions. The relationship did not show significant dependence on the Tcell conditions. summarizes the results of temperature experiments. Although substantial scatter was observed in the data, the dependence of sensitivity on Tcell appeared to be more significant than that on Plaser (Ttrap) for (NH4)2SO4. Higher Tcell conditions tended to exhibit higher sensitivity for (NH4)2SO4 particles. As mentioned in Section 2.3, major fragments of (NH4)2SO4 were observed at m/z 48 and 64, but detectable amounts of other sulfur-related signals, such as m/z 80 (SO3+) and 98 (H2SO4+), were not observed. The to
ratio can be used as a diagnostic for desorption because this ratio can vary depending on the chemical form of sulfur compounds (SO2, H2SO4, or others).
Figure 4. (a) Scatter plot of Ttrap versus Plaser from laboratory experiments. (b) Dependence of sulfate sensitivity on Plaser at various Tcell values. Tcell values for the experiment were selected as 280°C (filled circle), 250°C (triangle), and 200°C (open circle). Dashed lines denote the regression lines of each dataset in (b). Error bars depict the uncertainty originating from the random error of PT-LDMS (4%), CPC (2%), radiation thermometer (1%), and thermopile sensor (1%).
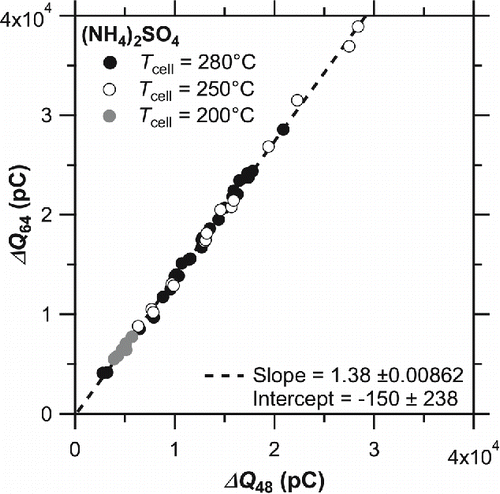
shows the scatter plot of versus
obtained during laboratory experiments. Previous studies have reported that the thermal decomposition products of (NH4)2SO4 mainly comprise SO2 above ∼400°C, and that the decomposition products vary below ∼400°C (Kiyoura and Urano Citation1970). According to the impact ionization (70 eV) mass spectrum database provided by the National Institute of Standard Technology (NIST) (http://webbook.nist.gov/chemistry/), the
to
ratio can differ significantly depending on the chemical form of sulfate compounds (SO2, H2SO4, etc.). The observed
value from (NH4)2SO4 was ∼1.4 with a high correlation coefficient (r2 ∼ 0.99). Although we did not directly identify the chemical form of sulfur compounds in the evolved gas, the tight correlation suggests that the thermal decomposition products of (NH4)2SO4 can be regarded as uniform.
Figure 5. Correlation plots of versus
from laboratory experiments (filled circle: Tcell = 280°C, open circle: Tcell = 250°C, and shaded circle: Tcell = 200°C). The data points for Tcell = 250°C include routine calibrations during ambient measurements. The dashed line denotes the regression line for entire data (r2 = 0.998).
3.2. Ambient measurements
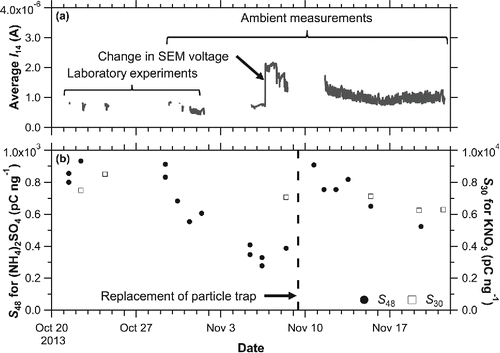
shows the time series of the average ion current at m/z 14 (reference signal) with the inlet open and the sensitivities for sulfate and nitrate during the laboratory experiment (October 2013) and ambient measurement (November 2013). The sensitivity of nitrate was determined using KNO3 as a calibration material (Section 2.3). The reference signal changed in accordance with the changes in SEM voltage. The sensitivity for sulfate exhibited a large decrease over the course of few days following the start of ambient measurements, while the sensitivity of nitrate exhibited a slight decreasing trend. The sensitivity recovered after the replacement of particle trap; however, it exhibited a similar decreasing trend again. Such a drastic change was not observed during the laboratory experiment or ambient measurement in March 2013 (Miyakawa et al. Citation2014). A scanning electron microscopy observation indicated that the Pt/W coating was partially detached from the particle trap surface (not shown), which could have caused decrease in sulfate sensitivity. The particle trap was replaced when the sensitivity of sulfate was decreased to ∼200 pC ng−1, although optimal timing of the replacement of particle trap is still under consideration.
Figure 6. Temporal changes in (a) the average of m/z 14 ion current, and (b) sensitivity of sulfate (filled circle) and nitrate (open square) during laboratory experiments and ambient measurements. The particle trap was replaced on November 9. The sensitivity data include laboratory experiments and routine calibrations. There was an overlap of ambient measurement and laboratory experiment periods because some laboratory experiments were performed during the ambient measurement period.
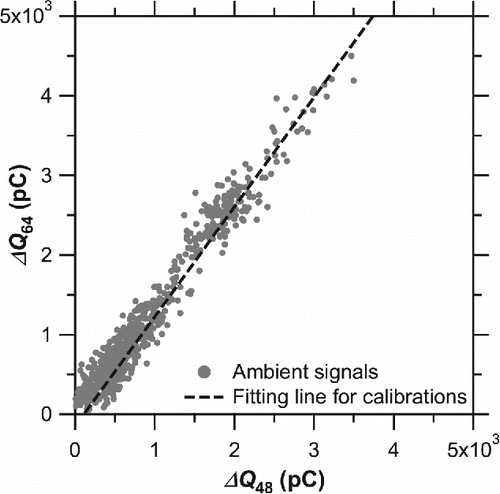
shows the scatter plot of versus
obtained during ambient measurements. For comparison, the regression line from the laboratory experiment is also shown. The
values for ambient aerosol particles showed good agreement with the laboratory data with a variation of 15%, indicating that the thermal desorption products from ambient sulfate aerosol particles were very similar to those from (NH4)2SO4 particles generated in the laboratory.
Figure 7. Correlation plots of versus
. The dashed line denotes the regression line shown in .
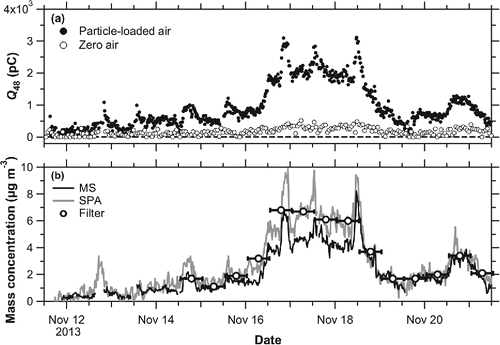
shows the time series of and
during ambient measurement period. The zero air signals were found to exhibit a positive correlation with ambient signals, suggesting that the zero air signals originated from residual sulfate compounds, which may have been incompletely vaporized by each laser shot. shows the time series of
,
, and
during the measurement period. Note that
is the ambient mass concentration measured by filter analysis. The
values were calculated using the data shown in and by interpolating the sensitivity data shown in . The
values were calculated using Equations (5)−(7), and the calibration was performed by the same method of PT-LDMS. During the period of high sulfate mass concentrations (November 17–19), the PT-LDMS data tended to be lower than the data obtained from SPA and filter analysis. On the other hand, the difference in the mass concentration of sulfate between PT-LDMS and SPA/filter analysis during the low mass concentration period (November 14 and 20) was not as large as the difference during the high sulfate period.
Figure 8. (a) Time series of and
. The filled and open circles denote the particle-loaded air signals and zero air signals, respectively. The dashed line represents the zero level. (b) Time series of
(solid line),
(shade line), and
(circle marker). Error bars of
depict the average time of filter sampling (12 h). Missing data points of PT-LDMS (e.g., November 12) were due to calibrations.
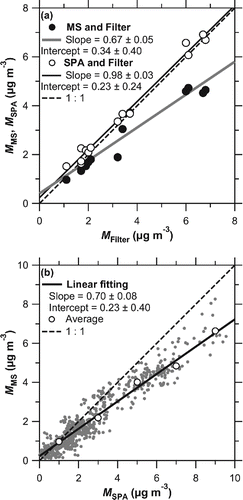
shows the scatter plot of ,
, and
and that of
and
. Good agreement was observed between
and
, with a regression slope of 0.98 and an r2 value of 0.99. Although
exhibited a close correlation with
(r2 = 0.96) and
(r2 = 0.95), the PT-LDMS tended to underestimate filter analysis (regression slope = 0.67) and SPA (regression slope = 0.70). It should be noted that the regression slope was largely affected by the data points in the higher mass concentration range. When focusing on the lower mass concentration range, the data points became closer to the 1:1 correspondence line.
Figure 9. Correlation plots of (a) and
versus
analysis, and (b)
versus
. The shaded and solid lines in (a) represent the regression lines of
versus
(r2 = 0.95) and
versus
(r2 = 0.99), respectively. The shaded points in (b) represent all data, while open circles represent averages in the
bin of 1 μg m−3. Solid line in (b) represents the regression line for averaged values (r2 = 0.96). Dashed lines in (a) and (b) denote 1:1 lines.
4. Discussion
4.1. Decrease in the sensitivity of sulfate
The observed decrease in sulfate sensitivity during ambient measurements is discussed. Decrease in sulfate sensitivity was not observed during the laboratory experiments but became evident after the start of ambient measurements (). The damage of particle trap may have been caused by the presence of refractory compounds in ambient aerosols that efficiently absorbed CO2 laser. This may have resulted in local and extreme heating of particle trap and partial detachment of coating. Further investigation and improvement are needed to increase the durability of trap.
4.2. Limit of detection
The limit of detection (LOD) for sulfate aerosols by PT-LDMS is now evaluated. The LOD can be estimated as the equivalent concentration at which the difference in the peak height of ion currents between particle-loaded and zero air is twice the standard deviation (2σ) of background level. The LOD value estimated by this method was 0.07 μg m−3, on average. Alternatively, the upper limit of LOD can be estimated by calculating variability in mass concentration in ambient air at sufficiently low mass concentrations (November 12–13 in ). The average and standard deviation of mass concentration during this period were 0.5 and 0.04 μg m−3, respectively, which yields an upper limit of 0.08 μg m–3 for LOD.
4.3. Possible uncertainties
4.3.1. PT-LDMS
Possible systematic uncertainties in the determination of sulfate mass concentrations by PT-LDMS are discussed based on theoretical expressions. Simply, we assume that was proportional to
.
can be theoretically expressed as follows:
[8] where A is a constant for conversion from mass to ion signals, TrX is the transmission efficiency of aerodynamic lens, CEX is the collection efficiency of particles, YX is the vaporization efficiency of particle compounds, αX is the adsorption efficiency of SO2 on cell, IEX is the ionization efficiency of the evolved gas, and
is the fragment ratio at m/z.
is the “true” mass concentration of sulfate aerosols, which is unknown in principle. Note that each parameter in Equation (8) is not explicitly determined by calibration.
Based on Equations (1)–(4) and (8), the relative error in the mass concentration of sulfate aerosols can be expressed as follows:[9]
The uncertainty in the mass concentration of sulfate determined by PT-LDMS depends on the difference in the following factors between ambient and calibration particles: transmission efficiency of aerodynamic lens (Tramb/Trcal), particle collection efficiency (CEamb/CEcal), vaporization efficiency of particles (Yamb/Ycal), adsorption efficiency of evolved gas ((1 − αamb)/(1 − αcal)), and ionization efficiency and fragmentation of evolved gas (IEamb/IEcal and framb/frcal, respectively). The uncertainty also depends on the accuracy of DMA sizing, CPC counting, and multiple charge correction, which can be cancelled out when taking into account the comparison between PT-LDMS and SPA. It is noted that these factors are also important for quantification using an Aerodyne AMS.
summarizes the estimates of systematic errors. The size cut of PT-LDMS was approximated as PM1 and limited by the transmission efficiency of aerodynamic lens, which is essentially the same as those used in Aerodyne AMS (Liu et al. Citation1995; Takegawa et al. Citation2009; Miyakawa et al. Citation2014). The transmission efficiency of particles with dm = 250 nm can be regarded as unity, but the transmission efficiency for multiple charged particles included in the DMA exit (dm = 412, 569 nm) may be less than unity. The overall Trcal was estimated to be ∼0.97 using the transmission efficiency curve given by Miyakawa et al. (Citation2014). The overall Tramb was estimated to be ∼0.85, assuming a typical size distribution of aerosol particles in Tokyo (see the SI). This yields an estimate of ∼0.88 for Tramb/Trcal in the case of PM2.5 ambient particles. Note that Tramb and Trcal values are strongly dependent on the accuracy of transmission efficiency of aerodynamic lens and the assumption of size distribution in ambient air.
Table 2. Possible error of each instrument.
The collection efficiency of ambient aerosol particles is also difficult to quantify because it depends on the size distribution and physical properties of aerosol particles. Compared with liquid particles, solid particles, such as dry (NH4)2SO4 particles, generated in the laboratory tend to exhibit a broader distribution on particle trap (Takegawa et al. Citation2012). Polydispersed, irregularly shaped particles, such as soot aggregate particles, may also exhibit a broader distribution than monodisperse, spherical particles because of the characteristics of aerodynamic lens (Zhang et al. Citation2004; Huffman et al. Citation2005; Salcedo et al. Citation2007). The degree of overlap between laser and particle beams can affect the temporal evolution of ion signals (Takegawa et al. Citation2012). A possible approach for estimating uncertainty is to compare the temporal evolution of ion signals at m/z 48 between ambient and calibration conditions, as shown in . The evolution profiles were found to be similar, suggesting that the distribution of collected particles did not significantly vary between ambient and calibration conditions, and that CEamb/CEcal can be assumed to be close to unity. This assumption does not mean that both CEamb and CEcal were unity but that these were comparable during the measurement period.
Figure 10. The evolution of mass normalized m/z 48 ion current for calibration (blue and turquoise lines) and ambient (red and orange lines) conditions. The accumulated mass of sulfate was 10.2 and 5.8 ng for calibration and ambient conditions, respectively. The ion current for ambient condition was scaled up by a factor of 1.76 (= 10.2/5.8) to clarify the comparison of two signals.
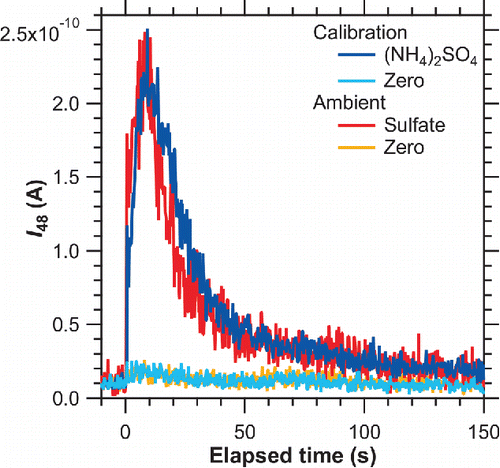
The Yamb/Ycal ratio is affected by two factors: incomplete vaporization of (NH4)2SO4 particles and contribution of refractory sulfate compounds, which were unlikely vaporized with their vaporization temperature exceeding Ttrap (Na2SO4 and K2SO4). The ratio of zero air to particle-loaded air signals was ∼0.13 for ambient particles and ∼0.03 for calibration particles. As a result, Yamb/Ycal originating from incomplete vaporization of (NH4)2SO4 was estimated to be ∼0.90.
Good agreement was observed for between ambient and calibration particles, suggesting that the major component of evolved gas in the case of ambient particles was very similar to calibration particles ( and ). Hence, we can assume that IEamb/IEcal,
, and (1 − αamb)/(1 − αcal) were close to unity.
The positive and negative signs of systematic errors in show that the mass concentration of sulfate in ambient air can be overestimated and underestimated, respectively. Errors for ambient and calibration conditions were partially cancelled out because of the similarities in the chemical form and size of sulfate aerosol particles between ambient and calibration conditions. Conversely, it is suggested that the choice of material and size of calibration particles is important for reducing measurement uncertainties.
4.3.2. SPA
Possible systematic uncertainties in the determination of sulfate mass concentrations by SPA are discussed. As mentioned in Section 2.4, was measured by using a HEPA filter, and
was measured by turning off the DMA voltage.
is expressed as follows:
[10] where B is a constant for conversion from the mass concentration of sulfate to the mixing ratio of SO2, ϵX is the conversion efficiency of sulfate to SO2, and
is the mixing ratio of gas-phase SO2 in sample air. As for calibration,
should be equal to
because there is no difference in the airflow line between particle-loaded and zero air (the zero level was measured by turning off DMA voltage). On the other hand,
and
may be different due to the adsorption of SO2 in HEPA filter, considering large surface area inside the filter. It is rather difficult to estimate the
value because the amount of SO2 adsorption on the filter may not correlate with the SO2 mixing ratio in the sample air (Eatough et al. Citation1995). Although
may also have been affected by the adsorption of SO2 on other surfaces, including conversion cell and tubing, we assumed that this effect was relatively minor.
Based on Equations (5)−(7) and (10), the mass concentration of sulfate aerosols determined by SPA can be expressed as follows:[11]
Uncertainty in the mass concentration of sulfate determined by SPA depends on two terms. The first term originates from the conversion of sulfate to SO2 and depends on conversion efficiency (ϵamb/ϵcal). The second term originates from SO2 in the ambient air and depends on the mixing ratio of ambient SO2.
The ϵamb/ϵcal ratio can be affected by the contribution of refractory sulfate compounds, which were unlikely vaporized with their vaporization temperature exceeding converter tube temperature of SPA (e.g., CaSO4).
As mentioned above, the uncertainty originating from gas-phase SO2 in ambient air was difficult to estimate because was not determined from measurements. If we assume an error of
to be 1 ppbv, for example, it corresponds to an error of 4 μg m−3 in the mass concentration of sulfate aerosols. This uncertainty could potentially become important with increasing mixing ratios of SO2.
4.4. Comparison between PT-LDMS and SPA
For comparison between PT-LDMS and SPA, it is necessary to consider difference in sulfate compounds measured by these instruments. The temperature of particle trap induced by CO2 laser was ∼500°C, while the temperature of the converter of SPA was set at 1000°C. Among various refractory sulfate compounds, Na2SO4 (dissociation temperature of 800°C) was likely to be the most relevant form of compound, which was detectable by SPA but not by PT-LDMS. Schwab et al. (Citation2006) reported that responses of SPA to Na2SO4 and K2SO4 were ∼20% and ∼60%, respectively. Based on their study, the contributions of Na2SO4 and K2SO4 were estimated to be ∼2% and ∼5%, respectively. Note that the contribution of Na2SO4 was estimated using Na+ and Cl− from the filter data assuming that Na+ originated from both sodium chloride (NaCl) and Na2SO4 (Takegawa et al. Citation2005). The contribution of K2SO4 was estimated, assuming that all K+ originated from K2SO4.
The combined uncertainty of the factors described in the previous section and the above factor was estimated to be ∼20%, which may potentially explain the underestimation by PT-LDMS to a large extent. Further studies are needed to improve the quantification of sulfate aerosol under various environments.
5. Summary
This study reports the quantification of sulfate aerosols by PT-LDMS. The instrument was modified compared with the previous version described by Takegawa et al. (Citation2012). Laboratory experiments were performed to investigate optimal conditions for ambient measurements and temperature dependence of sulfate sensitivity. The major decomposition product of (NH4)2SO4 was similar to that of ambient sulfate in PTLDMS. The results also showed that the sensitivity of (NH4)2SO4 particles tended to increase with Tcell, which may be explained by the decreased adsorption of the decomposition product of (NH4)2SO4 on cell surface at high temperatures.
Ambient measurements for the overall test of the instrument, as well as the intercomparison of PT-LDMS with other measurement methods, were performed in Tokyo. for ambient aerosol particles showed good agreement with the laboratory data, to be within 15%, indicating that the thermal desorption products from ambient sulfate aerosol particles were very similar to those from (NH4)2SO4 particles generated in the laboratory. The PT-LDMS exhibited tight correlation with SPA (r2 = 0.96) but tended to underestimate the mass concentration of sulfate (slope = 0.70).
Major uncertainties in PT-LDMS during the measurement period originated from the size cut of aerodynamic lens (∼12%) and the difference in the vaporization efficiency of (NH4)2SO4 particles between calibrations and ambient conditions (∼10%). In addition, uncertainty in the contribution of refractory sulfur compounds between PT-LDMS and SPA was estimated to be ∼6% in this case. These uncertainties were estimated by the consideration of numerous sources of systematic error and used for the interpretation of intercomparison results. The overall uncertainty due to above factors was estimated to be ∼20%, which may potentially explain the underestimation of PT-LDMS to a large extent. Further studies using polydisperse aerosol particles and other materials for calibrations are needed to improve the accuracy of quantification.
UAST_1139685_Supplementaly_Material.zip
Download Zip (482.9 KB)Acknowledgments
The authors thank Professor M. Koike of the University of Tokyo for useful comments.
Funding
This study was funded by the SENTAN program of the Japan Science and Technology Agency (JST) and the Grant-in-Aid for Scientific Research of the Japan Society for the Promotion of Science (25281004).
References
- Alfarra, M. R. (2004). Insights into Atmospheric Organic Aerosols Using an Aerosol Mass Spectrometer. PhD Thesis, University of Manchester Institute of Science and Technology.
- Allan, J. D., Jimenez, J. L., Williams, P. I., Alfarra, M. R., Brower, K. N., Jayne, J. T., Coe, H., and Worsnop, D. R. (2003). Quantitative Sampling Using an Aerodyne Aerosol Mass Spectrometer 1. Techniques of Data Interpretation and Error Analysis. J. Geophys. Res., 108(D3):4090. DOI: 10.1029/2002JD002358.
- Altshuller, A. P. (1973). Atmospheric Sulfur Dioxide and Sulfate Distribution of Concentration at Urban and Nonurban Site in United States. Environ. Sci. Tech., 7:8.
- Drewnick, F., Schwab, J. J., Hogrefe, O., Peters, S., Husain, L., Diamond, D., Weber, R., and Demerjian, K. L. (2003). Intercomparison and Evaluation of Four Semi-Continuous PM2.5 Sulfate Instruments. Atmos. Environ., 37:3335–3350.
- Eatough, D. J., Lewis, L. J., Eatough, M., and Lewis, E. (1995). Sampling Artifacts in the Determination of Particulate Sulfate and SO2 (g) in the Desert Southwest Using Filter Pack Samples. Environ. Sci. Technol., 29:787–791.
- Hogrefe O., Schwab J. J., Drewnick F., Lala G. G., Peters S., Demerjian K. L., Rhoads K., Felton H. D., Rattigan O. V., Husain L., and Dutkiewicz V. A. (2004). Semicontinuous PM2.5 Sulfate and Nitrate Measurements at an Urban and a Rural Location in NewYork: PMTACS-NY Summer 2001 and 2002 Campaigns. J. Air Waste Manag. Assoc., 54:1040–1060.
- Huffman, J. A., Jayne, J. T., Drewnick, F., Aiken, A. C., Onasch, T., Worsnop, D. R., and Jimenez, J. L. (2005). Design, Modeling, Optimization, and Experimental Tests of a Particle Beam Width Probe for the Aerodyne Aerosol Mass Spectrometer. Aerosol Sci. Technol., 39:1143–1163.
- Jayne, J. T., Leard, D. C., Zhang, X., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R. (2000). Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles. Aerosol Sci. Technol., 33(1–2):49–70.
- Khlystov, A., Wyers, G. P., and Slanina, J. (1995). The Steam-Jet Aerosol Collector. Atmos. Environ., 29:2229–2234.
- Kiyoura, R., and Urano, K. (1970). Mechanism, Kinetics, and Equilibrium of Thermal Decomposition of Ammonium Sulfate. Ind. Eng. Chem. Process Des. Dev., 9(4):489–494.
- Liu, P., Ziemann, P. J., Kittelson, D. B., and McMurry, P. H. (1995). Generating Particle Beams of Controlled Dimensions and Divergence: I. Theory of Particle Motion in Aerodynamic Lenses and Nozzle Expansions. Aerosol Sci. Technol., 22:293–313.
- Miyakawa, T., Kanaya, Y., Taketani, F., Tabaru, M., Sugimoto, N., Ozawa, Y., and Takegawa, N. (2015). Ground-Based Measurement of Fluorescent Aerosol Particles in Tokyo in the Spring of 2013: Potential Impacts of Non-Biological Materials on Autofluorescence Measurements of Airborne Particles. J. Geophys. Res. doi: 10.1002/2014JD022189.
- Miyakawa, T., Takeda, N., Koizumi, K., Tabaru, M., Ozawa, Y., Hirayama, N., and Takegawa, N. (2014). A New Laser Incandescence-Mass Spectrometric Analyzer (LII-MS) for Online Measurement of Aerosol Composition Classified by Black Carbon Mixing State. Aerosol Sci. Technol., 48:853–863.
- Pilinis, C., and Seinfeld, J. H. (1987). Continued Development of a General Equilibrium Model for Inorganic Multicomponent Atmospheric Aerosols. Atmos. Environ., 21:2453–2466.
- Salcedo, D., Onasch, T. B., Canagaratna, M. R., Dzepina, K., Huffman, J. A., Jayne, J. T., Worsnop, D. R., Kolb, C. E., Weimer, S., Drewnick, F., Allan, J. D., Delia, A. E., and Jimenz, J. L. (2007). Technical Note: Use of a Beam Width Probe in an Aerosol Mass Spectrometer to Monitor Particle Collection Efficiency in the Field. Atmos. Chem. Phys., 7:549–556.
- Saltzman, E. S., Brass, G. W., and Price, D. A. (1983). The Mechanism of Sulfate Aerosol Formation: Chemical and Sulfur Isotopic Evidence. Geophys. Res. Lett., 10(N7):513–516. DOI: 10.1029/GL010i007p00513.
- Schwab, J. J., Felton, H. D., and Demerjian, K. L. (2004). Aerosol Chemical Composition in New York State from Integrated Filter Samples: Urban/Rural and Seasonal Contrasts. J. Geophys. Res., 109:D16S05. DOI: 10.1029/2003JD004078.
- Schwab, J. J., Hogrefe, O., Demerjian, K. L., Dutkiewicz, V. A., Husain, L., Rattigan, O. V., and Felton, H. D. (2006). Field and Laboratory Evaluation of the Thermo Electron 5020 Sulfate Particle Analyzer. Aerosol Sci. Technol., 40:744–752
- Takegawa, N., Miyakawa, T., Nakamura, T., Sameshima, Y., Takei, M., Kondo, Y., and Hirayama, N. (2012). Evaluation of a New Particle Trap in a Laser Desorption Mass Spectrometer for Online Measurement of Aerosol Composition. Aerosol Sci. Technol., 46:428–443.
- Takegawa, N., Miyakawa, T., Watanabe, M., Kondo, Y., Miyazaki, Y., Han, S., Zhao, Y., van Pinxteren, D., Bruggemann, E., Gnauk, T., Herrmann, H., Xiao, R., Deng, Z., Hu, M., Zhu, T., and Zhang, Y. (2009). Performance of an Aerodyne Aerosol Mass Spectrometer (AMS) during Intensive Campaigns in China in the Summer of 2006. Aerosol Sci. Technol., 43:189–204.
- Takegawa, N., Miyazaki, Y., Kondo, Y., Komazaki, Y., Miyakawa, T., Jimenez, J. L., Jayne, J. T., Worsnop, D. R., Allan, J. D., and Weber, R. J. (2005). Characterization of an Aerodyne Aerosol Mass Spectrometer (AMS): Intercomparison with Other Aerosol Instruments. Aerosol Sci. Technol., 39:760–770.
- Takegawa, N., and Sakurai, H. (2011). Laboratory Evaluation of a TSI Condensation Particle Counter (Model 3771) under Airborne Measurement Conditions. Aerosol Sci. Technol., 45:272–283
- Voisin, D., Smith, J. N., Sakurai, H., McMurry, P. H., and Eisele, F. L. (2003). Thermal Desorption Chemical Ionization Mass Spectrometer for Ultrafine Particle Chemical Composition. Aerosol Sci. Technol., 37:417–475.
- Wang, X., Caldow, R., Sem, G. J., Hama, N., and Sakurai, H. (2010). Evaluation of a Condensation Particle Counter for Vehicle Emission Measurement: Experimental Procedure and Effects of Calibration Aerosol Material. J. Aerosol Sci., 41:306–318.
- Weber, R. J., Orsini, D., Daun, Y. Lee, Y.-N., Klotz, P. J., and Brechtel, F. (2001). A Particle-into-Liquid Collector for Rapid Measurement of Aerosol Bulk Chemical Composition. Aerosol Sci. Technol., 35:718–727.
- Zhang, Q., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L. (2005). Time- and Size-Resolved Chemical Composition of Submicron Particles in Pittsburgh: Implications for Aerosol Sources and Processes. J. Geophys. Res., 110:D07S09. DOI: 10.1029/2004JD004649.
- Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R. (2007). Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenic Ally-Influenced Northern Hemisphere Midlatitudes. Geophys. Res. Lett., 31:L13801. DOI: 10.1029/2007GL029979.
- Zhang, X., Smith, K. A., Worsnop, D. R., Jimenz, J. L., Jayne, J. T., Kolb, C. E., Morris, J., and Davidovits, P. (2004). Numerical Characterization of Particle Beam Collimation: Part II Integrated Aerodynamic-Lens-Nozzle System. Aerosol Sci. Technol., 38:619–638.