
ABSTRACT
Discrepancies between modeled and measured atmospheric organic aerosol (OA) have highlighted the need for in situ instrumentation to better characterize the sources, formation mechanisms, and atmospheric evolution of ambient OA. We have developed the Volatility and Polarity Separator (VAPS) for hourly measurements of volatility- and polarity-resolved OA detected using high-resolution time-of-flight mass spectrometry (HR-ToF-MS). Here, atmospheric OA is inertially impacted onto a collection cell, material is transferred onto a short transfer line located inside a gas chromatography (GC) oven, the oven is heated to provide a first-dimension separation of volatility, then thermally pulsed through a short polar GC column for a second-dimension polarity separation, and finally detected by HR-ToF-MS. This novel instrument increases the mass throughput of ambient OA in comparison to traditional GC due to shorter transfer paths and passivated coatings. Molecular separation resolution is partially sacrificed for this increased mass recovery, but the high-resolution mass spectral data recovers information such as chemical classes and even some individual compounds along with elemental composition to determine aerosol oxidation states. Different techniques for interpreting and representing VAPS data are considered and its applicability to positive matrix factorization (PMF) analysis is demonstrated.
Copyright © 2016 American Association for Aerosol Research
EDITOR:
1. Introduction
Globally, organic aerosol (OA) constitutes a large fraction of the submicron particle mass (Zhang et al. Citation2007; Jimenez et al. Citation2009). OA can be either directly emitted into the atmosphere as primary OA (POA) or formed as secondary OA (SOA) from oxidative gas-to-particle conversion processes (Kroll and Seinfeld Citation2008; Jimenez et al. Citation2009). Because of its ubiquity, it is important to understand the formation, composition, and evolution of OA, and in particular, the less characterized SOA component. While many improvements have been made in the past decade, current atmospheric chemistry models often still struggle to consistently predict OA concentrations and tend to under-predict measured OA loadings (Volkamer et al. Citation2006; Heald et al. Citation2011; Spracklen et al. Citation2011; Fast et al. Citation2014; Zhang et al. Citation2014). The uncertainties in atmospheric aerosol production and evolution only further amplify the large uncertainties that already exist when attempting to quantify the direct and indirect impact of aerosols on the climate system (IPCC Citation2013).
The chemical composition of OA is complex. Hundreds of organic compounds have been identified through traditional gas chromatography (GC) and mass spectrometry (MS) techniques and over 10,000 individual atmospheric organic compounds have been isolated in one PM study using two-dimensional chromatography (Hamilton et al. Citation2004). Even with the extensive separation and identification abilities offered by GC and MS techniques, the majority of the OA mass is typically not analyzable by direct speciation techniques due to poor mass recovery in chromatographic systems, especially of the more oxidized and polar material. This chemical complexity and these analytical restrictions present a challenge to the full characterization of organic particles and their sources and atmospheric processing.
Currently, analytical techniques designed to measure organic aerosol components fall into one of two general categories. The first is measurement of total OA mass without molecular identification, using methods such as total organic carbon measurements with the OC/EC aerosol analyzer (Sunset OC/EC) (Bauer et al. Citation2009) or total fine OA mass concentration on the Aerodyne aerosol mass spectrometer (AMS) (Decarlo et al. Citation2006). The second is measurement of trace organic functional groups or molecular marker compounds using methods such as batch sample collection with subsequent offline or online gas or liquid chromatography/mass spectrometry (GC/MS or LC/MS) analysis (Hohaus et al. Citation2010), chemical ionization mass spectrometry (CIMS) (Lopez-Hilfiker et al. Citation2014), as well as Fourier transform infrared (FTIR) spectroscopy (Russell et al. Citation2009), and nuclear magnetic resonance (NMR) spectroscopy (Paglione et al. Citation2014). Other related methods exist for single particle measurement (e.g., aerosol time of flight MS by Noble and Prather, Citation1996), nanoparticle characterization (thermal desorption chemical ionization MS by Smith et al. Citation(2004), or nano aerosol MS [Bzdek et al. Citation2011]), or specialized lab-based measurements (e.g., particle beam time of flight MS by Tobias and Ziemann [Citation1999], or electrospray ionization MS [Walser et al. Citation2008]). While these techniques all have their strengths, none of them are capable of in situ measurements of total OA mass with detailed molecular-level resolution. For example, the AMS analyzes total OA mass well but does not provide extensive chemical resolution, whereas an online GC instrument like the Thermal Desorption Aerosol Gas Chromatograph/Mass Spectrometer (TAG) (Williams et al. Citation2006; Goldstein et al. Citation2008) provides good molecular level chemical resolution but only analyzes a small fraction (estimated ∼20% on average) of total organic mass. The TAG system has been updated to include an online chemical derivatization process to analyze a larger portion of the organic mass (Isaacman et al. Citation2014).
While detailed chemical speciation is critical to solve chemical mechanism-specific questions, it may not be as important to answer key questions on OA sources and bulk transformation processes. It has recently been proposed that the most informative data for modeling of atmospheric OA may come in the form of two-dimensional mapping of some of the properties of OA that govern how it evolves in the atmosphere (Andreae Citation2009; Jiménez et al. Citation2009; Heald et al. Citation2010). Here we present development of an in situ instrument using a modified 2D GC technique with increased mass analysis that directly detects and reports the atmospheric evolution of volatility- and polarity resolved OA. The data obtained could improve inputs to atmospheric models and increase understanding of aerosol budgets, life cycles, and their effect on climate and air quality.
2. Methods
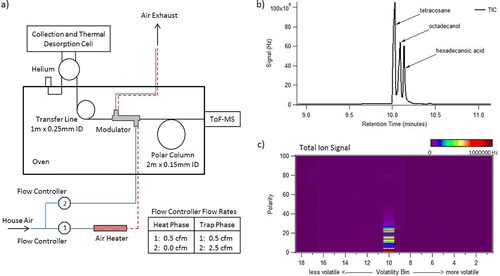
The volatility and polarity separator (VAPS) instrument is a new, fully automated, field-portable instrument for continuous hourly (sub-hour capability) chemical characterization of organic aerosols that achieves high organic mass detection capability as well as an adequate level of the speciation capability provided by truncated gas chromatography. The instrument collects and displays data in a two-dimensional framework that can be readily applied to aerosol models. Here, we refer to the 2D VAPS data figures as “vapograms.” shows a schematic of the analytical technique employed by the VAPS instrument. Following sample collection (described below), a thermal desorption step vaporizes and transfers the sample into a gas chromatograph system where a subsequent oven ramp coupled to a compressed air modulator separates discrete volatility bins to provide volatility separation. Effluent from the re-desorption of individual volatility bins is sent through a short polar chromatography column to provide polarity separation. An electron-impact ionization high-resolution time-of-flight mass spectrometer (HR-ToF-MS) is used as the detector. A flame ionization detector or quadrupole MS can also be used as more cost-effective detector options to yield 2D vapograms. However, the HR-ToF-MS allows for high mass resolution analysis to acquire improved compound identification and elemental abundances and will be the detector of focus in this article.
Figure 1. (a) Schematic of the VAPS modulator system. The heat exchanger provides a constant 0.5 cfm hot air flow to the modulator. A 2.5 cfm cold air flow is switched on during the trapping phase and off during the heating phase. (b) Chromatogram showing polarity resolution of three compounds of similar volatility and differing polarity. (c) 2D vapogram plot of same sample. Polarity units are shown as number of mass spectra acquired since start of modulator hot pulse, and the volatility axis bins are reversed from acquisition order to present with increasing volatility in moving from left to right.
2.1. Particle collection and thermal desorption
The particle collection and thermal desorption (CTD) system used for VAPS is based on the TAG CTD system. A detailed description of the TAG CTD system and its components can be found in Williams et al. Citation(2006) and Kreisberg et al. Citation(2009). Briefly, the aerosol stream is first sampled through a PM2.5 or PM1 cyclone to remove particles larger than 2.5 μm or 1.0 μm, respectively. The aerosol stream then undergoes slight humidification to prevent particle bounce and is sent through a 9-jet impactor and radially impacted onto a coated (Inertium, AMCX Inc.) stainless-steel collection cell. A thermal desorption step then volatilizes the collected sample into a helium stream which transfers the sample onto the head of a GC column for chemical separation.
The 9-jet impactor acts as a critical orifice and maintains a flow rate of 9 LPM (liters per minute). Typical ambient sample collection times are 30 min but this can vary depending on the concentrations of the particles in the aerosol stream. The CTD cell has an injection port for manual syringe injections of chemical standards or can be connected to an automated calibration injection system (Isaacman et al. Citation2011).
2.2. Selection of transfer materials and chromatography components
Although gas chromatography has proven very useful for characterization of thousands of individual organic compounds, highly oxidized and polar compounds have always provided a challenge. These species easily adsorb to uncoated sites in the sample path (e.g., cracks or chips in the GC column or stationary phase coating caused from installation or regular wear and tear) which act to retain polar species and prevent elution to the detector. Often times, analysts have to resort to methods such as chemical derivatization to alter these compounds into less polar and more volatile species that are easier to elute. These derivatization methods present analytical challenges in that the newly eluting compounds tend to be similar in mass spectral fragmentation features and are therefore more difficult to distinguish from each other. Also, derivatizing agents are specific to compound functionality, and a single derivatization agent does not cover all functional groups of interest when analyzing complex aerosol samples, and multiple agents would be necessary, adding further analysis complexity. Finally, derivatizing agents can reduce the lifespan of the chromatographic column or detector (Wenzl 2007; Lee and Khor Citation2015). VAPS aims to increase the mass throughput of the more oxidized compounds without the use of chemical derivatization. This is achieved primarily by using shorter sample paths. In addition, transfer line material and GC components are optimized as described below.
2.3. Volatility and polarity separation
Gas chromatography can be used to separate compounds in a sample by means of volatility or polarity differences. A vaporized sample is carried through a chromatography column by a mobile phase consisting of an ultrapure inert carrier gas such as helium. Differences in boiling point and in interactions between the sample components and the stationary phase, a viscous liquid coating on the interior of the column, cause the compounds to elute at varying retention times, resulting in chromatographic separation.
Higher peak capacity and greater selectivity can be achieved by utilizing two-dimensional gas chromatography (2D-GC). 2D-GC uses traditional gas chromatography technology but adds a second chromatography column (with a different stationary phase) in series with the first, which allows for orthogonal separation. The effluent from the first chromatography column is briefly retained and focused in a thermal modulation system before it is rapidly re-volatilized and injected as a narrow pulse into the second column for additional separation. The first column is typically a longer column that separates compounds based on their volatility, and the second column is typically a shorter column, which separates compounds based on their polarity. The resulting signal can be arranged to produce a three-dimensional plot of compound separation by volatility, compound separation by polarity, and electron-impact ion signal from individual compounds.
In the VAPS system, the thermally desorbed sample from the collection cell re-condenses on the head of a short chemically passivated transfer line at ambient temperature inside the GC oven. The short transfer line replaces the long nonpolar column traditionally used in 1D and 2D-GC. Volatility separation of the sample is achieved by a steady oven ramp which re-volatilizes the sample based on boiling point. Molecular separation is diminished, but the replacement of the long column with the short transfer line dramatically reduces the analysis time (from 2 h in 2D-GC to a faster 15 to 30 min in VAPS) and dramatically improves the mass recovery through the system, especially for polar compounds. Polarity-separation is achieved by sending volatility separated segments of sample into a modulator, collecting for a short period then rapidly desorbing onto a short polar column to achieve polarity-separation within each discrete volatility fraction. The modulator acts to concentrate a portion of the analytes eluting from the first-dimension transfer line and subsequently inject them as a narrow plug into the second-dimension column. The trapping and releasing function of the modulator is accomplished by cyclic heating and cooling of the modulator using hot and cold forced air and uses no cryogenics as is common in traditional 2D-GC.
2.4. ToFMS integration
The VAPS system has been connected with a high-resolution time-of-flight mass spectrometer (HR-ToF-MS; TofwerkAG, Thun, Switzerland) using an interface similar to that used by the TAG-AMS system described in detail by Williams et al. Citation(2014). A custom Inertium ® coated, stainless-steel, heated transfer line connects the effluent end of the polar column to the electron impact ionization region of the mass spectrometer. To eliminate the transmission of high concentrations of carrier gas ions, a quadrupole high-pass filter (TofwerkAG, Thun, Switzerland) was installed and set to deflect ions below m/z 10.
2.5. Data reduction and analysis
VAPS data analysis is performed using a custom built VAPS data panel based in Igor (Wavemetrics, Inc., Portland, OR, USA). VAPS data output is originally a single timeline of discrete volatility steps with polarity separation between steps (). A 2-D plotting function rearranges the data output and converts it into a 2-D data plot which is organized by volatility bins on the x-axis and polarity on the y-axis (). The user can choose the species of interest to be plotted which could be either total signal or an individual ion (unit mass or high resolution exact mass) of interest. In addition, a mass spectra search function allows for comparison of acquired mass spectra with a mass spectra database (e.g., NIST Mass Spectral Reference Library) for compound or source identification.
3. Method evaluation
3.1. Increased particle collection
Sample collection can be increased for a given collection time by increasing the flow rate of the sample stream. The original TAG collection system used a 9-jet impactor operating at a sample flow rate of 9 LPM under critical flow. A new 15-jet impactor with similar jet diameter (0.34 mm), jet-to-jet spacing (1.91 mm), and jet-to-impaction surface spacing (1.02 mm) was designed to provide an increased sample flow rate. Each of the 15 jets provides a 1 LPM critical flow rate for a total sample flow of 15 LPM. In laboratory tests, the new impactor maintained a particle size cutpoint of D50 = 80 nm, consistent with that of the original 9-jet impactor (Figure S1 in the online supplementary information).
3.2. Optimizing mass recovery
One key aim of the VAPS instrument is maximizing the mass throughput through the system, especially for analytically challenging polar compounds. Three locations within the system were identified where modifications or change of materials could reduce losses and improve analyte mass recovery. These were the unions and ferrules used to join columns and transfer lines, the first-dimension transfer line, and the second-dimension column. The maximum mass transfer through these components and their alternatives was characterized by injecting known quantities of a wide range of compounds into a heated GC injection port, and delivering the sample through the test component into a flame ionization detector (FID) for quantification. Injected samples included a pre-mixed alkane/PAH standard (AccuStandard, USA), a pre-mixed even-numbered alkane standard from C10-C40 alkanes (Accustandard, USA), an in-house prepared 15 component mixture of alkanes (C20, C24, C28), PAHs (chrysene, benzo(k)fluoranthene, benzo(ghi)perylene), alcohols (C14, C18, C22), monocarboxylic acids (C12, C16, C20), and dicarboxylic acids (C5, C8, C10), as well as in-house prepared standards of each of the individual compounds used in the 15 component mixture (Sigma Aldrich, USA).
Mass recovery can be limited by the type of connections used between GC columns and transfer lines. Two different types of union-ferrule sets: (1) stainless steel union with Vespel/Graphite ferrules (VICI), and (2) SilTite mini-unions (SGE), were tested to determine the best mass recovery for all compound classes tested. Differences in the thermal properties between the union (stainless steel) and the ferrule (Vespel/Graphite) for the VICI union often results in the development of leaks during continued thermal cycling. Also, the graphite in the ferrule may result in the ferrule having active spots that retain polar compounds. The SGE metal union and ferrule are both made of SilTite metal material, thereby eliminating leaks caused by differences in thermal properties of union and ferrule. In addition, the interior glass lining of the union and the lack of graphite in the ferrule could potentially reduce the activity of the SGE union and ferrule set. The results of the tests comparing these two unions are shown in Table S1. The SGE unions retained less of the injected compounds as seen through increased FID responses when using these unions compared to the response when using the VICI unions. For example, monocarboxylic acids saw a 9%, 22%, and 32% increase in mass recovery for C8-acid, C12-acid, and C18-acid when replacing two VICI unions with two SGE unions in otherwise identical systems. A significant improvement was also observed for the alkane/PAH mixture (19%), where a larger percentage of this increase was for the low-volatility species.
Most 2D-GC systems use a standard 30 m nonpolar chromatographic column for the first separation dimension. This long column provides a significant resistance to mass transfer as active polar compounds are easily attached and retained by imperfections and active spots located within the column. By replacing this first-dimension column with a short, chemically passivated transfer line, mass recovery of active compounds is significantly increased. Discrete volatility separation can still be achieved without the use of a chromatographic column by a programmed thermal ramp of the oven and use of a modulator. This is discussed in more detail in the next section.
With an interest in comparing potential VAPS improvements in mass recovery over traditional GC, the test compounds were first analyzed using a longer 30 m length of a low-polar column (Rxi-5ms, 5% diphenyl/95% dimethyl polysiloxane phase, 30 m × 0.25 mm × 0.25 μm), the same column that is typically applied in GC analysis of organic aerosols (Williams et al. Citation2006). The next sets of tests were done by replacing the 30 m column with various sample transfer lines of 35 cm in length, a shorter transfer line length used in the VAPS system. A wide range of potential transfer line materials were tested including untreated stainless steel (SS) (VICI, USA), custom coated stainless steel using Inertium (Inertium) (AMCX, USA), custom coated stainless steel using Ultra (Ultra) (AMCX, USA), Siltek/Sulfinert-coated stainless steel (Siltek) (Restek, USA), electroformed nickel (EN) (VICI, USA), fused silica Rxi deactivated guard column (0%) (Restek, USA), and low-polar fused silica (5%) (Rxi-5Sil-MS, Restek, USA). All transfer lines tested had an internal diameter of 0.25 mm and the 5% low-polar line had a 0.25 μm film thickness.
Although the second-dimension column, like the first, also provides losses in mass throughput, it cannot be removed as an active column phase is needed to provide the polarity separation desired in this dimension. The advantage here is that this column need not be as long as customary for the first-dimension column. Similar to the tests done for the transfer line material, two different polarity phases were tested to compare mass throughput. These were an intermediate polarity fused silica phase (50%) (BPX50, SGE, USA), and a polar polyethylene glycol wax (wax) (BP20, SGE, USA). Both columns sections tested had a 0.25 mm internal diameter and a 0.25 μm phase thickness.
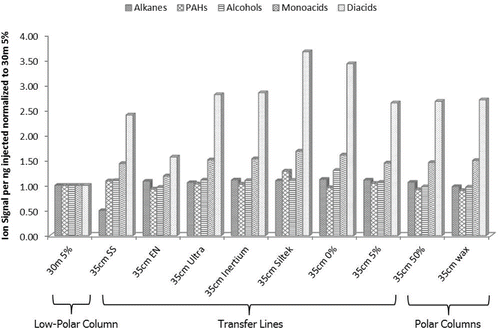
shows the results of these tests for 5-compound classes—alkanes, PAHs, alcohols, monocarboxylic acids, and dicarboxylic acids. Each class contains three species as previously listed. The left bracket in the figure identifies the 30 m column. The center bracket identifies the different sections of transfer line materials tested. Finally, the right bracket identifies two column phases tested for the second-dimension polar column. Response signal is per ng of compound injected and has been normalized to that of the 30 m column. There were no unions used in these tests, where traditionally the 30 m GC column would have at least two unions in place. The figure shows very little difference in the response for alkanes and PAHs for all sections and materials tested. These classes of compounds are normally well observed by the TAG system and other traditional GC techniques. The challenge species are usually the more polar compounds as previously discussed. There is a marked improvement in mass recovery for alcohols, a greater improvement for monocarboxylic acids, and an improvement approaching a factor of four for dicarboxylic acids by replacing the 30 m column with a shorter transfer line.
Figure 2. Results from testing transfer line materials. Each chemical class is comprised of 3 compounds covering a range of volatilities (see text for details). The z-axis displays materials tested for overall transfer. In the left bracket is a 30 m low-polar column typically used for traditional GC. The center bracket contains fixed length (35 cm, 0.25 mm ID) sections of transfer-line materials used to deliver sample between the CTD cell, through the modulator, and onto the polar column. Finally, the right bracket contains two polar column materials tested. All signals have been normalized to the response of the 30 m column.
Based on these tests, a Restek RXI deactivated guard column was chosen as the material for the first-dimension transfer line as it provided very good mass recovery of polar material. Both polar column materials tested provided similar results in terms of mass recovery of the tested compounds. The wax column was chosen for the second dimension as it was the most polar column material and thus provided the truest polarity separation. The disadvantage of the wax column is that its maximum temperature is only 260°C as compared to 330°C for the mid-polarity column. However, the technique employed by the VAPS allows measurement of compounds with volatility equivalent to a C34 alkane at 250°C, and larger molecules eluting later with a constant hold at maximum temperature, as is shown in the next section. As improvements in wax column temperature stability are made or an optional polar material with equivalent separation capacity and mass recovery is identified, the upper temperature limit of the VAPS system can be increased. However, as temperatures are increased, the potential for thermal decomposition of the more oxidized matter also increases. Evidence of thermal decomposition within the current settings are observed and highlighted in the factor analysis discussion presented in Section 3.6. We have recently reported observations of thermal decomposition of highly oxygenated organic aerosol as well as inorganic aerosol in the CTD cell during the original thermal desorption step (Williams et al. Citation2015). It is recommended in future work to acquire data during the “solvent delay” at the start of the analysis period that had traditionally been omitted to prevent detector damage from volatile solvents during standard injections or water during ambient aerosol injections. The thermal decomposition time period was not analyzed during VAPS development, but will be incorporated in future work.
3.3. Modulator design and volatility separation
Another critical component of the VAPS system is the modulator that serves as the interface between the transfer line and the polar column. The purpose of the modulator is to trap and focus discrete bins of analytes eluting from the first-dimension transfer line and subsequently desorb and inject them as a narrow plug into the second column for polarity separation. Traditional atmospheric chemistry GC x GC systems most often use cryogenic modulators (Hamilton Citation2010). However, these can become expensive and burdensome, especially during field deployments. It is therefore desirable to design a simple and robust modulator that uses minimal consumables. Worton et al. Citation(2012) designed a single stage modulator for the 2D-TAG system that cools by forced air convection and heats by capacitive electric discharge pulses. This single stage modulator was shown to be very effective with no analyte breakthrough. The modulator designed here uses forced air convection for both cooling and heating.
shows a schematic of the single stage modulator system employed by the VAPS system. It consists of a custom stainless steel modulator through which one end of the first-dimension transfer line passes through before it is connected to the second-dimension polar column at the outlet of the modulator. The stainless steel modulator is made of two 1/4″ Swagelok tees welded together, lengthwise. The ends orthogonal to the direction of the transfer line are connected to 1/4″ stainless steel tubing through which a convective air flow is passed countercurrent to the direction of the helium flow in the transfer line. The countercurrent flow provides more efficient heat transfer to the transfer line. The forced air passed through the modulator is vented as exhaust. The modulator and all stainless steel tubing connected to it are insulated to minimize interaction with the GC oven temperatures. Cooling and heating of the modulator is achieved by passing cold air or hot air, respectively, over the 12.5 cm section of transfer line tubing inside the modulator. The hot air is provided by a constant flow from a standard air heater (Tutco-Farnam, USA). The hot air flow to the modulator must remain on at all times due to the design of the heater, requiring constant flow to prevent overheating damage. When switching to the trapping phase, a higher rate of cold air flow (25°C) is turned on and allowed to flow with the hot air through the modulator. The higher cold flow rate is enough to significantly lower the hot air temperature. The cold air flow is again switched off during the next desorption period. The modulator heats up and remobilizes the trapped analytes and delivers them to the second-dimension column. This cycle of cooling and heating is repeated for the duration of the analysis program. The timing of each cycle of the modulator controls the volatility resolution. Very low volatility resolution (long trapping and heating cycles) results in a wide volatility range of species in each volatility bin (e.g., several n-alkanes all in one volatility bin). Very high volatility resolution (short trapping and heating cycles) could result in too low of a mass loading per desorption. Optimal flow rates for both the hot and cold air as well as duration of each trap and desorption period were evaluated by manual injections of an even alkane standard solution. Optimal performance of the modulator was obtained using a hot air flow of 0.5 cfm, and a cold air flow of 2.5 cfm with one minute cycles (55 seconds for cold trapping and 5 seconds for desorption). A typical temperature profile for the modulator is shown in . This temperature profile is measured by a thermocouple placed in the air stream inside the modulator.
Figure 3. (a) Transfer of an even alkane standard without the modulator. (b) Transfer of an even alkane standard with the modulator (18 bins) showing good separation and over 20 times more signal. Plotted in solid lines are MS signal at m/z 57 and temperature of oven ramp. Temperature of hot air flow and temperature of modulator are plotted in dashed lines.
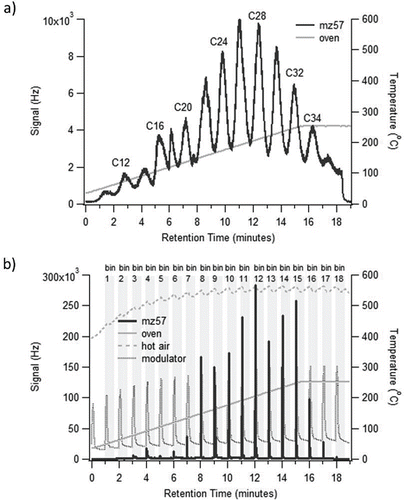
shows the effectiveness of this design in collecting, focusing, and delivering volatility separated sample bins without breakthrough into the second-dimension column. Changes in the aforementioned cooling and heating cycle times and modulator hot and cold air flow parameters would require a re-evaluation of analyte breakthrough. The chromatograms shown here are for a case without using the modulator () and a case with the modulator (). Here, a 1 m × 0.25 mm Restek RXI guard column was used for the first-dimension transfer line and a 2 m × 0.15 mm × 0.15 μm phase thickness Restek Stabiliwax column was used for the second-dimension polar column. The oven was held at 35°C during thermal desorption from the collection cell. It was then ramped at 14°C/min up to 250°C, and held at this temperature for 3.5 min. This results in a total analysis time of 19 minutes. Helium flow was set at a constant 2 ccm flow rate and the MS was set at 2 Hz acquisition. The trapping/desorption cycles and air flow rates for the modulator are as described previously (2.5 cfm cold flow, 0.5 cfm hot flow, 1 min cycles). When an even numbered alkane standard is desorbed and transferred through the short polar column without utilizing the modulator, the individual compounds are not well-focused but are still distinguishable from each other as shown in . It is important to note here that even though the maximum oven temperature was only 250°C, it is clear that we are able to resolve up to the range of a C36 alkane. When the modulator is used for the same standard, sharp distinct peaks with peak height signal amplification of over an order of magnitude are observed as shown in .
With traditional 2D-GC techniques, the second-dimension modulation occurs very fast (on the order of 6 s) and multiple data points are acquired across the width of a single compound slowly eluting from the first column. With slower modulation in the VAPS, a single compound is mostly eluted in a single modulation step, and there is potential for tailing into a second modulation period. By considering the bin where individual compounds elute, it is possible to relate VAPS volatility dimension to the volatility dimension of the volatility basis set (VBS) (Donahue et al. Citation2006; Presto et al. Citation2012). Table S2 lists the bin location where each of the even n-alkanes shown in elute and the corresponding VBS saturation concentration value. The original concept of this instrument is to no longer rely on individual compound analysis, but to easily group classes of compounds with similar mass spectral features and polarity as is often the final analysis point in many 2D-GC studies due to the difficulty in tracking the thousands of individual compounds present in ambient environmental samples. Changes in atmospheric sources or transformation processes can then be tracked in volatility- and polarity separated space similar to studies of aerosol evolution in the 2D-VBS space (Jimenez et al. Citation2009; Donahue et al. Citation2012) for example.
3.4. Polar separation and second-dimensional analysis
After effluent has been injected into the second-dimension polar column by the modulator, it is important that adequate polarity separation occurs so that a two-dimensional plot of volatility and polarity resolved aerosol mass can be produced and analyzed. It is expected that the use of a short column would result in reduced resolution. However, as the goal of the VAPS is not to obtained detailed speciation of organic compounds, but rather to obtain general volatility and polarity resolved information of a larger fraction of the aerosol mass as compared to traditional chromatography, a diminished molecular resolution is acceptable. shows the level of polarity resolution that can be achieved with the 2 m wax column described earlier. The figure shows the separation obtained from an injection of three compounds of similar volatility but with increasing polarity (tetracosane, octadecanol, hexadecanoic acid). shows a two-dimensional representation of the same chromatogram in . All three compounds elute in volatility bin 10 of the 2D plot and in the expected polarity order based on their dipole moments (0.000 Debye, 1.661 Debye, 1.739 Debye, respectively [Yaws Citation2014]). Good molecular resolution is possible on the VAPS system when evaluating a simple mixture. One concern is that very polar compounds could potentially be retained long enough that they may wrap around in the polarity dimension. The relatively long interval between hot pulses and the short polar column minimize this potential but a more detailed analysis using highly oxygenated or multifunctional standards is needed. While the second-dimension retention time is shown here as relative scan number within the volatility bin, the polarity dimension could be converted to something like Debye units by mapping known standard elution times onto the vapogram, similar to how the volatility dimension can be converted to vapor pressures or VBS-space using known standards.
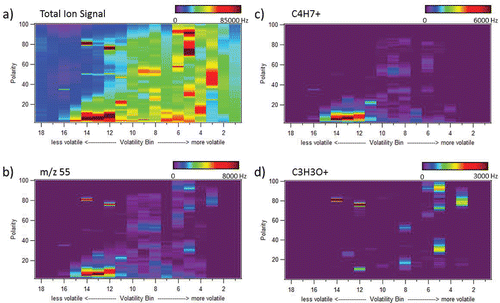
Another example of the 2D plots produced by the VAPS system, but for a more complex ambient sample, is shown in . Each point along the 2D space has a complete high resolution mass spectrum available. Depending on the interests of the user, this allows for several layers of information that could be obtained and plotted. shows a plot of the total signal. In this example, most of the two-dimensional space is occupied by detectable signal. In such a case, looking at individual unit mass ions could offer insight into the composition of the aerosol. shows a vapogram plot of the same sample, but now looking at only m/z 55. This ion is common to both primary and secondary organic aerosol species. Primary species, consisting mainly of hydrocarbons, typically contain a mass spectral fragment at m/z 55 corresponding to the C4H7+ ion. This ion is plotted in and shows a large signal in the low polarity (y-axis) region of bins 10 through 15, consistent with nonpolar hydrocarbons. Secondary organic aerosol species are oxidation products and have oxygen containing functional groups. Many of these species can have a mass spectral fragment at m/z 55 corresponding to the C3H3O+ ion. This ion is plotted in and shows several high intensity signals in the high polarity regions of the 2D plot. It is common for oxygenated molecules to possibly still have mass fragments that contain only carbon and hydrogen. It is observed in that some polar molecules do contain the hydrocarbon C4H7+ ion fragment.
Figure 4. Example 2D vapogram plot of an ambient sample (collected in Brent, AL) with 18 volatility bins on the x-axis and polarity on the y-axis. (a) Total ion signal, (b) m/z 55 ion signal, (c) C4H7+ high-resolution ion signal, and (d) C3H3O+ high-resolution ion signal.
3.5. System performance
System performance was evaluated in the laboratory using multi-level microliter injections of a four-compound standard. The four-compound standard consisted of compounds that span a wide range of polarities and O:C ratios. These include tetracosane (nonpolar; O:C = 0), hexadecanoic acid (mid-polarity; O:C = 0.125), cis-pinonic acid (high polarity; O:C = 0.3), and 2,2-dimethyl glutaric acid (high polarity; O:C = 0.57). lists these compounds along with the limit of quantitation (LOQ) and relative standard deviation (RSD) for each compound based on results of the system performance tests.
Table 1. Compounds in standard for testing of system performance.
The limit of quantitation for each compound in the standard was obtained by injecting successive dilutions of the standard to determine the point at which the ion signal for a compound was indistinguishable from the background noise. The LOQ for a single compound is defined here as the geometric mean mass of the lowest injected concentration with a quantifiable signal and the next lower dilution level with a visible but nonquantifiable signal. This definition and methodology for determining LOQ is similar to that employed by other online thermal desorption GC systems (Kreisberg et al. Citation2009). shows the LOQ for the four compounds in the standard. The LOQ for tetracosane is listed as less than 0.5 ng as that was the lowest concentration of this compound injected. The signal for this compound at this concentration was still well above the background noise suggesting the LOQ could be much lower. However, as this is not a challenge compound, further dilutions were not made and it is listed here mainly for an overall comparison. The LOQ for hexadecanoic acid and cis-pinonic acid was 1.8 ng for both compounds, whereas for 2,2-dimethyl glutaric acid, the LOQ was 14.1 ng. These LOQ values show the capability of VAPS for measuring high polarity compounds without the need for chemical derivatization.
The reproducibility for each compound was measured by triplicate injections at 4 different concentrations—4, 8, 16, and 24 ng for the alkane and 20, 30, 40, and 50 ng for the three acids. Single ion peaks were integrated and compared across injections. The resulting RSD of these peak areas is shown in . RSD for these four compounds ranges from 4.9% to 15.4%. The reproducibility seems to decrease as the polarity of the compound increases. This is consistent with reproducibility obtained by the TAG system (Williams et al. Citation2006). However, the reproducibility for the most polar compound, 2,2-dimethyl glutaric acid, is still 15%. This is a compound that would normally not be detected by GC systems unless a derivatization pre-treatment step was done prior to analysis. Linearity of the response of the system to these four compounds is shown in Figure S2. Good linearity is observed over the injection range for each compound with R2 values ranging from 0.97 to 0.99.
3.6. Ambient samples
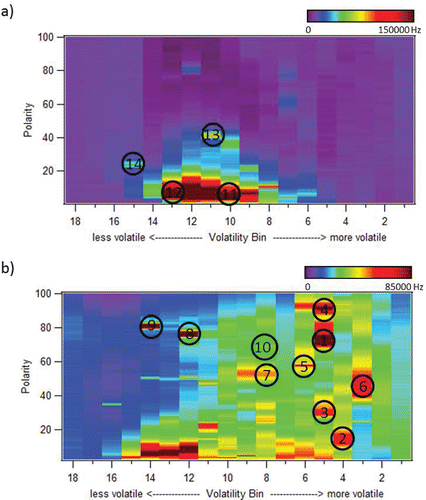
shows example vapograms from two different locations where the VAPS has been tested. It is expected that primary organic aerosol is dominated by hydrocarbons and has low polarity. As these particles age and are processed by the atmosphere, they are likely to undergo one of three reaction mechanisms. Functionalization adds oxygen containing functional groups and increases polarity while reducing volatility. Fragmentation results in smaller, more volatile molecules, whereas oligomerization produces products with decreased volatility. Gas phase emissions can undergo similar processing with their products partitioning to the particle phase as their volatility decreases, producing secondary organic aerosol (SOA). is an ambient sample from an urban environment taken at 9:00 AM on 23 August 2014 at Washington University in St. Louis. This sample is dominated by freshly emitted hydrocarbons that occupy the mid to lower volatility (volatility bins 6 to 14) and nonpolar regions of the vapogram. These hydrocarbons likely originated from vehicular emissions, with a major urban intersection located immediately adjacent to the building. is an ambient sample from a rural environment taken at 9:00 AM on 17 June 2013 in Brent, Alabama, during the Southern Oxidant and Aerosol Study (SOAS) campaign. In contrast to the St. Louis sample, this rural sample is dominated by SOA with increased oxygenated compound content. There is a substantial amount of semivolatile material in the more polar region of the vapogram. The distinction between two different types of aerosols is clearly highlighted by this measurement technique, even prior to identification of the exact molecules present. Table S3 lists a few of the individual resolved species present in the samples. These species have been identified by matching the mass spectrum with that of a NIST mass spectral database. The location of each of these compounds in the vapograms is identified by the number labels in the figure.
Figure 5. (a) Example vapogram of a sample from an urban site in St. Louis, MO. (b) Example vapogram of a sample from a rural site in Brent, AL. Numbered labels refer to individual resolved species described in Table S3.
In addition to the difference between vapograms of samples from different locations, it is also of interest to see how aerosol composition evolves over time at a single location. Figure S3 shows a series of ambient vapograms spanning several hours for two different overnight periods during SOAS. Figures S3a–d are from samples taken between 11:00 PM on 16 June, through 9:00 AM on 17 June. These figures show an oxidized aerosol throughout the night. There is no significant change observed in the overall composition of the aerosol but there is a decrease in mass by 7 AM as can be seen by the reduction in signal intensity. Figures S3e–h are from samples taken between 8:00 PM on 18 June through 6:00 AM on 19 June. The composition of this aerosol mass is not observed to change dramatically through the night, though differences in signal intensity show an increase in mass from 8:00 PM to 11:00 PM followed by a decrease in mass by 6:00 AM. The composition of this aerosol mass is different from that of the night shown in Figures S3a–d, but is still dominated by SOA oxygenated molecules.
Data obtained from VAPS is also suitable for other common types of analysis techniques such as positive matrix factorization (PMF). A new technique for separating chromatograms into bins and applying PMF to binned chromatography data proposed by Zhang et al. Citation(2014) lends itself well to VAPS data. With the binning technique, individual compound identification and integration is not required. Furthermore, this binning technique utilizes the entire eluting mass that encompasses both the resolved peaks and unresolved complex mixture (UCM) of which the latter could make up a large portion of the VAPS signal. The reader is directed to the paper by Zhang et al. Citation(2014) for further details on the binning method.
To demonstrate its capability, a PMF analysis was performed using binned mass spectra of VAPS samples obtained from 16–19 June during the SOAS field campaign. Each vapogram (while still in the originally acquired 1D format) was divided into 162 bins providing bin widths of 6.6 s (equal to the width of the narrowest peak in the vapogram). With the MS acquiring data at 2 Hz, each 6.6 s bin is composed of the sum of 13 individual mass spectra. The PMF analysis is able to separate the bulk data into individual compounds, homologous compound series, UCM, and column bleed. As described by Zhang et al. Citation(2014), the bin structure employed with this type of PMF analysis does not necessarily separate factors based on sources but on similarities in mass spectral features. A more extended PMF analysis and review of the SOAS data set will be presented in a separate paper. Here we highlight the versatility of the data obtained using VAPS and its applicability to PMF analysis.
shows 2D vapograms of each of the resulting factors from a 10-factor PMF solution. H:C and O:C ratios from elemental analysis of high resolution ions (Aiken et al. Citation2008; Canagaratna et al. Citation2015) are also listed for each factor in this figure. The average mass spectrum for each factor and a suggested composition based on vapogram signal intensity location, major peaks present, and match of mass spectrum to the NIST mass spectral database is shown in .
Figure 6. Vapogram of each factor for a 10-factor PMF analysis of data recorded by VAPS during the period 16–19 June 2013 during SOAS in Brent, AL. Also shown are H:C and O:C ratios for each factor from elemental analysis of high resolution ions.
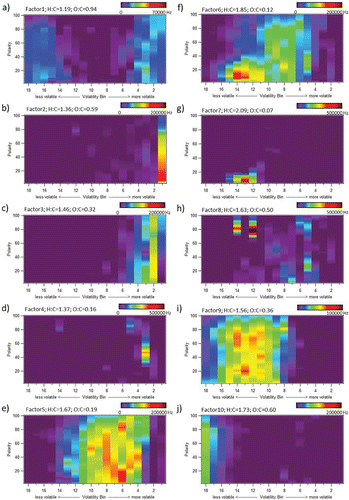
Figure 7. Average mass spectrum of each factor for the 10-factor PMF analysis. Each factor is identified on the basis of location of signal intensity in vapogram, major ions present in mass spectrum, and mass spectral match to NIST database.
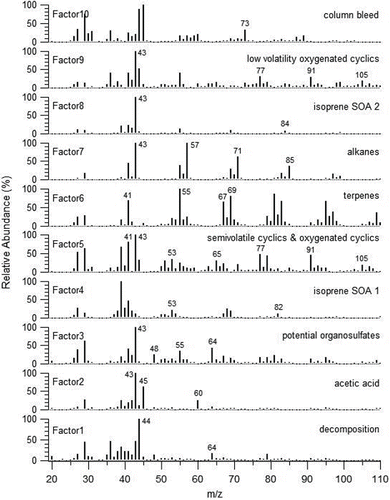
Factor 1 has less signal intensity than all other factors and its signal is concentrated at each volatility end of the vapogram. The mass spectrum for this factor has major signal at m/z 44 (CO2+) and m/z 64 (SO2+) and is likely a factor resulting from thermal decomposition of highly oxygenated organics and inorganic sulfate. The most volatile fraction of this factor was likely produced during initial thermal transfer from the CTD cell to the VAPS transfer line and tailed into the start of the vapogram acquisition. The low volatility fraction of this factor was likely produced during the second heating cycle and transfer of material from the transfer line to the detector. This is the most oxygenated component resulting from the PMF analysis (O:C = 0.94, ) and likely corresponds to the most oxygenated low volatility oxygenated organic aerosol component observed by the AMS measurements in most sampling locations, which is also dominated by the m/z 44 fragment ion (Ng et al. Citation2010).
The mass spectrum for Factor 2 is highly correlated (82% match as per NIST database search tool) to the mass spectral signature for acetic acid (high in m/z 43, 45, and 60). The fragment m/z 60 is often considered a tracer for biomass burning in AMS data, resulting from levoglucosan fragmentation, however, here we do not observe other fragments associated with levoglucosan. This factor is present in bin 1 of the vapogram, consistent with the higher volatility of acetic acid. It is not expected that a significant quantity of acetic acid would be present in the particle phase due to phase partitioning alone, and further lab studies would be needed to determine if the acetic acid is a result of aqueous uptake into the particle phase or if it is a thermal decomposition product of larger multi-functional compounds that was produced during initial thermal transfer from the CTD cell to the VAPS transfer line. Here it is observed that trapping of the most volatile components (like Factor 2) that elute in the first several volatility bins are not efficiently trapped with the cooling/heating trap parameters applied in the field during SOAS. Subsequent development in the laboratory has focused on improving modulator trapping efficiency for the most volatile component.
Factors 3 and 4 also elute early in the vapogram acquisition period (higher volatility). Here it is observed that by the third volatility bin, compound resolution is achieved, indicating that the modulator is sufficient for the semivolatile material eluting at this slightly higher temperature. Factor 3 has dominant signatures of the organic fragments m/z 43 and m/z 55 as well as the inorganic m/z 64 (SO2+) and m/z 48 (SO+), and therefore could contain organosulfates, although authentic standards would be required to confirm their presence. Current work is underway to determine the fate of organosulfates in the VAPS system and will be the focus of a future manuscript. Here, we tentatively identify this factor as potential organosulfates. Factor 4 is likely from isoprene-derived SOA as it has strong signals at m/z 53 (C4H5+) and m/z 82 (C5H6O+), consistent with an isoprene-derived SOA factor from PMF of AMS data collected during SOAS (Xu et al. Citation2015).
The following descriptions for Factors 5, 6, and 9 reference specific compounds available in the NIST mass spectral reference database and used here to indicate likely compound structures present in each factor, and are not meant to indicate that the referenced compounds are the exact molecules present in the samples. Factors 5, 6, and 9 are all UCM factors, with one major difference being the overall volatility of each individual factor as seen by their signal intensity variations in the vapograms. Factors 5 and 9 share many mass spectral fragments that are common to oxygenated cyclic compounds such as methyl chavicol, cumic aldehyde, cumic alcohol, anethol (e.g., m/z 43, 77, 91, 105). Factor 5 also contains ions common to primary cyclic compounds such as the terpenes α-pinene, β-pinene, limonene (e.g., m/z 27, 29, 39, 41, 53, 65), and has an overall O:C ratio (0.19) lower than that observed in Factor 9 (0.36). Here we identify Factor 5 as semivolatile cyclics and oxygenated cyclics (potentially containing terpenes, terpenoids, and their SOA products), and Factor 9 as low volatility oxygenated cyclics. It has been shown through SV-TAG measurements that a majority of monoterpene and sesquiterpene mass is found in the gas phase (Isaacman et al., Citationin review), and a minor fraction is found in the particle phase, as sampled here. Their oxidation products would have a relatively larger fraction in the particle phase. Factors 5 and 9 contain similar mass spectral signatures to the less-oxidized oxygenated organic aerosol (LO-OOA) component described by Xu et al. Citation(2015), as observed through PMF analysis of SOAS AMS data. Boyd et al. Citation(2015) indicate that this component may be from nighttime terpene + NO3 chemistry. The chemical composition of these spectrally similar VAPS PMF factors is the focus of current work and will be incorporated in a future report.
Factor 6 has a low overall O:C ratio (0.12), and in addition to many of the same primary cyclic ion fragments observed in Factor 5, contains ion fragments common to sesquiterpenes such as β-caryophyllene and β-gurjunene (e.g., m/z 41, 55, 67, 69, 79, 81). Factor 6 extends further into the low volatility region of the vapogram, further indicating that it may contain larger molecules than observed in Factor 5. Unlike Factors 5 and 9, Factor 6 does not contain the oxygenated fragments m/z 43 and m/z 44. Here we identify Factor 6 as terpenes. Again, this factor only detects the particle phase fraction of these molecules, which is likely a significantly smaller quantity than their gas-phase abundance.
Factor 7 is an alkane series factor, characterized by intense signal in the low polarity region of the vapogram and mass spectrum with a dominant ion fragment series typical of alkanes (i.e., m/z 43, 57, 71, and 85), and contains the lowest O:C ratio of all 10 factors (O:C = 0.07). In contrast, factor 8 is a more polar factor (O:C = 0.5), containing higher signal intensity in the higher polarity region of several volatility bins, further indicating a more oxidized component of the aerosol. Several of the molecules identified here (, Table S3) were also identified, or are structurally similar to, compounds measured in outflow air masses from the eastern U.S. during the ICARTT study in 2004 through TAG measurements, and were identified to be associated with isoprene oxidation SOA (Williams et al. Citation2007). Here, we label Factor 8 as Isoprene SOA 2, containing a distinct chemical class of molecules from Factor 4, but perhaps coming from a similar source. Future laboratory studies should include injection of previously identified isoprene SOA markers such as 2-methylglyceric acid, 2-methylthreitol, and 2-methylerythritol to test VAPS response to these markers that are typically chemically derivatized prior to injection. There is potential that these compounds are present, but unidentified within one of the isoprene SOA components observed here. These compounds may also thermally decompose in the CTD cell given their multifunctional structure.
Finally, Factor 10 is identified as a column bleed component. GC analyses produce a consistent signature associated with release of the stationary phase material at elevated oven temperatures. Here it can be seen that Factor 10 appears at the top oven temperatures and is composed of expected column bleed ion fragments (e.g., m/z 73, 133, 207).
4. Conclusions
We present a novel instrument for measuring and directly reporting the atmospheric evolution of volatility- and polarity resolved organic aerosol. A range of transfer materials and components were tested to obtain an inert transfer path that maximizes elution of oxidized effluent through the system. VAPS also improves on recovery of this oxidized material over traditional GC by using shorter transfer paths. A custom compressed air modulator coupled to an oven temperature ramp provides volatility separation of discrete volatility bins. A short polar column downstream of the modulator then provides polarity separation of each volatility segment. Although the minimized column length results in a loss of chemical resolution, the use of a HR-ToF-MS detector provides an additional dimension of information such as exact mass ions and elemental composition. In addition, the use of the modulator provides a boost in signal of up to 20 times the signal compared to when the modulator is not used. Data obtained from VAPS can be analyzed and displayed in several ways. A two-dimensional plot or vapogram of the bulk data directly recorded provides a visual representation of the evolution of atmospheric aerosols based on two important aerosol properties—volatility and polarity. Vapograms could also be made for individual unit or high resolution ions, classes, or individual compounds. Further, it was demonstrated that the data obtained from VAPS could be used in a factor analysis to deconvolve the structure of the vapograms into different classes and groups of compounds. The data obtained could improve inputs to atmospheric models and increase understanding of aerosol budgets, life cycles and their effect on climate and air quality. Future work will focus on a comprehensive laboratory evaluation of the instrument as well as continued field deployment and testing. In addition, future work will include development of a database of mass spectra as observed by VAPS from different individual sources and across a range of oxidative ages. This will allow for higher confidence in factor identification.
UAST_1147645_Supplementary_File.zip
Download Zip (773 KB)Funding
VAPS was developed through a Phase I and Phase II small business innovation research (SBIR) grant from the U.S. Department of Energy (contract# DE-SC0006192). Field deployment was supported by a grant from the U.S. Environmental Protection Agency's Science to Achieve Results (STAR) program. Although the research described in the article has been funded wholly or in part by the U.S. Environmental Protection Agency's STAR program through grant (R835402), it has not been subjected to any EPA review and therefore does not necessarily reflect the views of the Agency, and no official endorsement should be inferred.
References
- Aiken, A. C., DeCarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., et al. (2008). O/C and OM/OC Ratios of Primary, Secondary, and Ambient Organic Aerosols with High-Resolution Time-of-Flight Aerosol Mass Spectrometry. Environ. Sci. Technol., 42:4478–4485.
- Andreae, M. (2009). A New Look at Aging Aerosols. Science, 326:1493–1494.
- Bauer, J. J., Yu, X., Cary, R., Laulainen, N., and Berkowitz, C. (2009). Characterization of the Sunset Semi-Continuous Carbon Aerosol Analyzer. J. Air Waste Manag. Assoc., 59:826–833.
- Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L. (2015). Secondary Organic Aerosol (SOA) Formation from the β-Pinene + NO3 System: Effect of Humidity and Peroxy Radical Fate. Atmos. Chem. Phys. Discuss., 15:2679–2744.
- Bzdek, B. R., Zordan, C. A., Luther, G. W., and Johnston, M. V. (2011). Nanoparticle Chemical Composition During New Particle Formation. Aerosol Sci. Technol., 45:1041–1048.
- Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., et al. (2015). Elemental Ratio Measurements of Organic Compounds Using Aerosol Mass Spectrometry: Characterization, Improved Calibration, and Implications. Atmos. Chem. Phys., 15:253–272.
- DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., et al. (2006). Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer. Anal. Chem., 78:8281–8289.
- Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N. (2006). Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics. Environ. Sci. Technol., 40:2635–2643.
- Donahue, N. M., Kroll, J. H., Pandis, S. N., and Robinson, A. L. (2012). A Two-Dimensional Volatility Basis Set – Part 2: Diagnostics of Organic-Aerosol Evolution. Atmos. Chem. Phys., 12:615–634.
- Fast, J. D., Allan, J., Bahreini, R., Craven, J., Emmons, L., Ferrare, R., et al. (2014). Modeling Regional Aerosol and Aerosol Precursor Variability Over California and Its Sensitivity to Emissions and Long-Range Transport During the 2010 CalNex and CARES Campaigns. Atmos. Chem. Phys., 14:10013–10060.
- Goldstein, A. H., Worton, D. R., Williams, B. J., Hering, S. V., Kreisberg, N. M., Panic, O., and Gorecki, T. (2008). Thermal Desorption Comprehensive Two-Dimensional Gas Chromatography for in-situ Measurements of Organic Aerosols. J. Chromatograph. A, 1186:340–347.
- Hamilton, J. F., Webb, P. J., Lewis, A. C., Hopkins, J. R., Smith, S., and Davy, P. (2004). Partially Oxidised Organic Compounds in Urban Aerosol using GCxGC-TOF/MS. Atmos. Chem. Phys., 4:1279–1290.
- Hamilton, J. F. (2010). Using Comprehensive Two-Dimensional Gas Chromatography to Study the Atmosphere. J. Chromatograph. Sci., 48:274–282.
- Heald, C. L., Kroll, J. H., Jimenez, J. L., Docherty, K. S., DeCarlo, P. F., Aiken, A. C., et al. (2010). A Simplified Description of the Evolution of Organic Aerosol Composition in the Atmosphere. Geophys. Res. Lett., 37:L08803.
- Heald, C. L., Coe, H., Jimenez, J. L., Weber, R. J., Bahreini, R., Middlebrook, A. M., et al. (2011). Exploring the Vertical Profile of Atmospheric Organic Aerosol: Comparing 17 Aircraft Field Campaigns with a Global Model. Atmos. Chem. Phys., 11:12673–12696.
- Hohaus, T., Trimborn, D., Kiendler-Scharr, A., Gensch, I., Laumer, W., Kammer, B., et al. (2010). A New Aerosol Collector for Quasi On-Line Analysis of Particulate Organic Matter: The Aerosol Collection Module (ACM) and First Applications with a GC/MS-FID. Atmos. Measurement Techn., 3(5):1423–1436.
- Intergovernmental Panel on Climate Change (IPCC). (2013). Climate Change 2013: The Physical Science Basis. Cambridge University Press, UK.
- Isaacman, G., Kreisberg, N. M., Worton, D. R., Hering, S. V., & Goldstein, A. H. (2011). A Versatile and Reproducible Automatic Injection System for Liquid Standard Introduction: Application to in-situ Calibration. Atmos. Measur. Techn., 4:1937–1942.
- Isaacman, G., Kreisberg, N. M., Yee, L. D., Worton, D. R., Chan, A. W. H., Moss, J. A., et al. (2014). Online Derivatization for Hourly Measurements of Gas- and Particle-Phase Semi-Volatile Oxygenated Organic Compounds by Thermal Desorption Aerosol Gas Chromatography (SV-TAG), Atmos. Measur. Techn., 7:4417–4429.
- Isaacman, G., Kreisberg, N. M., Yee, L., Chan, A., Worton, D. R., Hering, S. V., Goldstein, A. H., et al. (in review). Hourly Measurement of the Concentration and Gas-Particle Partitioning of Oxygenated Organic Tracers in Ambient Aerosol.
- Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., et al. (2009). Evolution of Organic Aerosols in the Atmosphere. Science, 326:1525–1529.
- Kreisberg, N. M., Hering, S. V., Williams, B. J., Worton, D. R., and Goldstein, A. H. (2009). Quantitation of Hourly Speciated Organic Compounds in Atmospheric Aerosols, Measured by in-situ Thermal Desorption Aerosol Gas Chromatography (TAG). Aerosol Sci. Technol., 43:38–52.
- Kroll, J. H., and Seinfeld, J. H. (2008). Chemistry of Secondary Organic Aerosol: Formation and Evolution of Low-Volatility Organics in the Atmosphere. Atmos. Environ., 42:3593–3624.
- Lee, B. Q. and Khor, S. M. (2015). 3-Chloropropane-1,2-diol (3-MCPD) in Soy Sauce: A Review on the Formation, Reduction, and Detection of This Potential Carcinogen. Compr. Rev. Food Sci. Food Safety, 14:48–66.
- Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., et al. (2014). A Novel Method for Online Analysis of Gas and Particle Composition: Description and Evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO). Atmos. Measur. Techn., 7:983–1001.
- Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., et al. (2010). Organic Aerosol Components Observed in Northern Hemispheric Datasets from Aerosol Mass Spectrometry. Atmos. Chem. Phys., 10:4625–4641.
- Noble, C. A. and Prather, K. A. (1996). Real-Time Measurement of Correlated Size and Composition Profiles of Individual Atmospheric Aerosol Particles. Environ. Sci. Technol., 30:2667–2680.
- Paglione, M., Saarikoski, S., Carbone, S., Hillamo, R., Facchini, M. C., Finessi, E., et al. (2014). Primary and Secondary Biomass Burning Aerosols Determined by Proton Nuclear Magnetic Resonance (1H-NMR) Spectroscopy During the 2008 EUCAARI Campaign in the Po Valley (Italy). Atmos. Chem. Phys., 14(10):5089–5110.
- Presto, A. A., Hennigan, C. J., Nguyen, N. T., and Robinson, A. L. (2012). Determination of Volatility Distributions of Primary Organic Aerosol Emissions from Internal Combustion Engines Using Thermal Desorption Gas Chromatography Mass Spectrometry. Aerosol Sci. Technol., 46:1129–1139.
- Russell, L. M., Bahadur, R., Hawkins, L. N., Allan, J., Baumgardner, D., Quinn, P. K., and Bates, T. S. (2009). Organic Aerosol Characterization by Complementary Measurements of Chemical Bonds and Molecular Fragments. Atmos. Environ., 43:6100–6105.
- Smith, J. N., Moore, K. F., McMurry, P. H., and Eisele, F. L. (2004). Atmospheric Measurements of Sub-20 nm Diameter Particle Chemical Composition by Thermal Desorption Chemical Ionization Mass Spectrometry. Aerosol Sci. Technol., 38(2):100–110.
- Spracklen, D. V., Jimenez, J. L., Carslaw, K. S., Worsnop, D. R., Evans, M. J., Mann, G. W., et al. (2011). Aerosol Mass Spectrometer Constraint on the Global Secondary Organic Aerosol Budget. Atmos. Chem. Phys., 11:12109–12136.
- Tobias, H. J. and Ziemann, P. J. (1999). Compound Identification in Organic Aerosols using Temperature Programmed Thermal Desorption Particle Beam Mass Spectrometry. Anal. Chem., 71:3428–3435.
- Volkamer, R., Jimenez, J. L., San Martini, F., Dzepina, K., Zhang, Q, Salcedo, D., et al. (2006). Secondary Organic Aerosol Formation from Anthropogenic Air Pollution: Rapid and Higher Than Expected. Geophys. Res. Lett., 33:L17811.
- Walser, M. L., Desyaterik, Y., Laskin, J., Laskin, A., Nizkorodov, S. A. (2008). High-Resolution Mass Spectrometric Analysis of Secondary Organic Aerosol Produced by Ozonation of Limonene. Phys. Chem. Chemical Phys., 10:1009–1022.
- Wenzl, T., Lachenmeier, D. W., and Gökmen, V. (2007). Analysis of Heat-Induced Contaminants (acrylamide, chloropropanols and furan) in Carbohydrate-Rich Food. Anal. Bioanalyt. Chem., 389:119–137.
- Williams, B. J., Goldstein, A. H., Kreisberg, N. M., and Hering, S. V. (2006). An in-situ Instrument for Speciated Organic Composition of Atmospheric Aerosols: Thermal Desorption Aerosol GC/MS-FID (TAG). Aerosol Sci. Technol., 40:627–638.
- Williams, B. J., Goldstein, A. H., Millet, D. B., Holzinger, R., Kreisberg, N. M., Hering, S. V., et al. (2007). Chemical Speciation of Organic Aerosol During the International Consortium for Atmospheric Research on Transport and Transformation 2004: Results from in situ Measurements. J. Geophys. Res., 112:D10S26.
- Williams, B. J., Jayne, J. T., Lambe, A. T., Hohaus, T., Kimmel, J. R., Sueper, D., et al. (2014). The First Combined Thermal Desorption Aerosol Gas Chromatograph-Aerosol Mass Spectrometer (TAG-AMS). Aerosol Sci. Technol., 48:358–370.
- Williams, B. J., Zhang, Y., Zuo, X., Martinez, R. E., Walker, M. J., Kreisberg, N. M., Goldstein, A. H., Docherty, K. S., and Jimenez, J. L. (2015). Organic and Inorganic Decomposition Products from the Thermal Desorption of Atmospheric Particles, Atmos. Measure. Techn., 8:13377–13421.
- Worton, D. R., Kreisberg, N. M., Isaacman, G., Teng, A. P., McNeish, C., Gorecki, T., et al. (2012). Thermal Desorption Comprehensive Two-Dimensional Gas Chromatography: An Improved Instrument for In-Situ Speciated Measurements of Organic Aerosols. Aerosol Sci. Technol., 46:380–393.
- Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., et al. (2015). Effects of Anthropogenic Emissions on Aerosol Formation from Isoprene and Monoterpenes in the Southeastern United States. Proc. Natl. Acad. Sci., 112:37–42.
- Yaws, C. L. (ed.) (2014). Thermophysical Properties of Chemicals and Hydrocarbons (2nd ed.), William Andrew, Norwich, New York.
- Zhang, H., Chen, G., Hu, J., Chen, S., Wiedinmyer, C., Kleeman, M., and Ying, Q. (2014). Evaluation of a Seven-Year Air Quality Simulation using the Weather Research and Forecasting (WRF)/Community Multiscale Air Quality (CMAQ) Models in the Eastern United States. Sci. Total Environ., 473–474:275–285.
- Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., et al. (2007). Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenically-Influenced Northern Hemisphere Midlatitudes. Geophys. Res. Lett., 34:L13801.
- Zhang, Y., Williams, B. J., Goldstein, A. H., Docherty, K., Ulbrich, I. M., and Jimenez, J. L. (2014). A Technique for Rapid Gas Chromatography Analysis Applied to Ambient Organic Aerosol Measurements from the Thermal Desorption Aerosol Gas Chromatograph (TAG). Aerosol Sci. Technol., 48:1166–1182.