
ABSTRACT
The Aerodyne Aerosol Chemical Speciation Monitor (ACSM) is well suited for measuring non-refractory particulate matter up to approximately 1.0 µm in aerodynamic diameter (NR-sub-PM1). However, for larger particles the detection efficiency is limited by losses in the sampling inlet system and through the standard aerodynamic focusing lens. In addition, larger particles have reduced collection efficiency due to particle bounce at the vaporizer. These factors have limited the NR-sub-PM1 ACSM from meeting PM2.5 (particulate matter with aerodynamic diameter smaller than 2.5 µm) monitoring standards. To overcome these limitations, we have redesigned the sampling inlet, the aerodynamic lens, and particle vaporizer. Both the new lens and vaporizer are tested in the lab using a quadruple aerosol mass spectrometer (QAMS) system equipped with light scattering module. Our results show that the capture vaporizer introduces additional thermal decomposition of both inorganic and organic compounds, requiring modifications to the standard AMS fragmentation table, which is used to partition ion fragments to chemical classes. Experiments with mixed NH4NO3 and (NH4)2SO4 particles demonstrated linearity in the NH4+ ion balance, suggesting that there is no apparent matrix effect in the thermal vaporization-electron impact ionization detection scheme for mixed inorganic particles. Considering a typical ambient PM2.5 size distribution, we found that 89% of the non-refractory mass is detected with the new system, while only 65% with the old system. The NR-PM2.5 system described here can be adapted to existing Aerodyne Aerosol Mass Spectrometer (AMS) and ACSM systems.
Copyright © 2017 American Association for Aerosol Research
EDITOR:
1. Introduction
Aerosols play a significant role in influencing the Earth's radiation budget, altering atmospheric chemistry, and adversely affecting human health (Pöschl Citation2005; Pope and Dockery Citation2006; Solomon et al. Citation2007). These effects of aerosols are highly correlated with their physicochemical properties, such as size, chemical composition, and mass loading. Aerodyne Aerosol Mass Spectrometers (AMS) have been developed to quantify these properties (Jayne et al. Citation2000; Jimenez et al. Citation2003; Canagaratna et al. Citation2007). While the AMS provides invaluable information regarding size, mass loadings, and non-refractory chemical composition simultaneously with high time resolution (1 min or less), it is not well suited for routine measurements for air quality monitoring because of its high cost, complex operational and maintenance procedures, and multidimensional datasets. To address these issues, the Aerosol Chemical Speciation Monitor (ACSM) was developed (Ng et al. Citation2011), which has a lower cost, simplified data acquisition and analysis interface, and comparable composition measurement capabilities as the AMS. The ACSM does not measure particle size.
ACSMs have been deployed in monitoring stations and during field campaigns to measure the mass loading of the major non-refractory (NR) chemical species (organic matter, sulfate, nitrate, ammonium, and chloride) in particulate matter with aerodynamic diameter smaller than 1.0 µm (NR-sub-PM1) (Ng et al. Citation2011; Sun et al. Citation2012, Citation2013a,Citationb; Budisulistiorini et al. Citation2013; Crenn et al. Citation2015; Fröhlich et al. Citation2015). (For simplicity, we will use PM1 for NR-sub-PM1 and PM2.5 for NR-PM2.5 in the rest of this article.) The initial ACSM design was based on a quadrupole mass spectrometer (MS) detector. Recently, Fröhlich et al. (Citation2013) reported a Time-of-Flight MS based ACSM (ToF-ACSM) that has greater sensitivity and faster time response. ACSM systems have been widely deployed by the European Union project Aerosols, Clouds, and Trace gases Research InfraStructure (ACTRIS) to provide a long-term record of chemically speciated aerosol mass loading. In the fall of 2013, ACTRIS ACSM participants organized an intercomparison study with 13 quadrupole ACSMs (Q-ACSM), 1 ToF-ACSM, and 1 high-resolution ToF-AMS (HR-AMS). The results of this intercomparison are reported by Crenn et al. (Citation2015) who discussed the comparison between the ACSMs as well as the comparison with other measurement techniques, and Fröhlich et al. (Citation2015) who summarized source apportionment results with the application of positive matrix factorization (PMF) to these data sets.
While the standard ACSM has proved to be a successful speciation monitor for PM1, it has limited detection efficiency for larger particles. For general monitoring usage, an ACSM with PM2.5 (particulate matter with aerodynamic diameter smaller than 2.5 µm) measurement capability is required to meet mass-based air quality reporting standards. The low detection efficiency of super-micron particles is due to three factors: loss of particles due to impaction on sampling inlet surfaces, low transmission efficiency in the particle aerodynamic lens, which is a key particle delivery component of the ACSM and AMS, and low collection efficiency caused by particle bounce off the vaporizer surface. Throughout this work, we will make reference to the standard (PM1) ACSM, which is composed of the PM1 aerodynamic (standard) lens and the standard vaporizer.
Previous efforts have attempted to overcome the limitations of particle transmission in the aerodynamic lens. For example, a new inlet and high pressure aerodynamic lens (HPL) were developed for the transmission of particles between 80 nm and more than 3 µm in vacuum aerodynamic diameter (dva) (Williams et al. Citation2013), where dva is the diameter measured by the AMS defined as (Jayne et al. Citation2000)[1]
In Equation (Equation1[1] ), dmob is the mobility diameter measured by a differential mobility analyzer (DMA), ρ is the particle density, ρ0 is unit density, and S is the Jayne shape factor (Jayne et al. Citation2000; DeCarlo Citation2004). The relationships between dva, dmob, aerodynamic diameter (da), and physical diameter (dp) are explained in detail in DeCarlo et al. (Citation2004). The typical pressure in the standard PM1 lens is ∼173 Pa (1.3 Torr), while the pressure in the HPL is ∼1840 Pa (13.8 Torr). The higher pressure increases the drag force on particles, which focuses the larger particles to the center axis of the lens (Liu et al. Citation1995a,b; Schreiner et al. Citation1999; Williams et al. Citation2013). The experimental results were corroborated by computational fluid dynamics (CFD) modeling of particle trajectories (Williams et al. Citation2013). Although the HPL is a successful design for large particle transmission, the very high machining precision required for the HPL to perform well leads to a prohibitive lack of reproducibility in its manufacture (Williams et al. Citation2013).
Here, we present a new intermediate pressure lens (IPL) design that operates at a pressure of ∼507 Pa (3.8 Torr). The IPL, together with a new inlet particle delivery system, dramatically improves the transmission of large particles (1 to 2.5 µm). Analytical modeling of the IPL has been reported recently by our group (Peck et al. Citation2016). The IPL assembly can be machined with greater reproducibility than the HPL. However, since it operates at a lower pressure, the large particle transmission does not extend quite as far as the HPL.
To increase the particle collection efficiency at the detector, a new style of vaporizer, called the capture vaporizer (CV), was designed with an enclosed cavity, in contrast to the open cone of the standard vaporizer (SV) (). The combination of the IPL, the redesigned inlet system, and the capture vaporizer make up the PM2.5 ACSM system. We note that, while these developments are motivated by the goal of making the ACSM meet PM2.5 monitoring standards, the developments are also applicable to AMS systems. Most of the laboratory data presented here were acquired with an AMS (without the ACSM inlet system) rather than an ACSM to take advantage of particle-sizing, greater sensitivity, and faster data acquisition of the AMS.
Figure 1. Schematic of the experimental setup for (a) large particle size (>650 nm dva) lens transmission and particle bounce measurement, (b) and (c) small particle size side (<650 nm dva) lens transmission efficiency curve measurement without CPMA and with CPMA, respectively, and (d) schematic of an ACSM/AMS equipped with the new PM2.5 inlet, IPL, and capture vaporizer.
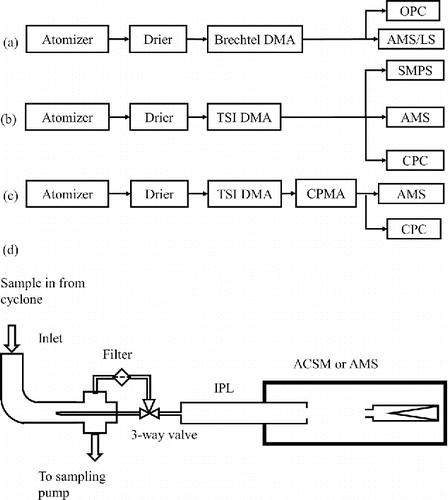
2. Experimental methods
2.1. Aerodynamic lens transmission efficiency measurements
The overall collection efficiency (CE) for the AMS or ACSM is a product of three terms, lens transmission efficiency (EL, which does not include sampling inlet transmission), collection efficiency due to particle bounce off the vaporizer (EB), and collection efficiency due to particle beam spread (ES) (Huffman et al. Citation2005),[2]
For the particle compositions used here, NaNO3, NH4NO3, (NH4)2SO4, and polystyrene latex spheres (PSL), ES is approximately 1, as independently determined in this work using a beam width probe (BWP, Huffman et al. Citation2005). The size-dependent transmission efficiency of the IPL was characterized using a quadrupole AMS (Q-AMS) with a light scattering (LS) module and standard vaporizer installed without an ACSM standard inlet. A schematic of the experimental setup was shown previously by Cross et al. (Citation2007), with the only difference here being that an IPL was installed. We used different methods for different particle size ranges. For larger size particles (400 nm to 2 µm dmob), we measured EL directly by comparing the number concentration of particles entering the Q-AMS with the number concentration of particles exiting the particle lens (). As a result, the size-dependent EL can be expressed as[3] where dva is the particle vacuum aerodynamic diameter; NLS is measured with the LS module and NOPC is measured with a GRIMM Technologies, Inc. model 1.109 optical particle counter (OPC). Particles larger than ∼ 1 µm in size were either NaNO3 or PSL generated using an atomizer or medical nebulizer and size selected using a Brechtel DMA because of its ability to select larger size particles (300 nm to 2 µm dmob) compared to the TSI DMA 3080L (10 nm to 1 µm dmob). The vaporizer temperature was set to 900°C for NaNO3, instead of the typical AMS vaporizer temperature of 600°C, because of its lower volatility than NH4NO3.
For particles with mobility diameter (dmob) between 250 nm and 450 nm, both LS and the mass-based method (Equations (Equation3[3] ) and (Equation4
[4] )) can be used to measure the lens particle transmission efficiency. In the mass-based method, the mass loading measured with the Q-AMS is compared to the mass loading calculated from the CPC counts. The Q-AMS operates in two modes, mass spectrum (MS) mode, in which the entire mass spectrum is collected for the ensemble of particles sampled, and particle time-of-flight mode (pToF), in which a single m/z is monitored for particles that have passed through a slit in a mechanical chopper wheel. The velocity of the particles (distance between the chopper and the detector/time of flight) is converted to particle size with a calibration curve.
For these smaller sizes, to obtain smaller dva based on Equation (Equation1[1] ), we used NH4NO3 particles, because of its lower density (1.72 g cm−3) compared with that of NaNO3 (2.26 g cm−3). Since NH4NO3 particles are known not to bounce off the vaporizer (EB = 1) (Middlebrook et al. Citation2011), the size-dependent CE = EL and EL can be expressed as
[4]
[5] where MassMS (dva) and MasspToF (dva) are the mass measured in either MS or pToF modes, and MassCPC (dva) is calculated from the CPC counts, dmob, ρ, and S. The ion signals in the Q-AMS are converted to mass loading using an experimentally determined ionization efficiency for NO3, mIENO3 (ions/pg), in which the sum of the signals (ions/s) at NO+ (m/z 30) and NO2+ (m/z 46) for size selected 300 nm NH4NO3 particles is ratioed to the mass calculated from the CPC counts, dmob, ρ, and S, and the flow rate into the instrument. For these measurements of EL, we measured NO3 mass with m/z 46 only using an effective mIE46, based on the fact that the signal intensity ratio of m/z 46/m/z 30 is independent of particle size. The advantage of using m/z 46 is its narrower pToF profile than that of m/z 30, which makes the pToF separation of singly and doubly charged particles much easier.
For particles smaller than 250 nm dmob, only the mass-based method was used for the transmission measurements, since LS module signals are too low for 100% detection efficiency (Cross et al. Citation2009). For these small sizes, care is required when using Equation (Equation4[4] ) for calculating EL, because small particles generated using an atomizer and a differential mobility analyzer (DMA) can have a significant number of multiply charged particles (larger particles with equivalent electrical mobility exiting the DMA). In MS mode, where only the integrated sum of all sizes of particles are measured, it is difficult to correct the total mass loading (numerator in Equation (Equation4
[4] )) for the presence of multiply charged particles. Instead, EL is calculated using Equation (Equation5
[5] ), in which the calculated input mass loading (MassCPC) is corrected for multiply charged particles using an SMPS-measured size distribution () and MasspToF (dva) represents only singly charged particles, since multiply and singly charged particles are clearly separated in the AMS pToF (dva) space. Alternatively, the multiply charged particles can be removed by adding a centrifugal particle mass analyzer (CPMA, Olfert and Collings Citation2005) after the DMA (). In the absence of multiply charged particles, both Equations (Equation4
[4] ) and (Equation5
[5] ) are applicable and the CPC calculated mass loading does not need to be corrected. Throughout most of this work, we used a DMA followed by a CPMA to eliminate multiply charged large diameter particles.
2.2. Particle bounce measurement of capture vaporizer
The bounce of particles off the vaporizer surface was measured using the same experimental setup as described by Cross et al. (Citation2009), shown here in . For large particles (dva > ∼400 nm), EB can be expressed as the ratio of particles counted by the MS after vaporization to the particles counted by LS before impacting the vaporizer[6] where NMS is the number concentration of single particle peaks in pToF space, and MassLS is obtained from NLS, and the single particle mass (calculated from mobility diameter, material density, and the shape factor, Jayne et al. Citation2000). The greater than or equal to sign represents the fact that there can be incomplete vaporization of single particles that hit the vaporizer resulting in an under measured mass compared to the individually counted particle. The mass ratio was used instead of the count ratio because, for the capture vaporizer, single particle vaporization events are not sharp in time (>250 µs, compared with 20–50 µs width for the standard vaporizer) and the threshold crossing based particle counting scheme in the Q-AMS cannot cleanly distinguish single particle events. This smearing of the single particle signal in time is an important aspect of the CV, which will be discussed later. In addition, for large particles, the counted number of particles may not represent the measured particle mass due to incomplete vaporization and multiple vaporization events for a single particle (bouncing). Large dva (>750 nm) NaNO3 particles were generated using a medical nebulizer and then size selected with a Brechtel DMA.
Since ambient particles are typically a mixture of chemical compounds, such as NH4NO3, (NH4)2SO4, and organics, it is desirable to measure the EB of internally mixed particles. Mixed inorganic particles were generated by atomizing solutions of NH4NO3 and (NH4)2SO4 with known composition ratios. Solutions with five different mass ratios, 30%, 43%, and 69% (NH4)2SO4, pure NH4NO3, and pure (NH4)2SO4, were used. Size- (DMA) and mass-(CPMA) selected 300 nm mixed particles were measured with the Q-AMS. The CE and the relative ionization efficiencies (RIE) for NH4 and SO4 (RIENH4 = mIENH4/mIENO3 and RIESO4 = mIESO4/mIENO3) were determined by comparing the measured NO3, SO4, and NH4 mass with the corresponding input mass, which were calculated using the CPC concentration, CPMA single particle mass, and composition ratios.
3. Results and discussion
3.1. Size-dependent transmission efficiency through the ACSM sampling system
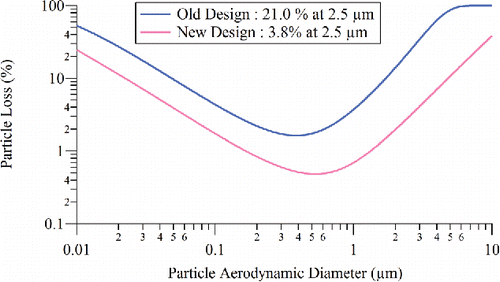
ACSM instruments, unlike AMS instruments, are delivered with an integrated sampling system, consisting of a cyclone (URG 3 LPM PM2.5), a 9.5 mm (0.375 in) inner diameter (ID) sampling line, an iso-kinetic sampler to draw the ACSM sample flow at 85 cm3/min, and a three-way valve to switch between ambient and filter sampling. The standard ACSM inlet after the iso-kinetic sampler consists of 3.2 mm (0.125 in) outer diameter (OD) and 1.4 mm (0.055 in) ID tubing with two 90° bends and limits the transmission of super-micron size particles, as does a 90° bend in the three-way valve. In this section, we describe a redesign of the ACSM sampling system to allow better transmission of super-micron sized particles (). A new three-way valve with a straight-through flow pattern for the sample position (and 90° bend only for the filter position), replaces the previous valve, in which the sample flow had a 90° bend (Ng et al. Citation2011). The new ACSM inlet line consists of a short (∼10 cm) straight tube with the same OD (3.2 mm or 0.125 in.) and ID (1.4 mm or 0.055 in.) as before. The size-dependent particle loss in the old and new designs of the ACSM inlet was calculated using a particle loss calculator (Von der Weiden et al. Citation2009), and the results are shown in . Particle loss at 2.5 µm da for the new inlet (3.8%) is much smaller than that for the old inlet (21%), which was mainly due to inertial deposition of large particles (da > 1.0 µm).
Figure 2. Calculated size-dependent particle loss for the old and new designs of the ACSM inlet.
3.2. Lens transmission results
shows the average transmission efficiency (EL) for four copies of the IPL measured independently with the Q-AMS. The error bars are the standard deviation (1σ) of four different lens measurements. The dva range from 50 to 650 nm is covered by NH4NO3 particles, using the DMA, SMPS, and CPC setup () and the mass method (Equation (Equation5[5] )), while the range above 650 nm is covered by NaNO3 and PSL particles, using the DMA and OPC setup () and the count method (Equation (Equation1
[1] )). The transmission efficiencies of PSLs generally agree with those of NaNO3, within experimental uncertainties. The current PM2.5 lens transmission efficiency is also compared with that of the standard PM1 lens () (Liu et al. Citation2007). The major change in the design of the IPL PM2.5 lens compared to the standard lens is the geometry of the exit nozzle, which has a diameter of 1.6 mm, compared with 0.9 mm in the HPL and 3.0 mm in the standard PM1 lens (Liu et al. Citation2007). The consequent intermediate pressure is a compromise between large particle transmission and less stringent machining tolerances required to produce the lens. The IPL lens transmission efficiency is about 50% at 3.5 µm vacuum aerodynamic diameter, which is approximately equal to a 2.7 µm aerodynamic diameter assuming an average ambient particle density of 1.6 g/cm3 (Hu et al. Citation2012). This agrees well with the transmission cutoff of the PM2.5 EPA Federal Reference Method (FRM) WINS (Well impactor ninety-six) separator, converted to vacuum aerodynamic diameter using an average ambient particle density of 1.6 g/cm3, also shown in (Vanderpool et al. Citation2001). The IPL exhibits slightly lower transmission efficiency for dva < ∼200 nm compared to the standard lens, but has much better transmission in the 1 to above 4 µm size range compared with the standard lens.
Figure 3. (a) Average PM2.5 lens transmission efficiency (EL) measured using NH4NO3, NaNO3, and PSL particles for different lens assemblies compared with that of PM1 lens from Liu et al. (Citation2007) (error bars are one standard deviation and 50% cutoff sizes are indicated by arrows). The transmission cutoff of the PM2.5 EPA Federal Reference Method (FRM) WINS (Well impactor ninety-six) separator, converted to vacuum aerodynamic diameter using an average ambient particle density of 1.6 g/cm3 (Hu et al. Citation2012), is shown for reference. (b) Velocity calibration of the IPL compared with those of the standard lens (PM1) and the high pressure lens (HPL). From top to bottom, the traces are HPL, IPL, and standard PM1 lens. Blue solid points are NH4NO3 results, while red solid points are NaNO3 results. The fit coefficients are given in .
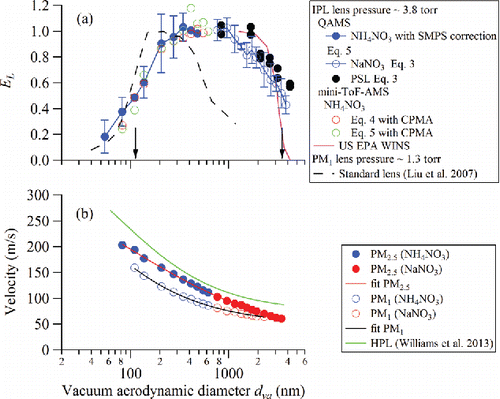
The smaller transmission efficiency of the IPL in the size range <∼200 nm, compared with that of the standard lens, might cause an underestimation of ambient hydrocarbon-like organic aerosol (HOA) mass loadings, since HOA particles from primary combustion emissions are generally in that aerodynamic size range. Based on a typical ambient PM2.5 size distribution (Chow and Watson Citation1998), the detected PM2.5 mass fraction increases from 65% to 89% when standard lens is replaced by IPL (Figures S1 and S2). Figure S2 shows that the decreased transmission efficiency of the standard lens and IPL at 200–300 nm only affects the measured PM2.5 mass to a small extent.
Using an IPL in an Aerodyne mini-ToF-AMS (a ToF-ACSM equipped with a pToF chopper for sizing), the particle transmission efficiency in the smaller size range (50 to 650 nm vacuum aerodynamic diameter dva) was measured using a DMA for size selection, a CPMA for mass selection () and both MS and pToF modes (Equations (Equation4[4] ) and (Equation5
[5] ), respectively; ). The particle transmission efficiency through the lens in the small size range agrees well for these two methods, and agrees with the Q-AMS measurements using Equation (Equation5
[5] ) with SMPS correction for multiply charged particles. The use of the CPMA filters out the multiply charged particles with larger diameter(or mass), which can bias the results for the MS method for sizes smaller than 300 nm dmob and eliminates the necessity of using an SMPS to measure and correct for multiply charged particles. In addition, the CPMA combined with a DMA is a more desirable instrument for performing the ionization efficiency calibration in ACSM or AMS systems since it sizes particles based on mass. Errors associated with assumptions and/or approximations needed for converting DMA mobility diameters to mass are eliminated.
We note here that the mini-ToF-AMS vacuum system is identical to the ToF-ACSM (Fröhlich et al. Citation2013). The only difference is the addition of the pToF module for sizing and a faster data acquisition card required to process pToF data. Although the ToF-ACSM vacuum system is designed around a custom four-stage split-flow turbo pump (Pfeiffer, SplitFlow 310), in contrast to discrete turbo pumps in other AMS and Q-ACSM systems, the particle transmission efficiency measured using the mini-ToF-AMS agrees well with the average transmission curve with the IPL installed in a Q-AMS (), demonstrating consistent performance across different vacuum systems.
Size-dependent velocities of particles that exit the aerodynamic lens can be used to convert particle flight time to vacuum aerodynamic diameter following calibration. Velocity calibration curves for the standard (PM1) lens, IPL, and HPL are shown in and the parameters for a semi-empirical fit are listed in (Allan et al. Citation2003). Higher lens pressure translates into larger ΔP and stronger expansion into the vacuum chamber, resulting in faster particle velocities. Furthermore, shows that the velocity curve of the IPL is steeper for particles larger than ∼1 µm (dva) compared to that of the HPL, which leads to better pToF resolution in systems using the IPL. For example, the difference in arrival time between 2.0 and 3.0 µm dva particles is 181 µs in the case of the HPL and 628 µs for the IPL (for a flight path length of 0.295 m). The size of each time-bin for pToF measurements, which determines the size resolution, is typically 10 µs in the Q-AMS and 32 µs with an HR-AMS. Based on the slope of the calibration curve, typically pToF length of 0.295 m, and pToF time-bin size of 10 µs, the maximum potentially achievable size resolutions at 1 µm are 111 and 66 for IPL and HPL, respectively. However, other factors, such as particle vaporization dynamics, have larger impact on the pToF resolution.
Table 1. Summary of velocity calibration fitting parameters for standard PM1, IPL, and HPL lenses. The formula used for fitting is , which is from Williams et al. (Citation2013).
3.3. Reduced bounce and capture vaporizer performance
Following improvements in the sampling system and the lens, we addressed the other important limiting aspect of the ACSM (and AMS) instruments, i.e., the bounce of particles impacting the particle vaporizer. It is well known that super-micron and sub-micron-sized particles can bounce off collection surfaces and this effect has been observed in the AMS (Matthew et al. Citation2008; Cross et al. Citation2009). On average, approximately half of ambient aerosol mass is not captured in the AMS and ACSM, requiring a collection efficiency correction (EB) due to particle bounce. The extent of particle bounce has been determined to be related to the particle phase and a phase-related/chemical composition-dependent correction for particle bounce has been reported by Middlebrook et al. (Citation2011). The standard vaporizer used in the ACSM, as well as AMS instruments, has a 60° angle inverted conical shape such that one additional collision is possible with the vaporizer if the particle bounces upon initial impact. In addition to the conical shape, the standard vaporizer impact surface is constructed of porous tungsten to provide a void volume to help trap particles. The porous structure is nominally 20% void space with 100–200 µm diameter pores. Some previous studies have investigated the effects of vaporizer material and geometry on particle bounce. For example, a study by Kang et al. (Citation2015) showed that particle bounce decreases with increased porosity of the material and a cavity-shaped vaporizer, but no significant differences among reverse-conical, trapezoidal, and reverse-T shapes were observed.
Following the work of Cross et al. (Citation2009), three types of single particle vaporization or detection events have been characterized following particle impaction on the standard vaporizer surface, (1) instantaneous and complete vaporization (), (2) partial vaporization, and (3) elastic bounce without any vaporization (null event) (). The first is the ideal case of complete flash vaporization on the first collision (occurring on a tens of µs time scale). For partial vaporization events (occurring over the ms to sec timescales), a bounced particle is trapped in the porous tungsten structure or lands on a nearby hot surface inside the ionization chamber and flash vaporizes or the materials in the particle slowly desorb. Detailed understanding of these processes remains to be completely unraveled and it is the partial vaporization case, which leads to the most ambiguity in the detailed interpretation of the mass concentration. There is also uncertainty regarding the effect of the porous tungsten structure. Earlier (unreported) measurements in our lab showed that porosity helped reduce bounce, in agreement with the published results of Kang et al. (Citation2015). However, the porous structure can also provide a physical mechanism for collection of lower volatility material that can slowly decompose, desorb and in the case of organic matter (OM), form char. This potential buildup of OM can lead to varying amounts of CO and CO2 formation when exposed to oxidizing material (Pieber et al. Citation2016). The use of tungsten for the standard vaporizer was chosen since porous structures are available in this refractory material. However, it is well known that tungsten has catalytic properties (especially when hot) (Bradley et al. Citation1969; Drewnick et al. Citation2015) and could lead to additional second-order chemical effects. According to Pieber et al. (Citation2016), a tungsten catalyzed reaction could happen on a time scale of tens of microseconds and could contribute to signal for both flash and slow vaporization of non-refractory materials.
Figure 4. Schematic diagram of particles impacting the standard vaporizer (a and b) and capture vaporizer (c), showing (a) flash vaporization, (b) bounce off the surface, and (c) trapped and delayed vaporization. Shading represents idealized vapor plume. Black arrow indicates attachment point of the thermocouple.
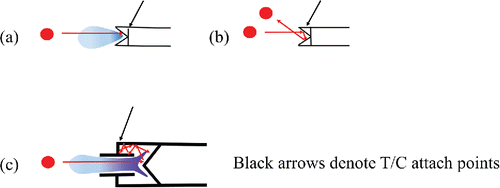
In this study, we describe a new particle vaporizer design, the capture vaporizer (CV), which addresses the bounce issue. A schematic of the CV is shown in (Jayne and Worsnop Citation2015). The internal shape of the vaporizer is designed such that particles striking the inner surface that do not vaporize on the first collision are trapped inside the cavity and undergo a series of collisions with the inner surfaces. This increases the probability of complete vaporization of particles (or EB = 1) and subsequent complete mass detection. Like the SV, the CV is inserted into the electron impact (EI) ionization source. But unlike the standard vaporizer, the impact surfaces of the capture vaporizer are solid molybdenum (Mo). Mo is less catalytic than tungsten (W) because Mo has higher ionization potentials (1st, 2nd) than W (Kramida et al. Citation2015). Future studies in our lab will investigate the CO, CO2 production mechanism reported by Pieber et al. (Citation2016) by comparing standard vaporizers made of Mo to tungsten.
The performance of the CV was compared with that of the standard inverted-conical vaporizer with test particles composed of NaNO3, NH4NO3, and (NH4)2SO4. The mass loading of NO3, SO4, and NH4 were calculated based on a modified fragmentation table for the capture vaporizer, (see Section 3.4). The fragmentation table is used to convert AMS or ACSM ion signals to species (Allan et al. Citation2004) and is dependent on the extent of thermal decomposition at the vaporizer. The mass loading calculations use the measured mIENO3 value of 8×103 ions/pg for the QAMS, an RIESO4 of 1.5, and an RIENH4 of 5.2. The RIESO4 of 1.5 is different from the typical value of 1.2 for AMS, which might be caused by the changes in the fragmentation pattern of sulfate in the CV. Determination of the RIE values is discussed below.
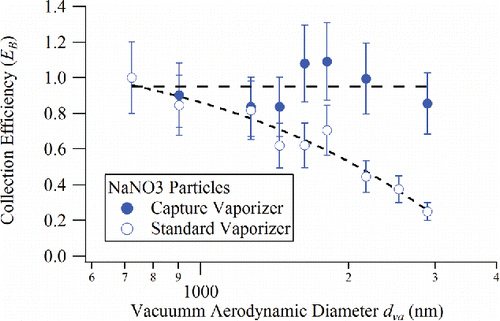
shows the size-dependent EB of NaNO3 particles for the standard vaporizer and the capture vaporizer calculated using Equation (Equation6[6] ) based on mass ratios for NO3 (both vaporizers are operated at about 900°C). For the standard vaporizer, the bounce factor (EB) decreases (bounce increases) with increasing particle size, indicating that larger NaNO3 particles have a higher propensity to bounce off the standard vaporizer surface. In contrast, for the capture vaporizer, EB stays constant with a value close to unity, independent of particle size, demonstrating that particles are indeed trapped in the cavity of the capture vaporizer and fully vaporize yielding the expected detectable mass.
Figure 5. Collection efficiency due to particle bounce, EB, plotted as a function of particle size on the standard vaporizer and capture vaporizer for NaNO3 particles. (Dashed lines are guides for the eye).
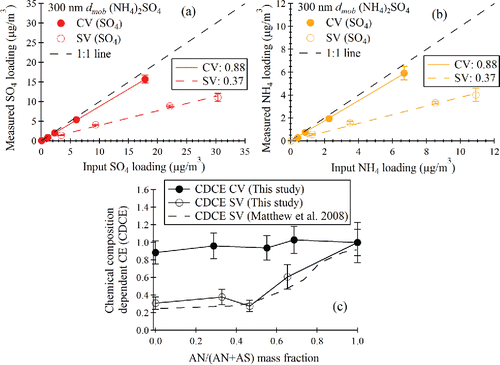
For pure (NH4)2SO4 particles, the SO4 and NH4 loadings as measured in MS mode of the Q-AMS were compared with the input loadings calculated from the CPC number concentration for size and mass selected 300 nm mobility diameter particles (). The slopes of the lines in give the collection efficiency. The CV data set shows that about 88% of the mass is collected and measured, while for the SV, only about 37% of the mass is reported. To further illustrate EB∼1 for the capture vaporizer, particles composed of mixtures of (NH4)2SO4 and NH4NO3 were prepared and sampled. shows the results of measurements of the chemical composition-dependent collection efficiency for the mixed particles for both the SV and CV, plotted as a function of NH4NO3 mass fraction (NH4NO3/(NH4NO3+ (NH4)2SO4), or AN/(AN+AS)). As shown in the figure, the collection efficiency is close to unity across the full composition range for the CV. The composition-dependent collection efficiency measured with standard vaporizer as well as that reported by Matthew et al. (Citation2008) are also plotted in .
Figure 6. (a) Q-AMS measured sulfate loading vs. CPMA and CPC calculated sulfate loading for standard and capture vaporizers. (b) Q-AMS measured NH4 loading vs. CPMA and CPC calculated NH4 loading for standard and capture vaporizers. (c) Chemical composition-dependent collection efficiency (CDCE) for capture vaporizer compared with that for standard vaporizer.
In order to test whether the RIEs measured for NH4 and SO4 are matrix dependent, calibration experiments were performed with mixed particles containing both NH4NO3 and (NH4)2SO4. For these experiments, the RIE values of NH4 and SO4 were simultaneously fit to two known constraints, i.e., the measured NH4 mass loading (Equation (Equation7a[7a] )) and the predicted NH4 mass loading based on the measured NO3 and SO4 mass loadings (Equation (Equation7b
[7b] )). The fit minimize the χ2 function shown in Equation (Equation7c
[7c] ). A 10% error is estimated in the predicted NH4 mass loading:
[7a]
[7b]
[7c] where NH4Meas and NH4Pred are the measured and predicted mass loading of NH4, respectively. The SigX is the measured signal intensity of X in ions/s, which is determined using the modified fragmentation tables for the CV as discussed in Section 3.4. The factor of 1.05 in the first term on the right-hand side of Equation (Equation7b
[7b] ) accounts for the fact that the mIENO3 is determined for m/z 30 and 46 only, while the fragmentation table generated signal for NO3 includes minor contributions (∼5%) at other m/z's, namely, N and O. The fit yields RIE values of 1.5 ± 0.3 and 5.2 ± 0.6 for SO4 and NH4, respectively (). These values are in good agreement with the RIE values of SO4 and NH4 obtained from a two-stage stepwise calibration using pure NH4NO3 and (NH4)2SO4 particles (Ng et al. Citation2011). In this method, the RIE of NH4 is first obtained relative to the calibrated NO3 ionization efficiency from NH4NO3. This RIE NH4 is then used to determine RIE SO4 from (NH4)2SO4 calibrations. RIE values of 1.5 ± 0.2 and 5.6 ± 0.4 are obtained for SO4 and NH4, respectively, in the CV measurements for single component particles.
Figure 7. (a) Δχ2 contour plot used for to determine RIE's from mixed NH4NO3 (AN)/(NH4)2SO4 (AS) particles. A Δχ2 value of 2.3 denotes the range of RIE values for 1 sigma uncertainties. (b) Measured AS/(AS + AN) mass fraction vs. input mass fraction in the atomizer solution. (c) Measured NH4 vs. NH4 predicted from measured NO3 and SO4. The fitting equation is NH4meas = b × NH4predict + a.
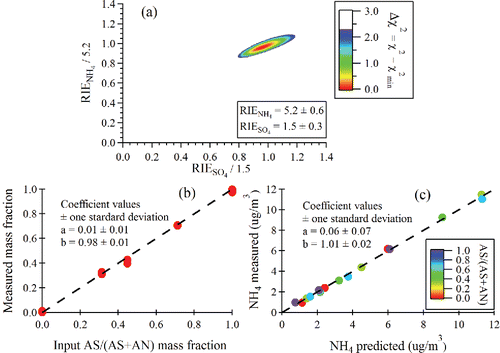
shows the input (NH4)2SO4 mass fraction (AS/(AS+AN)) compared with that calculated using the RIESO4 and RIENH4 values obtained from fitting Equation (Equation7c[7a] ). In , the measured NH4 mass loading is calculated using the newly obtained RIE value for NH4 (Equation (Equation7a
[7a] )) and compared with the predicted NH4 mass loading (Equation (Equation7b
[7b] )). is colored by the (NH4)2SO4 mass fraction in the mixture particles. The slopes are very close to unity in , indicating that the RIE values for SO4 and NH4 are appropriately determined in the fitting. An advantage of the mixed particle calibration is the fact that it allows identification of systematic biases in the measurement system, as compared to the single component calibration. In fact, a key observation from these graphs is the very good linearity with changing composition, demonstrating that detection and quantification of NO3, SO4, and NH4 with the capture vaporizer is independent of the presence of other components even though more thermal decomposition is observed.
3.4. Decomposition effects in the capture vaporizer
The results in show that the CV improves particle mass detection. This is a significant advance over the standard vaporizer, which detects only half of the mass for typical ambient aerosol (i.e., EB ∼ 0.5) (Middlebrook et al. Citation2011). However, as expected, the CV also alters the observed mass spectral patterns of ions, which is a result of increased thermal decomposition of species that experience longer residence times and multiple collisions within the hot (600°C) vaporizer. Increased decomposition is observed for both organic and inorganic species.
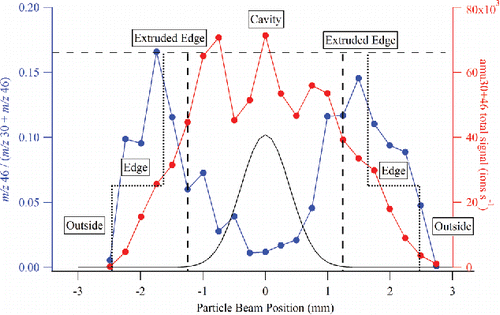
shows the m/z 46/(m/z 30 + m/z 46) ratio, representing (NO2+/(NO+ + NO2+)), for 300 nm size selected NH4NO3 particles as the aerodynamic lens focal position was translated vertically across the surface of the CV. When the particle beam strikes the opening of the capture vaporizer, less NO2+ fragment (m/z 46) is observed relative to NO+ fragment (m/z 30) indicating increased thermal decomposition. As the particle beam moves to the edge of the CV cage structure, the fragmentation pattern is more similar to that for the standard vaporizer, where particles experience only one (or two) collisions before vaporization and detection. Note that this experiment maps out the physical dimensions of the CV as indicated in and also shows that the particle beam is tightly focused (<1.0 mm diameter) for the 300 nm test aerosol, as confirmed by a beam width probe (Huffman et al. Citation2005) that was also installed in this instrument. Therefore, the spatial profile of the m/z 46 / (m/z 30 + m/z 46) ratio could be used to map out the 2-dimensional image of the vaporizer surface. The m/z 46 / (m/z 30 + m/z 46) ratio is also useful in aligning the particle beam to the center of capture vaporizer. For consistent results among different instruments or different field studies, special attention should be paid to lens alignment when using the CV design. A particle beam profile for typical ambient aerosol at the ACSM chamber length is also plotted in (Salcedo et al. Citation2007). It shows that CV collects 100% of the ambient particles. Using the particle beam profile and CV dimensions, the fractions of particles that hit the cavity, extruded edge, edge, and outside of the CV are simulated (Figure S3). The m/z 46/(m/z 30 + m/z 46) ratio is also simulated using 0.02, 0.33, 0.16, and 0 values for cavity, extruded edge, edge, and outside region, respectively (Figure S4). The simulated m/z 46/(m/z 30 + m/z 46) profile is comparable to the experimentally measured profile in .
Figure 8. Mapping of the capture vaporizer surface using the m/z 46 to m/z 30 + m/z 46 ratio and the total m/z 30 + m/z 46 signal. The edges of the capture vaporizer are indicated with short-dashed vertical lines, and the edges of the opening to the internal cavity are indicated with long-dashed vertical lines. The black trace is a typical ambient aerosol beam width profile (Salcedo et al. Citation2007) produced by a PM1 lens in an AMS, and projected on the dimension of the capture vaporizer at the ACSM chamber length.
To further study the fragmentation patterns of inorganic species due to the extended residence times in the CV, the spectrum of (NH4)2SO4 and NH4NO3 was measured in argon carrier gas. Argon gas was chosen to reduce the background signal levels at m/z 14 and m/z 32 in order to improve the detection of N from NO3 and NH4, and S from SO4, respectively. The following cascade of thermal decomposition processes was proposed for vaporization of (NH4)2SO4 and NH4NO3 from the SV by Drewnick et al. (Citation2015):[R1.1]
[R1.2]
[R1.3]
[R2.1]
[R2.2]
[R2.3] where (s) refers to the condensed phase particle entering the vaporizer, (g) refers to gas-phase products of vaporization, and the species not labeled (s) or (g) are most likely adsorbed to the vaporizer surface. A comparison among the fragmentation patterns of NO3, SO4, and NH4 in the CV and SV is shown in . The NH4 fragmentation patterns for SV and CV are very similar to each other likely because NH3 does not further thermally decompose at the vaporizer temperature. However, species such as NO3 and SO4 (that are detected after multiple decomposition steps) have obvious differences between their CV and SV fragmentation patterns. In both cases the same fragments are observed, but the CV fragmentation patterns (solid bars) tend to be more weighted toward the lower molecular weight products that are produced from more extensive thermal decomposition of the initial products. As a consequence, the fragmentation table values typically used in AMS or ACSM analysis need to be modified for the capture vaporizer and are shown in , although more work needs to be done to evaluate the variability of these fragmentation patterns under different operating conditions. Similar fragmentation pattern changes are expected for organics as well, especially for lower m/z's such as m/z 43 (C3H7+), and m/z's such as 44 (CO2+), and 18 (H2O+), which are produced by thermal decomposition of oxygenated organic molecules with acid and alcohol functional groups (Canagaratna et al. Citation2015). For the same organic compound, the f44 (defined as the ratio of m/z 44 signal to total organic signal) is expected to be higher in a CV system than that in an SV system. A more detailed discussion on the fragmentation changes for organic species in the CV due to prolonged vaporization will be the topic of future papers from our lab and Hu et al. (Citationin preparation).
Figure 9. Fragmentation patterns observed in the CV (solid) and the SV (shaded) for NH4 (left four bar pairs [yellow]), NO3 (middle bar pair [blue]), and SO4 (right four bar pairs [red]). The NH4 measurements are shown for both NH4NO3 and (NH4)2SO4.
![Figure 9. Fragmentation patterns observed in the CV (solid) and the SV (shaded) for NH4 (left four bar pairs [yellow]), NO3 (middle bar pair [blue]), and SO4 (right four bar pairs [red]). The NH4 measurements are shown for both NH4NO3 and (NH4)2SO4.](/cms/asset/9720999b-eda8-4b98-8ecd-fc8c684aa654/uast_a_1241859_f0009_oc.gif)
Table 2(a). Modified fragmentation table entries for sulfate for the CV. The frag_NO3 is unchanged, although the m/z 46/30 ratio has changed.
(b). Corresponding standard frag table for the SV.
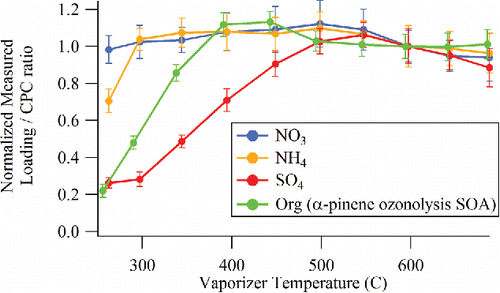
AMS instruments are typically operated with a vaporizer temperature of 600°C to maximize the efficiency of thermal vaporization of both inorganic and organic aerosol components. It is of interest to explore whether lower temperatures can be used with the CV to reduce the observed thermal decomposition. shows the CV signal to CPC ratio observed as a function of vaporizer temperature for input mass concentrations of 1:1 mixed NH4NO3 and (NH4)2SO4 particles as well as laboratory generated α-pinene ozonolysis secondary organic aerosol (SOA). SOA generated from a photochemical flow reactor (PAM) (Lambe et al. Citation2011) was utilized here because it is a complex mix of secondary oxidized species that is more representative of the complex mixtures observed in ambient organic aerosol particles than the particles composed of pure organic materials. The results shown in are consistent with previous temperature-dependent SV measurements of ambient aerosol (Docherty et al. Citation2011). In , the SO4 and organic signals reach a plateau at different temperatures compared with NH4 and NO3, consistent with the observation in Drewnick et al. (Citation2015). The data suggest that it would be possible to operate the CV at around 400°C when sampling pure organic aerosol particles or around 500°C when sampling mixed organic/inorganic particles. Future studies will examine the effect of CV temperature on quantification and thermal decomposition in more detail.
Figure 10. Normalized species concentrations measured at different CV temperatures for mixed NH4NO3 and (NH4)2SO4 particles and for organic (α-pinene ozonolysis SOA) particles.
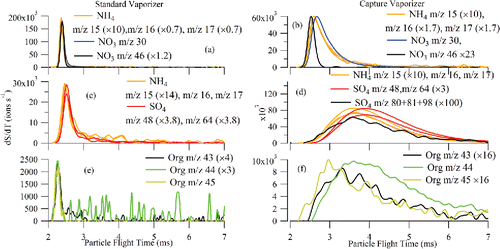
shows the normalized pToF profiles of 300 nm dmob NH4NO3, (NH4)2SO4, and glutaric acid particles for m/z 30 and 46 from NO3, m/z 48 and 64 from SO4, m/z 15, 16, and 17 from NH4, and m/z 43, 44, and 45 from glutaric acid for the CV and the SV. The pToF profiles observed for the CV are broader than for the SV. These broader profiles make it more difficult to measure particle size in the AMS. The detailed pToF profiles of the monodisperse particles shown in , however, give some insights into the dynamics of the escape of vapor from the cavity of CV as well as vapor decomposition kinetics. Since particle bounce is generally negligible for the case of NH4NO3 particles, the wider CV profiles of NH4NO3 (when compared to those obtained from the SV) are indicative of the increased residence time of the vaporized gases due to adsorption/desorption and thermal decomposition on the larger internal surface area of the CV. The slower escape of vapor from the CV (µs to ms as shown in ) is still faster than the MS Open/Closed time scale (typically 5 s). As a result, the background in the CV system is expected to be comparable with that in the SV system. The pToF profiles of (NH4)2SO4 and pure glutaric acid are even broader than those observed for NH4NO3 because they reflect an additional source of delay due to particle bounce.
Figure 11. pToF profiles for selected SO4-related ions (m/z 48 and 64), selected NH4-related ions (m/z 15, 16, and 17), selected NO3-related ions (m/z 30 and 46), and selected organic-related ions (m/z 43, 44, and 45) for (a) NH4NO3 with SV, (b) NH4NO3 with CV, (c) (NH4)2SO4 with SV, (d) (NH4)2SO4 with CV, (e) glutaric acid with SV, and (f) glutaric acid with CV (× x: x is the scaling factor).
The detailed pToF profiles of the different ions observed from NH4NO3 and (NH4)2SO4 are consistent with the decomposition mechanisms R1 and R2 in which (NH4)2SO4 and NH4NO3 evaporate to yield NH3 (g) rather than molecular salts as suggested in Murphy (Citation2016). Specifically, for NH4NO3, the ions produced from vaporized NH3 are observed to appear first along with a weak signal from m/z 46 (NO2+), which reflects ionization of the small fraction of the vaporized HNO3 molecules that do not undergo additional decomposition and interaction with the CV surfaces. The majority of the vaporized HNO3, however, thermally decomposes on the CV surfaces and is detected as m/z 30 (NO+). The delay between the appearance times of the m/z 30 and 46 ions is larger than the molecular-weight-dependent offset in ion travel times through the mass analyzer (Zhang et al. Citation2005) and reflects the time scales of the additional vapor decomposition kinetics in the CV. As in the case of NH4NO3, NH3 vaporized from (NH4)2SO4 appears first along with mass spectral ions such as m/z 80, 81, and 98 that are typically observed from H2SO4. However, NH3 signals from (NH4)2SO4 do not appear as rapidly as from NH4NO3 due to additional bouncing of the (NH4)2SO4 particles. The ions formed further along the thermal decomposition chain of H2SO4 (as shown in EquationR1.2[R1.2] and EquationR1.3
[R1.3] ) show delayed pToF appearance times. A similar delay in pToF appearance time is also observed for glutaric acid ions such as m/z 44 that are produced by thermal decomposition (Canagaratna et al. Citation2015).
4. Conclusion
A PM2.5 ACSM system consisting of a new inlet system, an intermediate pressure lens (IPL), and a capture vaporizer, has been designed and characterized. The new sampling inlet design presented here minimizes large size particle loss by straightening plumbing to the ACSM aerodynamic lens. The incorporation of a straight-through flow path filter switching valve is an important part of the new sampling inlet system. The IPL has more than 50% transmission efficiency between 110 nm and 3.5 µm in vacuum aerodynamic diameter. Compared with previous HPL and standard lens designs, the major change in the IPL is the dimension of the exit nozzle, resulting in a 507 Pa (3.8 Torr) of lens pressure, which is higher than that of the standard lens (173 Pa or 1.3 Torr) but lower than that of the HPL lens (1840 Pa or 13.8 Torr). The IPL is a compromise between the maximum large particle transmission size and the practical ability to manufacture the lens. The capture vaporizer (CV) introduced here addresses the bounce factor, which has limited complete collection of particle mass in previous ACSM and AMS systems. The CV also plays an important role for PM2.5 applications since, as shown here, super micron sized particles have a higher propensity to bounce necessitating the need to make EB corrections for composition as well as size with the standard vaporizer. The mass spectral patterns and pToF profiles together indicate that the CV generally produces more extensively fragmented mass spectra due to increased thermal decomposition. The mass spectral pattern of the species measured with the CV is shifted to lower m/z, indicating the need for a new fragmentation table for AMS and ACSM systems that incorporate a CV. Temperature-dependent mass measurements indicate that a CV operating temperature lower than 600°C would maintain sensitivity while possibly reducing fragmentation. Further work characterizing the species and temperature-dependent fragmentation patterns is in progress.
Despite the increased thermal decomposition introduced by the CV, the measurements are shown to yield quantitative species-dependent mass concentrations. Measurements of nitrate, sulfate, and ammonium using mixtures of (NH4)2SO4 and NH4NO3 also show that detection of these species with the CV is matrix-independent. In addition, it is also possible that the increased fragmentation reduces the organics information and potentially reduces the possibility to identify organic aerosol types with PMF.
The PM2.5 ACSM system enables real-time ambient PM monitoring of chemically speciated aerosols that can meet the PM2.5 monitoring standards for NR-PM. Considering a typical ambient PM2.5 size distribution, we found that 89% of the non-refractory mass is detected with the new system, while was only 65% with the old system. Field measurements are currently underway to compare measurements from a PM2.5 ACSM system with collocated instruments, such as the Tapered Element Oscillating Microbalance (TEOM), the Monitor for AeRosols and Gases in Ambient air (MARGA), and the real-time Organic/Elemental Carbon analyzer (OC/EC). Results from this intercomparison will be the subject of future papers. Finally, the PM2.5 system can also be adapted to AMS systems or other instruments that utilize a focused super micron particle beam.
UAST_1241859_Supplemental_File.zip
Download Zip (536.5 KB)Acknowledgments
The authors thank Andrew T. Lambe for helping carry out the Potential Aerosol Mass (PAM) Oxidation Flow Reactor (OFR) experiment to generate secondary organic aerosols (SOAs).
Funding
This work was supported by United States Department of Energy Small Business Innovative Research (SBIR) contract #DE-SC0001673 and the United States Environmental Protection Agency SBIR contract #EP-D-13-042. Support from these agencies is gratefully acknowledged.
ORCID
Douglas Worsnop http://orcid.org/0000-0002-8928-8017
Related Research Data
References
- Allan, J. D., Delia, A. E., Coe, H., Bower, K. N., Alfarra, M. R., Jimenez, J. L., Middlebrook, A. M., Drewnick, F., Onasch, T. B., Canagaratna, M. R., Jayne, J. T., and Worsnopf, D. R. (2004). A Generalised Method for the Extraction of Chemically Resolved Mass Spectra from Aerodyne Aerosol Mass Spectrometer Data. J. Aerosol Sci., 35:909–922.
- Allan, J. D., Jimenez, J. L., Williams, P. I., Alfarra, M. R., Bower, K. N., Jayne, J. T., Coe, H., and Worsnop, D. R. (2003). Quantitative Sampling using an Aerodyne Aerosol Mass Spectrometer - 1. Techniques of Data Interpretation and Error Analysis. J. Geophys. Res.-Atmos., 108:4090–4099.
- Bradley, J. N., Gilbert, J. R., and Park, A. J. (1969). Mass-Spectrometric Study of the Tungsten-Catalyzed Decomposition of Ammonia. Trans. Faraday Soc., 65:2772–2778.
- Budisulistiorini, S. H., Canagaratna, M. R., Croteau, P. L., Marth, W. J., Baumann, K., Edgerton, E. S., Shaw, S. L., Knipping, E. M., Worsnop, D. R., Jayne, J. T., Gold, A., and Surratt, J. D. (2013). Real-Time Continuous Characterization of Secondary Organic Aerosol Derived from Isoprene Epoxydiols in Downtown Atlanta, Georgia, Using the Aerodyne Aerosol Chemical Speciation Monitor. Environ. Sci. Technol., 47:5686–5694.
- Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R. (2007). Chemical and Microphysical Characterization of Ambient Aerosols with the Aerodyne Aerosol Mass Spectrometer. Mass Spectrom. Rev., 26:185–222.
- Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R. (2015). Elemental Ratio Measurements of Organic Compounds using Aerosol Mass Spectrometry: Characterization, Improved Calibration, and Implications. Atmos. Chem. Phys., 15:253–272.
- Chow, J. C., and Watson, J. G. (1998). Guideline on Speciated Particulate Monitoring, Report. Environment, Part A:1185–1201.
- Crenn, V., Sciare, J., Croteau, P. L., Verlhac, S., Fröhlich, R., Belis, C. A., Aas, W., Äijälä, M., Alastuey, A., Artiñano, B., Baisnée, D., Bonnaire, N., Bressi, M., Canagaratna, M., Canonaco, F., Carbone, C., Cavalli, F., Coz, E., Cubison, M. J., Esser-Gietl, J. K., Green, D. C., Gros, V., Heikkinen, L., Herrmann, H., Lunder, C., Minguillón, M. C., Močnik, G., O'Dowd, C. D., Ovadnevaite, J., Petit, J. E., Petralia, E., Poulain, L., Priestman, M., Riffault, V., Ripoll, A., Sarda-Estève, R., Slowik, J. G., Setyan, A., Wiedensohler, A., Baltensperger, U., Prévôt, A. S. H., Jayne, J. T., and Favez, O. (2015). ACTRIS ACSM Intercomparison – Part 1: Reproducibility of Concentration and Fragment Results from 13 Individual Quadrupole Aerosol Chemical Speciation Monitors (Q-ACSM) and Consistency with Co-located Instruments. Atmos. Meas. Tech., 8:5063–5087.
- Cross, E. S., Onasch, T. B., Canagaratna, M., Jayne, J. T., Kimmel, J., Yu, X. Y., Alexander, M. L., Worsnop, D. R., and Davidovits, P. (2009). Single Particle Characterization using a Light Scattering Module Coupled to a Time-of-Flight Aerosol Mass Spectrometer. Atmos. Chem. Phys., 9:7769–7793.
- Cross, E. S., Slowik, J. G., Davidovits, P., Allan, J. D., Worsnop, D. R., Jayne, J. T., Lewis, D. K., Canagaratna, M., and Onasch, T. B. (2007). Laboratory and Ambient Particle Density Determinations using Light Scattering in Conjunction with Aerosol Mass Spectrometry. Aerosol Sci. Technol., 41:343–359.
- DeCarlo, P. (2004). Particle Morphology and Density Characterization by Combined Mobility and Aerodynamic Diameter Measurements. Part 1: Theory. Aerosol Sci. Technol., 38:1185–1205.
- Docherty, K. S., Aiken, A. C., Huffman, J. A., Ulbrich, I. M., DeCarlo, P. F., Sueper, D., Worsnop, D. R., Snyder, D. C., Peltier, R. E., Weber, R. J., Grover, B. D., Eatough, D. J., Williams, B. J., Goldstein, A. H., Ziemann, P. J., and Jimenez, J. L. (2011). The 2005 Study of Organic Aerosols at Riverside (SOAR-1): Instrumental Intercomparisons and Fine Particle Composition. Atmos. Chem. Phys., 11:12387–12420.
- Drewnick, F., Diesch, J. M., Faber, P., Borrmann, S. (2015). Aerosol Mass Spectrometry: Particle–Vaporizer Interactions and Their Consequences for the Measurements. Atmos. Meas. Tech., 8:3811–3830.
- Fröhlich, R., Crenn, V., Setyan, A., Belis, C. A., Canonaco, F., Favez, O., Riffault, V., Slowik, J. G., Aas, W., Aijälä, M., Alastuey, A., Artiñano, B., Bonnaire, N., Bozzetti, C., Bressi, M., Carbone, C., Coz, E., Croteau, P. L., Cubison, M. J., Esser-Gietl, J. K., Green, D. C., Gros, V., Heikkinen, L., Herrmann, H., Jayne, J. T., Lunder, C. R., Minguillón, M. C., Močnik, G., O'Dowd, C. D., Ovadnevaite, J., Petralia, E., Poulain, L., Priestman, M., Ripoll, A., Sarda-Estève, R., Wiedensohler, A., Baltensperger, U., Sciare, J., and Prévôt, A. S. H. (2015). ACTRIS ACSM Intercomparison – Part 2: Intercomparison of ME-2 Organic Source Apportionment Results from 15 Individual, Co-located Aerosol Mass Spectrometers. Atmos. Meas. Tech., 8:2555–2576.
- Fröhlich, R., Cubison, M. J., Slowik, J. G., Bukowiecki, N., Prévôt, A. S. H., Baltensperger, U., Schneider, J., Kimmel, J. R., Gonin, M., Rohner, U., Worsnop, D. R., and Jayne, J. T. (2013). The ToF-ACSM: A Portable Aerosol Chemical Speciation Monitor with TOFMS Detection. Atmos. Meas. Tech., 6:3225–3241.
- Hu, M., Peng, J., Sun, K., Yue, D., Guo, S., Wiedensohler, A., and Wu, Z. (2012). Estimation of Size-Resolved Ambient Particle Density Based on the Measurement of Aerosol Number, Mass, and Chemical Size Distributions in the Winter in Beijing. Environ. Sci. Technol., 46:9941–9947.
- Hu, W., Campuzano-Jost, P., Day, D. A., Croteau, P., Canagaratna, M. R., Jayne, J. T., and Jimenez, J. L. (2016). Intercomparison of Capture and Standard Vaporizers in the Aerodyne Aerosol Mass Spectrometer (AMS): Inorganic. Atmos. Chem. Phys., in press.
- Huffman, J. A., Jayne, J., Drewnick, F., Aiken, A. C., Onasch, T., Worsnop, D., and Jimenez, J. (2005). Design, Modeling, Optimization, and Experimental Tests of a Particle Beam Width Probe for the Aerodyne Aerosol Mass Spectrometer. Aerosol Sci. Technol., 39:1143–1163.
- Jayne, J. T., Leard, D. C., Zhang, X. F., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R. (2000). Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles. Aerosol Sci. Technol., 33:49–70.
- Jayne, J. T., and Worsnop, D. R. (2015). Particle Capture Device Aerodyne Research, Inc. US Patent 20,150,040,689 A1.
- Jimenez, J. L., Jayne, J. T., Shi, Q., Kolb, C. E., Worsnop, D. R., Yourshaw, I., Seinfeld, J. H., Flagan, R. C., Zhang, X., Smith, K. A., Morris, J. W., and Davidovits, P. (2003). Ambient Aerosol Sampling using the Aerodyne Aerosol Mass Spectrometer. J. Geophys. Res., 108:8413–8425.
- Kang, M., Cho, H.-J., Kwak, H., and Park, K. (2015). Evaluation of Particle Bounce in Various Collection Substrates to be Used as Vaporizer in Aerosol Mass Spectrometer. Aerosol Sci. Technol., 49:332–339.
- Kramida, A., Ralchenko, Y., Reader, J., and Team, N. A. (2015). NIST Atomic Spectra Database (version 5.3). Available at http://physics.nist.gov/asd
- Lambe, A. T., Ahern, A. T., Williams, L. R., Slowik, J. G., Wong, J. P. S., Abbatt, J. P. D., Brune, W. H., Ng, N. L., Wright, J. P., Croasdale, D. R., Worsnop, D. R., Davidovits, P., and Onasch, T. B. (2011). Characterization of Aerosol Photooxidation Flow Reactors: Heterogeneous Oxidation, Secondary Organic Aerosol Formation and Cloud Condensation Nuclei Activity Measurements. Atmos. Meas. Tech., 4:445–461.
- Liu, P., Ziemann, P. J., Kittelson, D. B., and McMurry, P. H. (1995a). Generating Particle Beams of Controlled Dimensions and Divergence: I. Theory of Particle Motion in Aerodynamic Lenses and Nozzle Expansions. Aerosol Sci. Technol., 22:293–313.
- Liu, P., Ziemann, P. J., Kittelson, D. B., and McMurry, P. H. (1995b). Generating Particle Beams of Controlled Dimensions and Divergence: II. Experimental Evaluation of Particle Motion in Aerodynamic Lenses and Nozzle Expansions. Aerosol Sci. Technol., 22:314–324.
- Liu, P. S. K., Deng, R., Smith, K. A., Williams, L. R., Jayne, J. T., Canagaratna, M. R., Moore, K., Onasch, T. B., Worsnop, D. R., and Deshler, T. (2007). Transmission Efficiency of an Aerodynamic Focusing Lens System: Comparison of Model Calculations and Laboratory Measurements for the Aerodyne Aerosol Mass Spectrometer. Aerosol Sci. Technol., 41:721–733.
- Matthew, B. M., Middlebrook, A. M., and Onasch, T. B. (2008). Collection Efficiencies in an Aerodyne Aerosol Mass Spectrometer as a Function of Particle Phase for Laboratory Generated Aerosols. Aerosol Sci. Technol., 42:884–898.
- Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R. (2011). Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data. Aerosol Sci. Technol., 46:258–271.
- Murphy, D. M. (2016). The Effects of Molecular Weight and Thermal Decomposition on the Sensitivity of a Thermal Desorption Aerosol Mass Spectrometer. Aerosol Sci. Technol., 50:118–125.
- Ng, N. L., Herndon, S. C., Trimborn, A., Canagaratna, M. R., Croteau, P. L., Onasch, T. B., Sueper, D., Worsnop, D. R., Zhang, Q., Sun, Y. L., and Jayne, J. T. (2011). An Aerosol Chemical Speciation Monitor (ACSM) for Routine Monitoring of the Composition and Mass Concentrations of Ambient Aerosol. Aerosol Sci. Technol., 45:780–794.
- Olfert, J. S., and Collings, N. (2005). New Method for Particle Mass Classification—The Couette Centrifugal Particle Mass Analyzer. J. Aerosol Sci., 36:1338–1352.
- Peck, J., Gonzalez, L. A., Williams, L. R., Xu, W., Croteau, P., Timko, M. T., Jayne, J. T., Worsnop, D., Miake-Lye, R., and Smith, K. A. (2016). Development of an Aerosol Mass Spectrometer Lens System for PM2.5. Aerosol Sci. Technol., 50(8):781—789.
- Pieber, S. M., El Haddad, I., Slowik, J. G., Canagaratna, M. R., Jayne, J. T., Platt, S. M., Bozzetti, C., Daellenbach, K. R., Fröhlich, R., Vlachou, A., Klein, F., Dommen, J., Miljevic, B., Jiménez, J. L., Worsnop, D. R., Baltensperger, U., and Prévôt, A. S. H. (2016). Inorganic Salt Interference on CO2+ in Aerodyne AMS and ACSM Organic Aerosol Composition Studies. Environ. Sci. Technol., 50(19):10494–10503.
- Pope, C. A. I., and Dockery, D. W. (2006). Health Effects of Fine Particulate Air Pollution: Lines that Connect. J. Air Waste Manage. Assoc., 56:709–742.
- Pöschl, U. (2005). Atmospheric Aerosols: Composition, Transformation, Climate and Health Effects. Angew. Chem. Int. Edit., 44:7520–7540.
- Salcedo, D., Onasch, T. B., Canagaratna, M. R., Dzepina, K., Huffman, J. A., Jayne, J. T., Worsnop, D. R., Kolb, C. E., Weimer, S., Drewnick, F., Allan, J. D., Delia, A. E., and Jimenez, J. L. (2007). Technical Note: Use of a Beam Width Probe in an Aerosol Mass Spectrometer to Monitor Particle Collection Efficiency in the Field. Atmos. Chem. Phys., 7:549–556.
- Schreiner, J., Schild, U., Voigt, C., and Mauersberger, K. (1999). Focusing of Aerosols into a Particle Beam at Pressures from 10 to 150 Torr. Aerosol Sci. Technol., 31:373–382.
- Solomon, S., Qin, D., Manning, M., Chen, Z., Marquis, M., Averyt, K. B., Tignor, M., and Miller, H. L. (2007). Climate Change 2007: The Physical Science Basis. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA.
- Sun, Y., Wang, Z., Dong, H., Yang, T., Li, J., Pan, X., Chen, P., and Jayne, J. T. (2012). Characterization of Summer Organic and Inorganic Aerosols in Beijing, China with an Aerosol Chemical Speciation Monitor. Atmos. Environ., 51:250–259.
- Sun, Y., Wang, Z., Fu, P., Jiang, Q., Yang, T., Li, J., and Ge, X. (2013a). The Impact of Relative Humidity on Aerosol Composition and Evolution Processes During Wintertime in Beijing, China. Atmos. Environ., 77:927–934.
- Sun, Y. L., Wang, Z. F., Fu, P. Q., Yang, T., Jiang, Q., Dong, H. B., Li, J., and Jia, J. J. (2013b). Aerosol Composition, Sources and Processes During Wintertime in Beijing, China. Atmos. Chem. Phys., 13:4577–4592.
- Vanderpool, R. W., Peters, T. M., Natarajan, S., Tolocka, M. P., Gemmill, D. B., and Wiener, R. W. (2001). Sensitivity Analysis of the USEPA WINS PM2.5 Separator. Aerosol Sci. Technol., 34:465–476.
- Von der Weiden, S.-L., Drewnick, F., and Borrmann, S. (2009). Particle Loss Calculator – A New Software Tool for the Assessment of the Performance of Aerosol Inlet Systems. Atmos. Meas. Tech., 2:479–494.
- Williams, L. R., Gonzalez, L. A., Peck, J., Trimborn, D., McInnis, J., Farrar, M. R., Moore, K. D., Jayne, J. T., Robinson, W. A., Lewis, D. K., Onasch, T. B., Canagaratna, M. R., Trimborn, A., Timko, M. T., Magoon, G., Deng, R., Tang, D., de la Rosa Blanco, E., Prévôt, A. S. H., Smith, K. A., and Worsnop, D. R. (2013). Characterization of an Aerodynamic Lens for Transmitting Particles Greater than 1 Micrometer in Diameter into the Aerodyne Aerosol Mass Spectrometer. Atmos. Meas. Tech., 6:3271–3280.
- Zhang, Q., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J.-L. (2005). Time- and Size-Resolved Chemical Composition of Submicron Particles in Pittsburgh: Implications for Aerosol Sources and Processes. J. Geophys. Res.: Atmos., 110:D07S09.