
ABSTRACT
Viscosity of atmospheric aerosol spans at least 15 orders of magnitude, from thin liquids to glassy solids, with possible concomitant impact on multiple processes of meteorological and/or climatological concern. Recently there has been interest in aerosol phase assessment techniques based upon dimer coalescence. Theoretical treatment suggests discernible reductions in dimer diameter begin when viscosity ∼108 Pa·s and the dimer is spherical at ∼105 Pa·s for submicron particles, or the middle range of the semisolid regime. A method using nanoparticle dimers synthesized by utilizing differential mobility analyzers of opposite polarity to produce monomers of opposite charge that subsequently undergo electrostatically mediated coagulation has been developed and is detailed in this work. This method was used to assess the aerosol phase state of several atmospherically relevant organic species and inorganic salts at relative humidity (RH) values ranging between 10% and 100%. Ammonium sulfate, monosodium α-ketoglutaric acid, sodium chloride, and sucrose all displayed RH-dependent phase state. These observed viscous transitions occurred at RH values less than existing deliquescence RH data, a result consistent with existing literature reports of RH-induced structural rearrangements. Fully coalesced and fully uncoalesced diameters could be fitted to single values, indicating that the presented technique is absolute. The method was also used to assess the phase state of dry sucrose aerosol at temperatures between 20°C and 70°C. A phase transition was noted at 63.7°C ± 4.4°C, near the glass transition temperature, suggesting the presented method may also be useful for probing phase responses to temperature perturbations.
Copyright © 2016 American Association for Aerosol Research
EDITOR:
Introduction
Organic compounds are a major constituent of atmospheric aerosol in both fractional abundance (Volkamer et al. Citation2006) and number of species (Goldstein and Galbally Citation2007). Much of this organic aerosol is thought to form a liquid phase (Marcolli et al. Citation2004; Cappa and Ravishankara Citation2008; Bateman et al. Citation2015; Cheng et al. Citation2015). However, there is evidence that a significant fraction can also form amorphous solids (Zobrist et al. Citation2008; Mikhailov et al. Citation2009; Virtanen et al. Citation2010). These glassy particles lack traditional crystalline structure, but are of sufficiently high viscosity to behave mechanically like solids. Typically, the threshold viscosity for a glass is defined as 1012 Pa·s (Debenedetti and Stillinger Citation2001), nearly 15 orders of magnitude greater than the viscosity of water. Particle phase transitions can occur through changes in either temperature (Koop et al. Citation2011; Dette and Koop Citation2015) or relative humidity (RH) (Mikhailov et al. Citation2009; Koop et al. Citation2011; Bateman et al. Citation2015; Montgomery et al. Citation2015). High particle viscosity hinders uptake of chemical species (Roth et al. Citation2005; Pfrang et al. Citation2011; Shiraiwa et al. Citation2011; Chan and Chan Citation2013; Gržinič et al. Citation2015; Davies and Wilson Citation2015), influences ice nucleation pathways (Wang et al. Citation2012; Baustian et al. Citation2013; Schill and Tolbert Citation2013; Berkemeier et al. Citation2014), and may kinetically limit water uptake (Tong et al. Citation2011; Zobrist et al. Citation2011; Bones et al. Citation2012; Lu et al. Citation2014).
Existing approaches for assessing particle phase utilize impactor bounce (Virtanen et al. Citation2010), quartz crystal balances (Arenas et al. Citation2012), circulating beads (Renbaum-Wolff et al. Citation2013), poke testing (Renbaum-Wolff et al. Citation2013), and fluorescent rotors (Hosny et al. Citation2013). Alternatively, a number of techniques have recently been developed based upon coalescence state of aggregate particles. Power et al. (Citation2013) used optical tweezers to induce coalescence between 10-μm droplets then observed the amount of time required for full coalescence via light microscopy. Järvinen et al. (Citation2016) correlated measurements of optical depolarization to asphericity of naturally coagulated chamber secondary organic aerosol (SOA). Zhang et al. (Citation2015) similarly investigated asphericity of agglomerates by comparing mass and mobility diameters of SOA undergoing Brownian coagulation in a smog chamber.
Coagulation-based methods are desirable because they measure viscosity directly by probing relaxation times due to flow and because they can characterize high viscosities in the 105–1010 Pa·s regime. One current limitation of coagulation-based methods is that the controlled synthesis and isolation of coagulated dimers is challenging. The coagulation rate (−dN/dT) is related to the particle concentration (Seinfeld and Pandis Citation2006):[1] where N1 and N2 are number concentrations of particles having diameters D1 and D2 and k12 is the rate constant. For monodisperse submicron particles and N < 105 cm−3 coagulation rates are negligible at the time scale of typical laboratory experiments. Coagulation rates can be enhanced or suppressed through electrostatic interactions of charged particles (Zebel Citation1958). This effect has been used by several groups to prepare aggregated particles. Borra et al. (Citation1999) developed a technique for synthesizing powders via coagulation of particles produced through two opposite polarity electrosprays. In this technique, uncoagulated particles retain a charge and thus can be readily removed by application of an electrostatic filter. Maisels et al. (Citation2000) demonstrated nanoparticle dimers could be similarly synthesized using two differential mobility analyzers (DMAs) of opposite polarity to create monodisperse particles of opposite charge. DMAs are ubiquitous in aerosol research and can select particles with diameters ∼10 nm; accordingly, the Maisels et al. (Citation2000) approach is an attractive option for synthesis in laboratory settings of nanoparticle dimers such as utilized by the Zhang et al. (Citation2015) phase method.
This work presents a technique for assessment of particle phase that applies a variant of the Zhang et al. (Citation2015) asphericity method for assessing viscosity to dimers that were synthesized following the Maisels et al. (Citation2000) approach. It is demonstrated that this technique detects phase transitions arising due to changes in RH or temperature. This method is applied to assess the aerosol phase state of several pure inorganic and organic compounds used as atmospheric proxies across a range of relative humidities and the phase state of sucrose upon exposure to a heating (melting) cycle.
Methods
Viscosity estimation
This section describes how the measured dimer mobility diameter (Dp) can be used to determine if the particle has fully or partially coalesced, or remains uncoalesced. Consider two particles with mobility diameters D1 and D2, respectively, comprising a dimer. The sphere equivalent diameter of the dimer (Dse) is[2]
Dimers that coalesce into a sphere have a dynamic shape factor of unity and the measured Dp equals Dse. This idealized limit is denoted as fully coalesced dimer diameter (Dc). Dimers that do not coalesce approximate a rod-shaped particle and have a dynamic shape factor of ∼1.1 (Hinds Citation1999). Particles with higher dynamic shape factor experience an increased drag force and thus have larger mobility diameters. This observed upper limit of the shaped particle is denoted as the fully uncoalesced dimer diameter (Duc). Experimentally determined Dp values are bound by[3] and can be used to bound the particle viscosity.
Viscosity as a function of mobility diameter measurements is modeled over the transition regime using a combination of assumptions similar to those in the supplement of Zhang et al. (Citation2015) and predicted geometries from modified Frenkel theory (Pokluda et al. Citation1997). To illustrate the relationship between viscosity (η), residence time (τ), surface tension (σ), monomer diameter (Dmono), dimer mobility diameter (Dp), uncoalesced dimer diameter (Duc), and coalesced dimer diameter (Dc), the following analytical approximation was derived[4]
[5] where ξ is the particle geometry factor. Residence time refers to the characteristic time period over which dimer relaxation can occur. For the purposes of this work, this is typically the average residence time within a thermal conditioning unit (conditioning unit volume / flow rate), although in some circumstances, the average time necessary for a particle to traverse the distance between the entry to the coagulation chamber and the inlet to the particle sizer may be more relevant. Dimer mobility diameter is the size of the dimer as measured by a scanning mobility particle sizer (SMPS) after any relaxation has occurred. A complete derivation is provided in the online supplemental information (SI). Equation (Equation4
[4] ) shows that the implied viscosity is directly proportional to time available for the particles to coalesce and tension of the surface. The solution is singular at Dp equal to Duc and asymptotic as Dp approaches Dc. Viscosity values can be determined for partially coalesced particles and are bounded for fully coalesced and fully uncoalesced particles. The bounds of viscosity can be varied over several orders of magnitude by changing residence time. The bounds of viscosity are only weakly sensitive to realistic values of surface tension and insensitive to the range between fully uncoalesced and fully coalesced dimer diameters. The closed form solution provided by Equation (Equation4
[4] ) is only accurate to ∼1 order of magnitude due to simplifying assumptions. A more accurate numerical representation of Frenkel theory (SI) is used for actual model calculations.
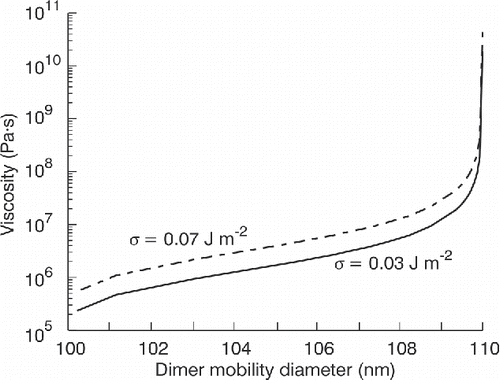
Example numerical model calculations are shown in for Dmono = 80 nm, Duc = 110 nm, Dc = 100 nm, and τ = 5 s. These values are representative of some of the experiments presented in this work. Calculations were performed at two different surface tensions (0.03 and 0.07 J m−2), characteristic values for pure organics and water (Seinfeld and Pandis Citation2006). From it can be inferred that η ≥ 108 Pa·s when the dimer is fully uncoalesced, η ≤ 105 Pa·s when the dimer is fully coalesced, and 105 ≤ η ≤ 108 Pa·s as the dimer is transitioning from uncoalesced to coalesced.
Figure 1. Modeled viscosity as a function of dimer diameter for Dmono = 80 nm, Duc = 110 nm, Dc = 100 nm, and τ = 5 s, values characteristic of some experiments presented in this work. Two surface tensions are depicted, one characteristic of pure organics (0.03 J m−2), one characteristic of a fully deliquesced particle (0.07 J m−2).
Experiment types
Experiments performed for this work can be sorted into two classes: cooling cycle and heating cycle (melting), where dimers are briefly exposed to varying levels of RH and temperature, respectively. Most of the experiments were of the former type, although a melting experiment was performed for sucrose. During each experiment, data were collected at ∼10 nominal RH values between 20% and 100% RH or nominal temperatures between 0°C and 70°C. Typical spacing was 10% RH or 10°C well away from any observed humidity- or temperature-induced shifts in measured coagulated diameters and 5% RH or 5°C near such shifts. Typically, 3–5 SMPS scans were performed at each nominal conditioner RH or temperature.
Chemicals used
The following chemical reagents were used: adipic acid (99%, Sigma Aldrich, St. Louis, MO, USA), ammonium sulfate (99.9%, Fisher, Hampton, NH, USA, Calcium nitrate tetrahydrate (Sigma-Aldrich, 99%), Monosodium α-ketoglutaric acid (Sigma-Aldrich, 98%), polyethylene glycol 600 (Fisher; henceforth PEG-600), polyethylene glycol 1000 (Alfa-Aesar; henceforth PEG-1000), polyethylene glycol 10000 (Alfa-Aesar., Haverhill, MA, USA; henceforth PEG-10000), sodium chloride (Fisher, 99%), sodium dodecyl sulfate (Sigma-Aldrich, 99%), succinic acid (Sigma-Aldrich, 99.0%), and sucrose (Sigma-Aldrich, 99.5%). All reagents were used as provided without further purification. Sample solutions were prepared to 2% (w/w) in HPLC grade water. For the sucrose melting experiment a 0.1% (w/w) solution of sucrose in a mixture of methanol and a small amount of HPLC grade water [∼2% (v/v)] was used.
Experimental design
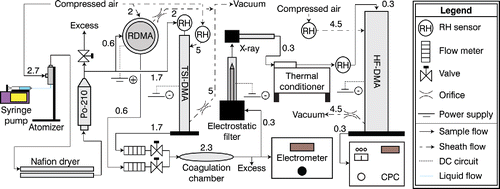
The experimental setup is shown in . Particles were generated via a constant output atomizer (TSI 3076). Solutions were routed to the atomizer using a syringe pump. The particle stream was routed through two Nafion gas dryers (PD-50T-24MSS, PermaPure, Lakewood Township, NJ, USA) in series to remove the solvent then exposed to radiation from a 210Po source to charge equilibrate the aerosol. The dried and neutralized particles were split between two DMAs, one with cylindrical geometry (TSI 3081; henceforth TSI-DMA; TSI Inc., Shoreview, MN, USA) connected to a negative-polarity power supply and selecting only positively charged particles, the other with radial geometry (Zhang et al. Citation1995; henceforth RDMA) connected to a positive-polarity power supply and selecting only negatively charged particles. Each DMA was configured to select particles of a single monomer diameter: nominally 80 nm in cooling cycle experiments, 50 nm in the melting experiment. Sheath flow in the TSI-DMA and RDMA were 5 L and 2 L min−1, respectively. Sample flows through the DMAs were controlled through valves connected to their respective sample outlets, plus an excess release valve positioned immediately after the neutralizer outlet. Rates of 1.7 L min−1 through the TSI-DMA and 0.6 L min−1 through the RDMA were typical. The high sheath-to-sample flow rates result in a broad size cut and were chosen to maximize the transmitted particle concentration. The DMA sample outflows were merged and routed into a 0.3 L mixing chamber where coagulation was allowed to occur. For a typical flow of 2.3 L min−1, this implies an average chamber residence time of 8 s. The flow was split again after the mixing chamber with 0.3 L min−1 directed toward a custom-built electrostatic filter where uncoagulated particles were removed and the remainder directed toward either an aerosol electrometer (TSI 3068; cooling cycle experiments) or condensation particle counter (CPC) (TSI 3772; sucrose melting experiment). The particle counters were used to monitor particle generation and overall flow stability in real time, but were not utilized in phase analysis. The electrostatic filter consisted of a negatively charged ¼″ diameter center electrode maintained at 2 kV electrical potential and a grounded 1″ diameter outer tube. Removal of 100% of charged particles was confirmed experimentally. Particles that dimerized by coagulation of +1 and −1 charges were charge neutral and transmitted through the electrostatic filter. The transmitted uncharged dimerized particles were passed through an X-ray source (TSI 3088), briefly exposed to variations in temperature and/or humidity that may trigger viscous relaxation and subsequent coalescence, and routed through a SMPS. The SMPS consisted of a high flow DMA column (Stolzenburg et al. Citation1998; henceforth HF-DMA) using a CPC as detector (TSI 3020). Sheath and sample flow rates were 4.5 and 0.3 L min−1, respectively. Signal from the TSI 3020 was acquired from the +5 V counter trigger and the instrument was operated in single-particle counting mode, valid of concentrations <2000 cm−3. The SMPS was configured to scan across a range of voltages corresponding to a range of diameters that included the expected coagulated peak. Details on the configuration of the column and SMPS configuration are described elsewhere (Suda and Petters Citation2013; Nguyen et al. Citation2014). All sheath flows were controlled using critical orifices on the outlet and dry, compressed, particle free zero air on the inlet. The RH of zero air was below the detection limit of the RH sensors.
Figure 2. Schematic of the experimental system including typical flow rates in units of L min−1. The setup differed slightly for the sucrose melting experiment as a condensation particle counter was utilized to monitor particle generation instead of an electrometer.
Temperature and humidity control
A series of temperature and humidity sensors (Rotronic HygroClip2) were installed in the system to monitor conditions. Humidity is introduced into the system by incomplete solvent removal by the Nafion membrane driers. The most relevant humidity for interpretation of the experiments is that inside the thermal conditioner, which was maintained under near-isodrosothermal conditions. In cooling cycle experiments dimers were briefly exposed to variations in RH by chilling the flow inside the thermal conditioner. The conditioner consisted of a 14″ long, 3/8″ inner diameter aluminum sample line embedded within an aluminum plate itself in thermal contact with a Peltier-type thermoelectric heat exchanger (CP-200TT, TE Technology, Traverse City, MI, USA). Temperature was measured using a thermistor (TE Technology MP-3193) in physical contact with the metal plate of the conditioner unit. System dew point was measured using a RH sensor positioned within the flow between the outlet of the thermal conditioner and the inlet of the final DMA. The conditioner was able to swing temperatures ±45°C relative to ambient. Thus, the lower range of temperatures during cooling cycle experiments was approximately −20°C, sufficient to saturate the sample. During melting experiments, dimers were briefly exposed to variations in temperature by ramping the conditioner up to as high as 70°C. At 0.3 L min−1 sample flow the average residence time within the conditioner was 5 s.
The thermal conditioning approach for RH control was adopted because it obviates the need for an additional humidification and drying step. However, because viscosity is also a function of temperature, the utilized approach for RH control introduced a temperature dependence viz-à-viz a true humidification system. Specifically, reductions in temperature increase viscosity, potentially counteracting the plasticizing effects of increased RH. For the purposes of this work—demonstrating that RH-induced phase transitions can be detected by the presented coagulation technique—this is an acceptable limitation, although some implications are considered in discussion of the results. During the melting experiment, RH values were <15% at 20°C, so it is expected the temperature dependence of viscosity dominated.
RH sensor calibration and uncertainty
The dew point derived from the RH sensor adjacent to the thermal conditioner was calibrated as follows. Temperature within the conditioner was reduced until a continuous reduction in measured dew point was observed—that is, saturation was reached and conversion of vapor to condensed phase was triggered. Under these conditions, it was assumed that the conditioner temperature was the actual dew point. Deviations between the dew point reported by the sensor and inferred from the conditioner temperature were observed for 2-min intervals. This procedure was repeated at several temperatures between −15°C and −19°C. The observed deviations were progressively more negative at lower temperatures, reaching −1.8°C at −19°C, and the overall trend did not readily fit to either a linear or exponential curve. Accordingly, a piecewise constant-adjustment calibration was implemented. With this approach, we believe that the dew point measurements are in the worst case within 0.3°C of actual. Under typical system conditions (Td > −20°C), this translates to an absolute uncertainty of ±3% in measurements of conditioner RH (Lawrence Citation2005). Total absolute uncertainty in conditioner RH for a single SMPS scan was calculated from the quadrature combination of this value and twice the standard deviation of the RH values observed over the course of the scan. The latter contribution was often negligible compared to the former.
Data collection and processing
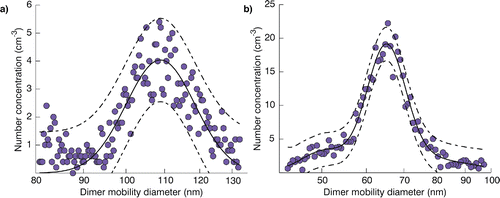
Size spectra measured by the SMPS were fitted to a lognormal distribution. The fitted curve had either one () or three modes (), depending upon the range of voltages utilized in the final DMA scan. The mode diameter of the one mode fit and of the central mode of the three mode fit are interpreted as corresponding to +1 charged particles, taken to be equal to the dimer mobility diameter (Dp). The 95% confidence interval radius for this fitted parameter was taken as the uncertainty in dimer diameter measurement. Confidence intervals of ±1 nm were typical.
Figure 3. Example single-mode (a) and three-mode (b) lognormal fits to SMPS measurements of coagulated dimers for nominal 80 nm ammonium sulfate (a) and 50 nm sucrose (b) monomers (solid lines) with associated 95% observational prediction intervals (dashed lines).
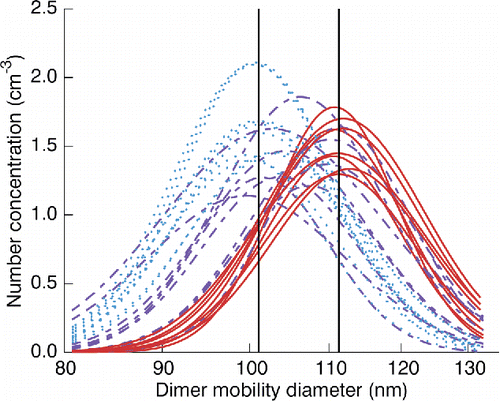
Experiments with a discernible diameter shift due to humidification or heating were fitted to a logistic equation of the form[6] where x is the independent variable of interest (RH for cold cycle experiments, temperature for melting experiments), x0 is a critical value of the independent variable characteristic of coalescence (henceforth relaxation RH [RHr] or relaxation temperature [Tr] as appropriate), and k is a steepness parameter. The 95% confidence interval radius of the x0 parameter was interpreted as the uncertainty in the critical value. The complete set of lognormal fits for the SMPS scans comprising a representative experiment that had a discernible diameter shift is shown in .
Figure 4. Fitted one-mode lognormal curves for all SMPS scans comprising a cooling-cycle experiment for nominal 80 nm sucrose monomers. Curves are coded according to the RH value of the particular SMPS scan: <48.4% (solid), 48.4%–68.4% (dashed), and >68.4% (dotted). These thresholds were selected so as to depict the relation of each scan to the fitted RHr value of 58.4% ± 0.9% and corresponding expected coalescence states (i.e., uncoalesced, partially coalesced, or fully coalesced). The left and right vertical lines correspond to fitted Dc (101.1 nm ± 0.6 nm) and Duc (111.3 nm ± 0.7 nm) values, respectively.
Qualitative characterization of dimer coalescence degree
Cooling cycle experiments for several reagents did not display significant variations in Dp over the entire range of tested RH values. In these cases, a logistic fit was not possible and calibration data was required to assess phase state. For nonvolatile particles, one approach was to compare the diameter to the fitted bounds of a second compound that did display a phase transition. Alternatively, the diameter was compared to the measured dry Duc and Dc values for compounds that were predictably solid or liquid at ambient conditions. Ammonium sulfate and PEG-600 were utilized, respectively. The former readily crystalizes at RH < 40% (Ciobanu et al. Citation2010), well above the RH at the entrance of the monomer selection DMAs (typically 10–20%). The latter has a liquid viscosity of 0.135 Pa·s at 25°C (Zhang et al. Citation2011), well below the threshold for coalescence. PEG-600 begins transitioning from a waxy solid to liquid phase at 32°C with maximum melting at 22°C (Sarier and Onder Citation2008) and PEG-600 always peaked near the expected Dc. For the results presented in this article, the first calibration method was preferred as it minimized the number of species-specific non-idealities introduced, although the second technique was utilized with one experiment where a suitable reference experiment did not exist.
With semivolatile compounds, an alternative approach was utilized. Monomer size measurements were measured using the volatility tandem DMA approach (Rader and McMurry Citation1986). For these measurements, particles were generated sequentially with both source DMAs, the electrostatic filter was turned off, and the evaporated size based on the residence time in our system was observed using the SMPS. Results were used to calculate an expected Dc via Equation (Equation2[2] ). Duc was estimated as 1.1 times this value based upon the expected shape factor.
In the absence of a detectable phase transition measured Dp values slightly larger than Dc or slightly smaller than Duc must be handled with care. In principle, they may correspond to transition viscosity values. However, given the sensitivity of Dp to η in the transition regime, if an analyte has consistent Dp values across a full range of RH values, we believe it likely was either fully coalesced or fully uncoalesced at all RH and the differences in measured mobility diameters arose due to slight differences in deviation from ideal spherical geometry or dry diameter in the size-selecting DMAs.
Experimental results
Cooling cycle (RH) experiments
Results for example compounds from cooling cycle experiments are summarized in . Results for the remaining compounds studied are shown in supplementary Figures S2–S7. Adipic acid () and sodium dodecyl sulfate (Figure S2) consistently peaked at the fully uncoalesced diameter (η ≥ 108 Pa·s) for all RH. Conversely, calcium nitrate (), PEG-1000 (Figure S3), and PEG-10000 (Figure S4) consistently peaked at the fully coalesced diameter (η ≤ 106 Pa·s) for all RH. Note the specified viscosity upper bound is an order of magnitude larger than what would be suggested by . This adjustment is necessary because in the absence of at least one uncoalesced data point it is possible that the time scale between the coagulation event and the dimer reaching the SMPS inlet (an order of magnitude longer than the thermal conditioning time scale assumed by ) was itself sufficient to allow for full coalescence. Finally, sucrose (), ammonium sulfate (Figure S5), mono-sodium α-ketoglutaric acid (Figure S6), and sodium chloride (Figure S7) showed RH-dependent phase state. These results along with corresponding DRH values from literature are summarized in . Observed coagulated diameters in the fully uncoalesced and fully coalesced regimes fell within the 95% observational prediction interval of the fitted logistic curves with few exceptions. This suggests the uncoalesced diameter is roughly constant except for a narrow range of viscosities near the coalescence point, consistent with . Similarly, this suggests that incomplete coalescence is only possible over a narrow range of viscosities, with full coalescence occurring otherwise.
Figure 5. Measured dimer mobility diameters for nominal 80 nm adipic acid (a) and calcium nitrate (b) monomers versus relative humidity with lines indicating fully uncoalesced (top dashed lines) and fully coalesced (bottom dashed lines) diameters as predicted from calibration data.
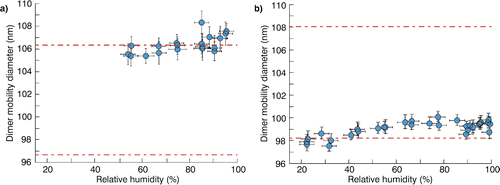
Figure 6. (a) Measured dimer mobility diameters for nominal 80 nm sucrose monomers versus relative humidity, fitted lognormal curve (solid line) with associated 95% observational prediction interval (dashed lines), and viscosity estimates for selected diameters in the transition regime assuming a surface tension of 0.03 J m−2. (b) Comparison of sucrose humidity-dependent viscosity estimates from (a) (line) to measurements reported by Power et al. (Citation2013) (filled circles). The dashed portion of the line indicates onset of coalescence and corresponds to diameters shifts that are within the measurement uncertainty for individual data points. Measurements in panel (a) were collected at temperatures between −3 and −11°C, whereas the Power et al. (Citation2013) measurements were collected at room temperature.
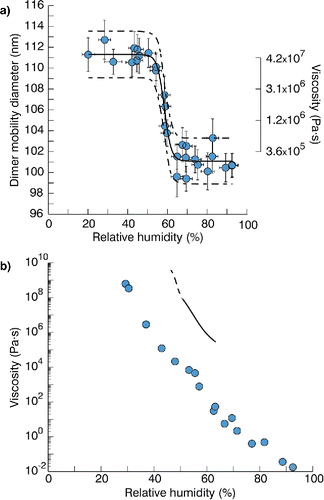
Table 1. Observed relaxation RH valuesFootnotea and literature deliquescence RH values for reagents considered in this work.
For species with discernible phase transitions, RHr was consistently below the DRH for that compound (). Because of the temperature dependence intrinsic to our RH methodology, the difference between the RHr and the DRH may actually be understated by the data presented here. In general, DRH increases with decreasing temperature (Brooks et al. Citation2002). This is because solubility generally decreases with decreasing temperature and DRH and solubility are inversely related (Equation (10.70) in Seinfeld and Pandis Citation2006). Calcium nitrate was found already at reduced viscosity at the lower limits of RH supported by our particle generation system, again well below the reported literature DRH. Adipic acid, which deliquesces under supersaturated conditions (Christensen and Petters Citation2012), did remain uncoalesced at RH values up to 100%. For the inorganics ammonium sulfate, sodium chloride, and calcium nitrate, the reduced viscosity observed prior to deliquescence implies water adsorption onto the crystal can lead to plasticization of the particle and initiate physical restructuring prior to bulk water uptake. This is consistent with reported restructuring artifacts observed from fluorescence microscopy (Montgomery et al. Citation2015) and tandem differential mobility analyzer measurements (Mikhailov et al. Citation2009; Montgomery et al. Citation2015). For sucrose, RHr (58.4% ± 0.9%) is much lower than the DRH (85%) (Salameh and Taylor Citation2006). However, DRH is referenced against a crystallized sucrose particle. Dried sucrose particles do not readily crystallize and form amorphous solids. For example, Robinson et al. (Citation2014) observed that the onset RH for hygroscopic growth was 31%, well below the RHr reported here. This suggests that water adsorption acts as a gradual plasticizer with amorphous solids in addition to crystalline instances. Again, this is consistent with reported restructuring observed in hygroscopicity measurements (Mikhailov et al. Citation2009).
Estimates of viscosity for sucrose at various RH values versus data measured by Power et al. (Citation2013) are plotted in . The trend in our viscosity estimates is similar to that reported by Power et al. (Citation2013), although our estimates are 3–4 orders of magnitude higher. This was likely because our data was collected at much colder temperatures (∼ −7°C vs. room temperature) due to our cold-cycle-based system of RH control. For example, the viscosity of sorbitol (a C6 sugar), increases from ∼105 to ∼1010 Pa·s when cooled from 25°C to 0°C (interpolated from Angell et al. [Citation1982] and glass transition data in Nakanishi and Nozaki [Citation2011]). Therefore, the strong temperature dependence of viscosity at values approaching the glass transition is important for accurate interpretation of the cooling experiments.
Sucrose heating cycle (melting) experiment
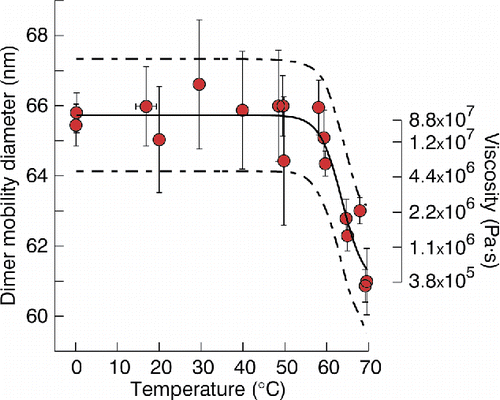
shows example “melting” data for pure sucrose aerosol. In this experiment, Dmono = 50 nm, Dc = 60.9 nm ± 2.5 nm observed at T ∼70°C, Duc = 65.7 nm ± 0.5 nm observed at T < 50°C, and transition diameters corresponding to partially coalesced particles are observed between 50°C and 70°C. The relaxation temperature (Tr) was measured as 63.7°C ± 4.4°C. This temperature is higher than the 60°C glass transition temperature (Tg) of amorphous sucrose estimated from differential scanning calorimetry data (Simperler et al. Citation2006), as would be expected for a less viscous particle. However, the observed difference is within measurement uncertainties in either measurement. This raises the question of how readily Tr and Tg can be distinguished in practice. Because the coalescence observed by our method is estimated to occur at a viscosity spanning from four to seven orders of magnitude less than the glass transition, such a differentiation would be useful in applications modeling the temperature dependence of viscosity. In this particular experiment, limits in our thermal control system prevented collection of data at temperatures well beyond the point of full coalescence. Accordingly, the fit of Dc was imprecise, if still distinct from Duc. If Dc was 1–2 nm too high, the reported Tr will accordingly be too low. Thus, it is quite possible the actual difference between Tg and Tr was greater than presented here. More generally, while to the best of our knowledge little laboratory data exists on the temperature dependence of viscosity for metastable organic semi-solids, available data does suggest there is at least 20°C of difference between η ∼107 and η ∼1012 Pa·s for several compounds containing hydroxyl groups (Hutzler et al. Citation1972). Thus, while the difference between Tr and Tg may not be resolvable for all systems, we expect that they will be for a significant fraction of such systems, and thus the method here is useful for elucidating temperature-viscosity dependency over part of the metastable range, even if the specific numerical result presented here is of limited utility.
Figure 7. Measured dimer mobility diameters for nominal 50 nm sucrose aerosol monomers versus temperature, fitted lognormal curve (solid line) with associated 95% observational prediction interval (dashed lines), and viscosity estimates for selected diameters in the transition regime assuming a surface tension of 0.03 J m−2.
Discussion
Diameter shifts have been observed for Dp ≥ 60 nm. Diameters for fully coalesced and fully uncoalesced particles were broadly consistent with those estimated from the sphere equivalent diameter of a shaped particle. Thus, the method is absolute, i.e., a single measurement of particle size (coupled with suitable verification of dry and coagulated diameters) is enough to classify a particle's viscosity as greater than the threshold (∼108 Pa·s), less than the threshold (∼105 Pa·s), or of a transition value. This may prove especially useful for qualitatively identifying an aerosol as not being in a glassy phase as the threshold viscosity is well below the glassy range. However, actual viscosity estimates are limited to a few orders of magnitude in the middle of the semisolid regime for a given set of experimental parameters. The viscosity corresponding to the midpoint between the fully uncoalesced and fully coalesced particle is ∼2 × 106 Pa·s. Since the inferred viscosity depends on the relaxation time, increasing the residence time inside the conditioner from 5 s to 500 s would increase this value to ∼2 × 108 Pa·s. Similarly, increasing monomer size from 80 to 800 nm would decrease this value to ∼2 × 105 Pa·s. Thus, some additional viscosity estimates can be made by altering experimental parameters, but viscosity measurements across the entire 15 orders of magnitude observed in atmospheric aerosol would require unrealistic combinations of residence time and monomer size. Droplet-based techniques can measure aerosol viscosity on an order of magnitude basis over the entire range of interest (Power et al. Citation2013), but are currently only applicable to micron-diameter particles. Thus, this technique could be applied in validation of (semi-)empirical size corrections mapping micron particle viscosity data to the nanoscale.
There are several additional limitations to the methodology. First, if equilibrium vapor pressure (p°) is sufficiently large that monomers partially evaporate, erroneous conclusions about particle phase state may be reached. For example, evaporation on experimental time scales was observed with succinic acid (p° ≈ 1.5 × 10−3 Pa at 25°C [Koponen et al. Citation2007]). In this vapor pressure range, Duc with evaporation may appear identical to Dc without evaporation. Such cases can be partially corrected for by measuring the selected monomer size using volatility tandem DMA that is implicitly available within the experimental setup. However, the strong dependence of vapor pressure on temperature and evaporation rates on residence time and build up in the vapor phase will reduce the precision of the measurement. Second, the preparation of very high concentrations of charged monomers requires working with broad size cuts and with relatively high aerosol generation flow rates. Coagulated signal can be enhanced by increasing the residence time in the chamber, however, this comes at cost of increased noise due to monomers that undergo spontaneous loss of charge and pass through the electrostatic filter. The atmospheric discharge rate is ∼30 min (Hoppel Citation1985). At the current 8 s configuration and assuming ambient charge fields, up to 0.4% of particles could spontaneously lose their charge inside the coagulation chamber. Actual discharge rate will depend on shielding and location. Third, stable, highly efficient solvent removal without incurring large particle losses is challenging. This may affect the initial particle phase state after drying and before entering the monomer DMAs. Operating the current setup below 10%–20% RH will require modifications to the drying setup. Fourth, nonspherical particle shape will result in deviations from expected Dc values. For example, the measured ammonium sulfate Dc was consistently slightly larger than the theoretical Dse. This is consistent with a slight dynamic shape factor of 1.01–1.04 of crystallized ammonium sulfate (Biskos et al. Citation2006; Zelenyuk et al. Citation2006). Somewhat larger deviations were seen for sodium chloride, which has a dynamic shape factor of ∼1.08 in its cubic crystalline form (Zelenyuk et al. Citation2006; Wang et al. Citation2010; Petters and Petters Citation2016). More strongly shaped crystalline particles may bias viscosity estimates if Dc and Duc are not both known. Finally, inferred viscosity estimates are uncertain due to the need to assume a surface tension. Spanning the realistic range between 0.03 and 0.07 J m−2 results in variation of 0.4 in log10 η values estimated through the modified Frenkel (Pokluda et al. Citation1997) sintering model.
Summary
We have presented a system for classifying particle phase based upon mobility diameter measurements of dimers synthesized by coagulation of charged monomers produced by DMAs of opposite polarities. The phase measurement system is capable to classify particles as either η ≥ 108 Pa·s or η ≤ 105 Pa·s. The measurement is absolute, that is, a single measurement of particle size is enough to determine if the dimer has Duc or Dc, provided that the selected monomer is nonvolatile and approximately spherical. For select conditions, a viscosity can be inferred in the transition regime. Phase transitions can be observed by either humidifying particles subjecting them through a cold cycle, or melting the particle subjecting them through a heat cycle. RH-induced transitions were observed at lower humidities than deliquescence, consistent with existing evidence for RH-induced structural reorganizations. We expect the melting transition to be a useful complement to the glass transition and to occur under similar conditions.
UAST_A_1221050_Supplement.docx
Download MS Word (964.5 KB)Acknowledgments
We thank Darrel Baumgardner and Patrick Chuang for use of the RDMA and HF-DMA, respectively. The authors thank Timothy Wright for technical assistance with the instrumentation.
Funding
This research was funded by United States Department of Energy, Office of Science, Biological and Environment Research, Grant number DE-SC 0012043.
Related Research Data
References
- Angell, C. A., Stell, R. C., and Sichina, W. (1982). Viscosity-Temperature Function for Sorbitol from Combined Viscosity and Differential Scanning Calorimetry Studies. J. Phys. Chem, 86:1540–1542.
- Arenas, K. J. L., Schill, S. R., Malla, A., and Hudson, P. K. (2012). Deliquescence Phase Transition Measurements by Quartz Crystal Microbalance Frequency Shifts. J. Phys. Chem. A, 116:7658–7667.
- Bateman, A. P., Bertram, A. K., and Martin, S. T. (2015). Hygroscopic Influence on the Semisolid-to-Liquid Transition of Secondary Organic Materials. J. Phys. Chem. A, 119:4386–4395.
- Baustian, K. J., Wise, M. E., Jensen, E. J., Schill, G. P., Freedman, M. A., and Tolbert, M. A. (2013). State Transformations and Ice Nucleation in Amorphous (Semi-)Solid Organic Aerosol. Atmos. Chem. Phys., 13:5615–5628.
- Berkemeier, T., Shiraiwa, M., Pöschl, U., and Koop, T. (2014). Competition Between Water Uptake and Ice Nucleation by Glassy Organic Aerosol Particles. Atmos. Chem. Phys., 14:12513–12531.
- Biskos, G., Paulsen D., Russell, L. M., Buseck, P. R., and Martin, S. T. (2006). Prompt Deliquescence and Efflorescence of Aerosol Nanoparticles. Atmos. Chem. Phys., 6:4633–4642.
- Bones, D. L., Reid, J. P., Lienhard, D. M., and Krieger, U. K. (2012). Comparing the Mechanism of Water Condensation and Evaporation in Glassy Aerosol. Proc. Natl. Acad. Sci. U. S. A., 109:11613–11618.
- Borra, J. P., Camelot, D., Chou, K. L., Kooyman, P. J., Marijnissen, J. C. M., and Scarlett, B. (1999). Bipolar Coagulation for Powder Production: Micro-Mixing Inside Droplets. J. Aerosol Sci., 30:945–958.
- Brooks, S. D., Wise, M. E., Cushing, M., and Tolbert, M. A. (2002). Deliquescence Behavior of Organic/Ammonium Sulfate Aerosol. Geophys. Res. Lett., 29:1917.
- Cappa, C. D., Lovejoy, E. R., and Ravishankara, A. R. (2008). Evidence for Liquid-Like and Nonideal Behavior of a Mixture of Organic Aerosol Components. Proc. Natl. Acad. Sci. U. S. A., 105:18687–18691.
- Chan, L. P., and Chan, C. K. (2013). Role of the Aerosol Phase State in Ammonia/Amines Exchange Reactions. Environ. Sci. Technol., 47:5755–5762.
- Cheng, Y., Su, H., Koop, T., Mikhailov E., and Pöschl, U. (2015). Size Dependence of Phase Transitions in Aerosol Nanoparticles. Nat. Commun., 6:5923.
- Christensen, S. I., and Petters, M. D. (2012). The Role of Temperature in Cloud Droplet Activation. J. Phys. Chem. A., 116:9706–9717.
- Ciobanu, V. G., Marcolli, C., Krieger, U. K., Zuend, A., and Peter, T. (2010). Efflorescence of Ammonium Sulfate and Coated Ammonium Sulfate Particles: Evidence for Surface Nucleation. J. Phys. Chem. A, 114:9486–9495.
- Davies, J. F., and Wilson, K. R. (2015). Nanoscale Interfacial Gradients Formed by the Reactive Uptake of OH Radicals Onto Viscous Aerosol Surfaces. Chem. Sci., 6:7020–7027.
- Debenedetti, P. G., and Stillinger, F. H. (2001). Supercooled Liquids and the Glass Transition. Nature, 410:259–267.
- Dette, H. P., and Koop, T. (2015). Glass Formation Processes in Mixed Inorganic/Organic Aerosol Particles. J. Phys. Chem. A, 119:4552–4561.
- Goldstein, A. H., and Galbally, I. E. (2007). Known and Unexplored Organic Constituents in the Earth's Atmosphere. Environ. Sci. Technol., 41:1514–1521.
- Gržinić, G., Bartels-Rausch, T., Berkemeier, T., Türler, A., and Ammann, M. (2015). Viscosity Controls Humidity Dependence of N2O5 Uptake to Citric Acid Aerosol. Atmos. Chem. Phys., 15:13615–13625.
- Hinds, W. C. (1999). Aerosol Technology: Properties, Behavior, and Measurement of Airborne Particles (2nd ed.). John Wiley & Sons, New York, pp. 52.
- Hings, S. S., Wrobel, W. C., Cross, E. S., Worsnop, D. R., Davidovits, P., and Onasch, T. B. (2008). CCN Activation Experiments with Adipic acid: Effect of Particle Phase and Adipic Acid Coatings on Soluble and Insoluble Particles. Atmos. Chem. Phys., 8:3735–3748.
- Hoppel, W. A. (1985). Ion-Aerosol Attachment Coefficients, Ion Depletion, and the Charge Distribution on Aerosols. J. Geophys. Res., 90:5917–5923.
- Hosny, N. A., Fitzgerald, C., Tong, C., Kalberer, M., Kuimova, M. K., and Pope, F. D. (2013). Fluorescent Lifetime Imaging of Atmospheric Aerosols: A Direct Probe of Aerosol Viscosity. Faraday Discuss., 165:343.
- Hutzler, J. S., Colton, R. J., and Ling, A. C. (1972). Viscosities of Some Organic Glasses Used as Trapping Matrices. III. J. Chem. Eng. Data, 17:324–327.
- Järvinen, E., Ignatius, K., Nichman, L., Kristensen, T. B., Fuchs, C., Hoyle, C. R., Höppel, N, Corbin, J. C., Craven, J., Duplissy, J., Ehrhart, S., El Haddad, I., Frege, C., Gordon, H., Jokinen, T., Kallinger, P., Kirkby, J., Kiselev, A., Naumann, K., Petäjä, T., Pinterich, T., Prevot, A. S. H., Saathoff, H., Schiebel, T., Sengupta, K., Simon, M., Slowik, J. G., Tröstl, J., Virtanen, A., Vochezer, P., Vogt, S., Wagner, A. C., Wagner, R., Williamson, C., Winkler, P. M., Yan, C., Baltensperger, U., Donahue, N. M., Flagen, R. C., Gallagher, M., Hansel, A., Kulmala, M., Stratmann, F., Worsnop, D. R., Möhler, O., Leisner, T., and Schnaiter, M. (2016). Observation of Viscosity Transition in α-Pinene Secondary Organic Aerosol, Atmos. Chem. Phys., 16:4423–4438.
- Kelly, J. T., and Wexler, A. S. (2006). Water Uptake by Aerosol: Water Activity in Supersaturated Potassium Solutions and Deliquescence as a Function of Temperature. Atmos. Environ., 40:4450–4468.
- Koop, T., Bookhold, J., Shiraiwa, M., and Pöschl, U. (2011). Glass Transition and Phase State of Organic Compounds: Dependency on Molecular Properties and Implications for Secondary Organic Aerosols in the Atmosphere. Phys. Chem. Chem. Phys., 13:19238–19255.
- Koponen, I. K., Riipinen, I., Hienola, A., Kulmala, M., and Bilde, M. (2007). Thermodynamic Properties of Malonic, Succinic, and Glutaric Acids: Evaporation Rates and Saturation Vapor Pressures. Environ. Sci. Technol., 41:3926–3933.
- Lawrence, M. G. (2005). The Relationship Between Relative Humidity and the Dew Point Temperature in Moist Air: A Simple Conversion and Applications. Bull. Amer. Meteorol. Soc., 86:225–233.
- Lu, J. W., Rickards, A. M. J., Walker, J. S., Knox, K. J., Miles, R. E. H., Reid, J. P., and Signorell, R. (2014). Timescales of Water Transport in Viscous Aerosol: Measurements on Sub-Micron Particles and Dependence on Conditioning History. Phys. Chem. Chem. Phys., 16:9819–9830.
- Maisels, A., Kruis, F. E., Fissan, H., Rellinghaus, B., and Zähres, H. (2000). Synthesis of Tailored Composite Nanoparticles in the Gas Phase. Appl. Phys. Lett., 77:4431–4433.
- Marcolli, C., Luo, B. P., and Peter, T. (2004). Mixing of the Organic Aerosol Fractions: Liquids as the Thermodynamically Stable Phases. J. Phys. Chem. A, 108:2216–2224.
- Mikhailov, E., Vlasenko, S., Martin, S. T., Koop, T., and Pöschl, U. (2009). Amorphous and Crystalline Aerosol Particles Interacting with Water Vapor: Conceptual Framework and Experimental Evidence for Restructuring, Phase Transitions, and Kinetic Limitations. Atmos. Chem. Phys., 9:9491–9522.
- Montgomery, J. F., Rogak, S. N., Green, S. I., You, Y., and Bertram, A. K. (2015). Structural Change of Aerosol Particle Aggregates with Exposure to Elevated Relative Humidity. Environ. Sci. Technol., 49:12054–12061.
- Nakanishi, M., and Nozaki, R. (2011). Systematic Study of the Glass Transition in Polyhydric Alcohols. Phys. Rev. E, 83:051503.
- Nguyen, T. K. V., Petters, M. D., Suda, S. R., Guo, H., Weber, R. J., and Carlton, A. M. (2014). Trends in Particle Phase Liquid Water During the Southern Oxidant and Aerosol Study. Atmos. Chem. Phys., 14:10911–10930.
- Petters, S. S., and Petters, M. D. (2016). Surfactant Effect on Cloud Condensation Nuclei for Two-Component Internally Mixed Aerosols. J. Geophys. Res. Atmos., 121:1878–1895.
- Petters, M. D., Kreidenweis, S. M., Snider, J. R., Koehler, K. A., Wang, Q., Prenni, A. J., and DeMott, P. J. (2006). Cloud Droplet Activation of Polymerized Organic Aerosol. Tellus B, 58:196–205.
- Pfrang, C., Shiraiwa, M., and Pöschl, U. (2011). Chemical Ageing and Transformation of Diffusivity in Semi-Solid Multi-Component Organic Aerosol Particles. Atmos. Chem. Phys., 11:7343–7354.
- Pokluda, O., Bellehumeur, C., and Vlachopoulos, J. (1997). Modification of Frenkel's Model for Sintering. AIChE J., 43:3253–3256.
- Power, R. M., Simpson, S. H., Reid, J. P., and Hudson, A. J. (2013). The Transition from Liquid to Solid-Like Behaviour in Ultrahigh Viscosity Aerosol Particles. Chem. Sci., 4:2597–2604.
- Rader, D. J., and McMurry, P. H. (1986). Application of the Tandem Differential Mobility Analyzer to Studies of Droplet Growth or Evaporation. J. Aerosol Sci., 17:771–787.
- Renbaum-Wolff, L., Grayson, J. W., Bateman, A. P., Kuwata, M., Sellier, M., Murray, B., Shilling, J. E., Martin, S. T., and Bertram, A. K. (2013). Viscosity of α-Pinene Secondary Organic Material and Implications for Particle Growth and Reactivity. Proc. Natl. Acad. Sci. U. S. A., 110:8014–8019.
- Robinson, C. B., Schill, G. P., and Tolbert, M. A. (2014). Optical Growth of Highly Viscous Organic/Sulfate Particles. J. Atmos. Chem., 71:145–156.
- Roth, C. M., Goss, K.-U., and Schwarzenbach, R. P. (2005). Sorption of a Diverse Set of Organic Vapors to Urban Aerosols. Environ. Sci. Technol., 39:6638–6643.
- Salameh, A. K., and Taylor, L. S. (2006). Role of Deliquescence Lowering in Enhancing Chemical Reactivity in Physical Mixtures. J. Phys. Chem. B, 110:10190–10196.
- Sarier, N., and Onder, E. (2008). Thermal Insulation Capability of PEG-Containing Polyurethane Foams. Thermochimica Acta, 475:15–21.
- Schill, G. P., and Tolbert, M. A. (2013). Heterogeneous Ice Nucleation on Phase-Separated Organic-Sulfate Particles: Effect of Liquid vs. Glassy Coatings. Atmos. Chem. Phys., 13:4681–4695.
- Seinfeld, J. H., and Pandis, S. N. (2006). Atmospheric Chemistry and Physics: From Air Pollution to Climate Change (2nd ed.). John Wiley & Sons, Hoboken, pp. 454, 464, 599.
- Shiraiwa, M., Ammann, M., Koop, T., and Pöschl, U. (2011). Gas Uptake and Chemical Aging of Semisolid Organic Aerosol Particles. Proc. Natl. Acad. Sci. U. S. A., 108:11003–11008.
- Simperler, A., Kornherr, A., Chopra, R., Bonnet, P. A., Jones, W., Motherwell, W. D. S., and Zifferer, G. (2006). Glass Transition Temperature of Glucose, Sucrose, and Trehalose: An Experimental and in Silico Study. J. Phys. Chem. B, 110:19678–19684.
- Stolzenburg, M. R., Kreisberg, N. M., and Hering, S. V. (1998). Atmospheric Size Distributions Measured by Differential Mobility optical Particle Size Spectrometry. Aerosol Sci. Technol., 29:402–418.
- Suda, S. R., and Petters, M. D. (2013). Accurate Determination of Aerosol Activity Coefficients at Relative Humidities up to 99% Using the Hygroscopic Tandem Differential Mobility Analyzer Technique. Aerosol Sci. Technol., 47:991–1000.
- Tong, H.-J., Reid, J. P., Bones, D. L., Luo, B. P., and Krieger, U. K. (2011). Measurements of the Timescales for the Mass Transfer of Water in Glassy Aerosol at low Relative Humidity and Ambient Temperature. Atmos. Chem. Phys., 11:4739–4754.
- Virtanen, A., Joutsensaari, J., Koop, T., Kannosto, J., Yli-Pirilä, P., Mäkelä, J. M., Holopainen, J. K., Pöschl, U., Kulmala, M., Worsnop, D. R., and Laaksonen, A. (2010). An Amorphous Solid State of Biogenic Secondary Organic Aerosol Particles. Nature, 467:824–827.
- Volkamer, R., Jimenez, J. L., San Martini, F., Dzepina, K., Zhang, Q., Salcedo, D., Molina, L. T., Worsnop, D. R., and Molina, M. J. (2006). Secondary Organic Aerosol Formation from Anthropogenic Air Pollution: Rapid and Higher than Expected. Geophys. Res. Lett., 33:L17811.
- Wang, B., Lambe, A. T., Massoli, P., Onasch, T. B., Davidovits, P., Worsnop, D. R., and Knopf, D. A. (2012). The Deposition Ice Nucleation and Immersion Freezing Potential of Amorphous Secondary Organic Aerosol: Pathways for Ice and Mixed-Phase Cloud Formation. J. Geophys. Res. Atmos., 117:1–12.
- Wang, Z., King, S. M., Freney, E., Rosenoern, T., Smith, M. L., Chen, Q., Kuwata, M., Lewis, E. R., Pöschl, U., Wang, W., Buseck, P. R., and Martin, S. T. (2010). The Dynamic Shape Factor of Sodium Chloride Nanoparticles as Regulated by Drying Rate. Aerosol Sci. Technol., 44:939–953.
- Woods, E., Kim, H. S., Wivagg, C. N., Dotson, S. J., Broekhuizen, K. E., and Frohardt, E. F. (2007). Phase Transitions and Surface Morphology of Surfactant-Coated Aerosol Particles. J. Phys. Chem. A., 111:11013–11020.
- Zebel, G. (1958). Zur Theorie des Verhaltens Elektrisch Geladener Aerosole. Kolloid-Zeitschrift., 157:37–50.
- Zelenyuk, A., Cai, Y., and Imre, D. (2006). From Agglomerates of Spheres to Irregularly Shaped Particles: Determination of Dynamic Shape Factors from Measurements of Mobility and Vacuum Aerodynamic Diameters. Aerosol Sci. Technol., 40:197–217.
- Zhang, K., Yang, J., Yu, X., Zhang, J., and Wei, X. (2011). Densities and Viscosities for Binary Mixtures of Poly(ethylene Glycol) 400 + Dimethyl Sulfoxide and Poly(ethylene Glycol) 600 + Water at Different Temperatures. J. Chem. Eng. Data, 56:3083–3088.
- Zhang, S., Akutsu, Y., Russell, L. M., Flagan, R. C., and Seinfeld, J. R. (1995). Radial Differential Mobility Analyzer. Aerosol Sci. Technol., 23:357–372.
- Zhang, Y., Sanchez, M. S., Douet, C., Wang, Y., Bateman, A. P., Gong, Z., Kuwata, M., Renbaum-Wolff, L., Sato, B. B., Liu, P. F., Bertram, A. K., Geiger, F. M., and Martin, S. T. (2015). Changing Shapes and Implied Viscosities of Suspended Submicron Particles. Atmos. Chem. Phys., 15:7819–7829.
- Zobrist, B., Soonsin, V., Luo, B. P., Krieger, U. K., Marcolli, C., Peter, T., and Koop, T. (2011). Ultra-Slow Water Diffusion in Aqueous Sucrose Glasses. Phys. Chem. Chem. Phys., 13:3514–3526.
- Zobrist, B., Marcolli, C., Pedernera, D. A., and Koop, T. (2008). Do Atmospheric Aerosols form Glasses? Atmos. Chem. Phys., 8:5221–5244.