
ABSTRACT
The aerosol mass spectrometer (AMS) and aerosol chemical speciation monitor (ACSM) are widely used for quantifying aerosol composition. The quantification uncertainty of these instruments is dominated by the collection efficiency (CE) due to particle bounce. A new “capture vaporizer” (CV) has been recently developed to achieve unit CE. In this study, we examine the performance of the CV while sampling ambient aerosols. AMS/ACSMs using the original standard vaporizer (SV) and CV were operated in parallel during three field studies. Concentrations measured with the CV (assuming CE = 1) and SV (using the composition-dependent CE of Middlebrook et al.), as well as SMPS and PILS-IC are compared. Agreement is good in all cases, verifying that CE ∼ 1 in the CV when sampling ambient particles. Specific findings include: (a) The fragmentation pattern of ambient nitrate and sulfate species observed with the CV was shifted to smaller m/z, suggesting additional thermal decomposition. (b) The differences in fragmentation patterns of organic vs. inorganic nitrate and sulfur species are still distinguishable in the CV, however, with much lower signal-to-noise compared to the SV. (c) Size distribution broadening is significant, but its impact is limited in field studies since ambient distributions are typically quite broad. Consistent size distributions were measured with the SV and CV. (d) In biogenic areas, UMR nitrate is overestimated based on the default fragmentation table (∼factor of 2–3 in SOAS) for both vaporizers, due to underestimation of the organic interferences. We also report a new type of small interference: artifact chloride signal can be observed in the AMS when high nitrate mass concentration is sampled with both the SV (∼0.5% chloride/nitrate) or CV (∼0.2% chloride/nitrate). Our results support the improved quantification with the CV AMS and characterize its chemical detection properties.
Copyright © 2017 American Association for Aerosol Research
EDITOR:
1. Introduction
Aerosol mass spectrometers (AMS) and aerosol chemical speciation monitors (ACSM) commercialized by Aerodyne Research Inc. (ARI) are used widely to measure the mass concentrations and size distributions of non-refractory species in submicron-particles (Jayne et al. Citation2000; Allan et al. Citation2003a,Citationb; Canagaratna et al. Citation2007; Jimenez et al. Citation2009; Ng et al. Citation2011; Crenn et al. Citation2015, and references therein). With the vaporizer that is installed in almost all commercial instruments to date (referred to as “standard,” or SV in this work), the quantification of ambient aerosol mass concentration requires the use of a collection efficiency (CE) to correct for the fraction of particles that bounce off the vaporizer and are therefore not efficiently detected (Huffman et al. Citation2005; Quinn et al. Citation2006; Matthew et al. Citation2008; Middlebrook et al. Citation2012).
CE in the SV depends on aerosol phase, aerosol acidity/neutralization of sulfate content, ammonium nitrate (NH4NO3) fraction, and typically contributes most to the uncertainty in the quantification of AMS concentration measurements (Huffman et al. Citation2005; Bahreini et al. Citation2009; Middlebrook et al. Citation2012). Bahreini et al. (Citation2009) estimated an uncertainty of AMS OA concentrations around ±38% and ±34% for inorganic aerosols. Middlebrook et al. (Citation2012) developed a formulation to estimate CE for ambient particles from their chemical composition by comparing the mass concentration measured by AMS with independent measurements. Note that this CE cannot be used for the aerosol quantification from chamber studies or pure compounds or mixtures in the laboratory. A separate determination of CE is needed for such studies (Docherty et al. Citation2012; Robinson et al. Citation2016). Multiple studies using the Middlebrook CE formulation have shown good agreement with other instruments (e.g., Jimenez et al. Citation2016 and references therein). However, the remaining uncertainty impedes progress on other quantification issues and obscures detection of other instrument or analysis problems, as anomalous results that are often incorrectly attributed to an anomalous CE.
To improve AMS and ACSM quantification, a new “capture” vaporizer (CV) has been designed to reduce or eliminate the CE correction (Jayne and Worsnop Citation2016). Instead of an inverted cone as with the SV, the CV was designed to have a cage with a narrow entrance in order to increase the number of collisions and hence the residence time of particles in contact with the hot vaporizer surfaces, thus reducing the fraction of particles that bounce without evaporation (). Compared to almost all the SV in commercialized instruments are made of porous tungsten, the CV is made of solid molybdenum instead. When using the new PM2.5 lens developed for AMS systems (Peck et al. Citation2016), use of the CV to minimize loss due to bounce for supermicron particles will be more important (Xu et al. Citation2017). Previously, the CV performance was examined in laboratory studies with several standard compounds (Hu et al. Citation2016b; Xu et al. Citation2017). Hu et al. (Citation2016b) and Xu et al. (Citation2017) independently found identical CE (= 1) for purely flash-evaporated species NH4NO3 and higher CE (>0.7) for less volatile ammonium sulfate [(NH4)2SO4] and sodium nitrate (NaNO3) in the CV vs. SV, demonstrating the improved particle collection of the CV. The fragmentation patterns for pure nitrate and sulfate salt aerosols are shifted to smaller ions in the CV due to the additional thermal decomposition, arising from longer residence time and increased collisions on the vaporizer surfaces.
Figure 1. (a) Schematic of the standard vaporizer (SV) and capture vaporizer (CV). (b) Setup used for the ambient comparisons of two AMSs equipped with SV and CV, respectively, in the field studies. All the sampled air was dried to be below 30% RH before reaching the instruments with Nafion dryers.
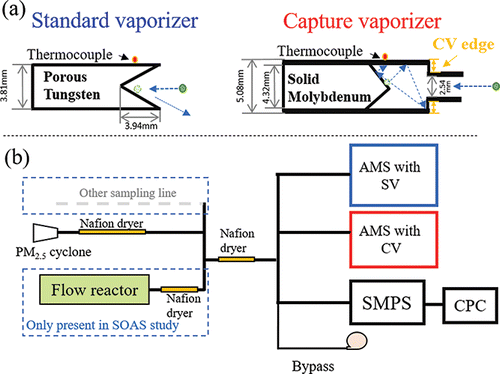
For the SV, ambient CE is often higher for ambient particles than for pure laboratory particles (Matthew et al. Citation2008; Middlebrook et al. Citation2012). Thus, a characterization of CV quantification for ambient particles is needed. It is also important to assess how the change in mass spectral patterns affects quantification, species identification, and potential interferences in the field. Finally, the performance of the AMS particle sizing measurement with the CV is evaluated. To test the performance of the CV, we operated AMS/ACSMs with SV and CV in parallel during three field studies under different ambient conditions, including one where aerosol was sampled from the output of an oxidation flow reactor (OFR) that processed ambient aerosols (Hu et al. Citation2016a). The performance of the CV in those studies is the focus of this manuscript.
2. Experiment and instrumentation
2.1. Field measurement setup
The three field studies analyzed in this work are introduced here:
The Southern Oxidant and Aerosol Study (SOAS) field study was carried out in the SE US in the summer (1 June to 15 July) of 2013 (Carlton et al. Citation2017). Results shown in this study were from the SEARCH Centreville supersite (CTR) in a semi-polluted mixed forest in Alabama (32.95° N, 87.13°W) (Hu et al. Citation2015, Citation2016a). A schematic plot of the instrumental setup is illustrated in . Two AMSs equipped with an SV and CV and a scanning mobility particle sizer (SMPS, model: 3080, TSI, Shoreview, MN, US) were operated in parallel and sampled ambient air and OFR-processed air alternating every 4 min.
Both ambient and OFR sampling lines were dried by Nafion driers (e.g., MD-110-24S-4, Perma Pure LCC) to keep RH below 30% before gas and aerosol entered instruments. The purpose of the drying is to ensure dry particles were measured with the AMS or SMPS, which is especially critical for estimating total dry volume from SMPS data, and to ensure that the Middlebrook et al. (Citation2012) parameterization of CE in the SV, which was derived for dry ambient particles, can be applied to this dataset. The OFR data used in this study are from ambient air after exposure to high levels of nitrate (NO3) radical. NO3 radicals were generated through rapid thermal dissociation of injected N2O5 (Brown and Stutz Citation2012; CitationPalm et al. Citation2017). The NO3-OFR was developed to investigate secondary organic aerosol (SOA) formation from biogenic VOCs with NO3 radicals. However, the feature of the OFR data that make them useful for this work is that the processed air exhibited sporadic rapid changes in aerosol nitrate fraction, due to formation of high concentrations of NH4NO3 particles formed from nitric acid (HNO3) that was either injected along with the N2O5 or formed by hydrolysis of N2O5 on wetted OFR walls in some cases (Palm et al. Citation2017). This provided an excellent test case for the application of composition-dependent CE in the SV by comparing its time series with those from the SMPS and CV.
Two additional studies were conducted by sampling outdoor air through inlets at the University of Colorado (Boulder, CO, USA) and Aerodyne Research (Billerica, MA, USA). Instrumentation setup in those two studies was similar to SOAS (), except that SMPS and OFR data were not collected. In the Boulder study, substantial amount of OA from primary sources was observed. This is in contrast to SOAS, where biogenic SOA was dominant (Hu et al. Citation2015; Xu et al. Citation2015). In the Billerica study, ACSMs instead of AMSs were used. A summary of field sampling locations, times, and instrumentation details is given in .
Table 1. Brief overview of three field experiments shown in this study.
2.2. AMS/ACSM measurements
In the SOAS study, a high-resolution time-of-flight AMS (HR-ToF-AMS; DeCarlo et al. Citation2006) equipped with an SV was used for the entire campaign. The CV data for the SOAS study was obtained with a compact time-of-flight AMS (C-ToF-AMS; Drewnick et al. Citation2005) for most of the campaign, and with a second HR-ToF-AMS for a few days (7–10 June). The Boulder field study was conducted with two HR-ToF-AMS instruments. In the Billerica study, two Aerosol Chemical Speciation Monitors (ACSM; Ng et al. Citation2011) were used, which provides a good opportunity to evaluate the performance of CV using unit mass resolution (UMR) analysis with different aerosol/background method and sequence than in the AMS. Concentrations and size distributions of aerosol species collected with the AMS were analyzed with standard ToF-AMS analysis software packages (Squirrel version >1.52M and PIKA version >1.12). The ACSM datasets were analyzed with ACSM analysis software (version = 1.5.5.0).
Before each study, the alignment of the particle beam (“Aerodynamic lens alignment”) was verified on each AMS/ACSM to ensure the focused particle beam impacted in the center of the vaporizers. For AMS, aerosol mass concentrations were calculated using the standard method for ambient studies, i.e., as the difference signal between ambient particles (particle beam “open”: 6 or 5 s) and background signal (particle beam “closed”: 4 or 5 s) in mass spectrum (MS) mode (Jimenez et al. Citation2003). ACSM does not have an internal mechanism to open/close the particle beam, so the background signal is instead measured by sampling ambient filtered air (Ng et al. Citation2011). This design feature, together with the slower time response of the ACSM mass spectrometer, results in a longer time step for ambient/background switching (∼30 s open/30 s filtered in this study). The ionization efficiency (IE) in the AMS was calibrated with monodisperse 400 nm dried NH4NO3 particles every few days during the field studies. Both brute-force single particle (BFSP, DeCarlo et al. Citation2006) and condensation particle counter (CPC) calibrations (DeCarlo et al. Citation2006; Ng et al. Citation2011) were performed for the SV-AMS while only the latter method was applied to the CV-AMS and ACSM. The BFSP method, while preferable, is not possible for a CV-AMS since single particle events are not always detectable due to the temporal broadening of single particle signals in the CV (Hu et al. Citation2016b). The relative IE (RIE) of ammonium and sulfate in the AMS was measured with pure NH4NO3 and (NH4)2SO4 particles (Canagaratna et al. Citation2007). Compared to the default SO4 RIE of 1.2, the CV-AMS always exhibited higher sulfate RIEs (1.7–2.4); however, ∼1 was found in the CV-ACSM. This difference between CV-AMS and -ACSM may be due to the different background method, or possibly to differences in ionizer and extractor size and geometry.
The HR and UMR fragmentation table treatment of m/z 18 (H2O+) and m/z 32 (S+) from sulfate in AMS with CV was adjusted according to the ratio reported in Hu et al. (Citation2016b). Other fragmentation table entries were unchanged from the default table applied for analysis of SV-AMS datasets. The default RIE of ambient OA (1.4) and chloride (1.3) were used in all the datasets for both vaporizers. The RIE of chloride in both vaporizers from HR-ToF-AMS (∼1.3–1.4) was calibrated in the laboratory after the field campaigns, which are similar to the default value. The particle size measured by the AMS was calibrated with dried monodisperse pure NH4NO3 or (NH4)2SO4 particles from 70 to 550 nm, size-selected with a differential mobility analyzer (DMA).
2.3. SMPS measurement
The sizing measurement of the SMPS was verified with monodisperse polystyrene latex spheres (PSLs; Duke Scientific, Palo Alto, CA, US) with nominal standard diameters of 250–400 nm. The flow rates of the sheath and sample flows of the SMPS were also verified. The submicron volume concentration was estimated from SMPS measurement by integration of the measured number distributions. The mass concentration was calculated by multiplying the volume concentration by a bulk aerosol density estimated from the AMS chemical composition (assuming internally mixed particles). The aerosol density can be calculated by Equation (Equation1[1] ) (DeCarlo et al. Citation2008):
[1] where [OA] is the mass concentration of organic aerosol, and
is the density of each component (g cm−3).
was estimated from its oxidation level as measured by the SV-AMS (Kuwata et al. Citation2012). The elemental ratios used for the density estimation were based on the updated calibrations of Canagaratna et al. (Citation2015). The calculated density from the updated calibration method actually shows slightly better agreement with measured density of chamber SOA (Slope = 0.98) than when using the original Aiken method (Slope = 0.95; Aiken et al. Citation2008), as shown in Figure S1. A time series of OA density (changing with the ambient OA elemental composition) was used in Equation (Equation1
[1] ). In SOAS, the average estimated ambient OA density was 1.34 ± 0.005 g cm−3 (variability in time, ±1σ std. err.). The inorganic densities used in Equation (Equation1
[1] ) were estimated from Haynes (Citation2015) and held constant. The 1σ uncertainty of the total aerosol density (12%) was estimated by Montecarlo error propagation, assuming 15% (5%) error in OA (inorganics) density. The total uncertainty of the SMPS mass concentration was estimated to be 28% by propagating the uncertainties from aerosol density (12%) and SMPS volume estimation (25%).
2.4. Organic nitrate detection
To interpret the fragmentation patterns of ambient inorganic nitrate, it is necessary to first characterize the detection of organic nitrates in the CV, since those species were significantly present during, e.g., SOAS (Ayres et al. Citation2015; Lee et al. Citation2016).
The main fragment ions of nitrate for NH4NO3 particles are NO+ and NO2+, which typically contribute >95% of the total nitrate signal in both vaporizers (Hogrefe et al. Citation2004; Hu et al. Citation2016b). However, nitrate groups (−ONO2) from organic nitrate (RONO2) can generate the same ions (Farmer et al. Citation2010). For the same instrument and operating conditions, the NO2+/NO+ ratio for organic nitrate is typically a factor of ∼2–3 lower than for pure NH4NO3 (Rollins et al. Citation2009; Farmer et al. Citation2010; Fry et al. Citation2011, Citation2013). RONO2 is thought to decompose thermally at the AMS vaporizer into gas-phase NO2 and RO. NO2 can then thermally decompose to gas-phase NO. Thus, the gas-phase products of thermal decomposition of RONO2 are a combination of NO2 and NO. For NH4NO3, the products are thought to be gas-phase HNO3, NO2, and NO in the SV (Drewnick et al. Citation2015; Hu et al. Citation2016b; Pieber et al. Citation2016). The low NO2+/NO+ for organic nitrate is likely due to the dominance of gas-phase NO2 and NO, which produce much lower NO2+/NO+ (∼0.3–0.5 and zero, respectively) than HNO3 (∼1.17–2) after 70eV EI ionization (Friedel et al. Citation1959; Hu et al. Citation2016b; Linstrom and Mallard Citation2016). Thus, using measured NO2+/NO+ ratios from pure NH4NO3 and ambient NO2+/NO+ ratios measured in the AMS, NH4NO3, and organic nitrate fractions can be estimated (Fry et al. Citation2013). Note that to use this method, other inorganic nitrate salts (e.g., NaNO3 or Ca(NO3)2) need to be negligible within the AMS submicron size ranges, since they show comparably low NO2+/NO+ ratios (0.01–0.1) as organic nitrate (Allan et al. Citation2004a; Farmer et al. Citation2010; Hu et al. Citation2016b).
Furthermore, we investigated whether those differences of NO2+/NO+ ratios between pure NH4NO3 and organic nitrate can still be discerned with the CV. In chamber studies where large amount of organic nitrates (>1–8 µg m−3 –ONO2 functional group mass) were formed through α-pinene+NO3 radical chemistry, the NO2+/NO+ ratio in the CV (∼0.0045) were 10 times lower than from pure NH4NO3 (∼0.06, ). This suggests that organic nitrate and pure NH4NO3 in CV can be still separated in the mixed nitrate aerosol based on their differences of NO2+/NO+ ratios. However, the application of this method is more likely to be limited by the signal-to-noise levels at low concentrations, compared to using the method for SV data. Based on the results in , we estimated that an order-of-magnitude higher nitrate concentration (∼100 ng m−3) is needed for the CV than for the SV (10 ng m−3) to distinguish NO2+/NO+ from NH4NO3 vs. organic nitrate, as shown in .
Figure 2. (a) Time series of NO2+/NO+ ratios for CV and SV, total nitrate for secondary organic aerosol produced from α-pinene oxidation by NO3 radical in environmental chamber experiments. The nitrate present was entirely organic. The difference of total nitrate signal between the SV and CV at high nitrate concentration loadings was likely caused by the different AMS aerodynamic lens inlet size cuts as the particles grew very large at the end of the experiment. (b) 1-min measurement noise in NO2+/NO+ ratios, quantified as the standard deviation of 1-min data points during periods of constant concentration in (a). Regression lines for each trace show that RONO2 can be theoretically identified with one vaporizer until the noise level becomes comparable with the actually NH4NO3-RONO2 difference, which corresponds to ∼10 ng m−3 for SV and 100 ng m−3 for CV. Note that the identification can be more difficult in field data for a variety of reasons.
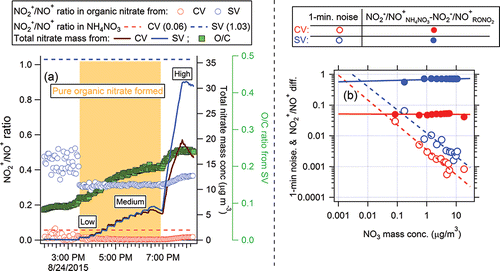
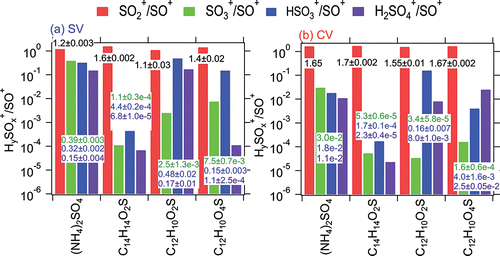
Figure 3. Ratios of intensity for major sulfur-containing fragment ions (SO2+, SO3+, HSO3+, and H2SO4+) to SO+, for ammonium sulfate and three sulfone molecules in (a) the SV and (b) the CV. Sulfones tested were benzyl sulfone (C14H14O2S), diphenyl sulfone (C10H12O2S), and bis (4-hydroxy-phenyl) sulfone (C10H12O4S). The uncertainty of each ratio was obtained from the regression slope uncertainty when analyzing multiple 20 s laboratory measurements.
2.5. Organic sulfur detection
Ambient aerosols are known to sometimes contain organic sulfur species, and their presence might be detectable using fragment ratios, analogously to organic nitrates. It is of interest to evaluate the ability of the SV or CV to allow such investigations. As an example, we examined organic sulfones (R-S(=O)2, e.g., dimethyl sulfone or bis-hydroxymethyl sulfone). Sulfones have been reported to be oxidation products of dimethyl sulfide (DMS) or SO2 from power plant and urban plumes (Eatough and Hansen Citation1984; Berresheim et al. Citation1993). We examined the HySOx+/SO+ ratios in three different organic sulfone standards in the laboratory, i.e., benzyl sulfone (C14H14O2S), diphenyl sulfone (C12H10O2S), and bis (4-hydroxy-phenyl) sulfone (C12H10O4S), together with inorganic (NH4)2SO4 in both the SV and CV (). The SO3+/SO+ ratios from different types of organic sulfone (10−3) were found to be more than 2–3 orders of magnitude lower than those in (NH4)2SO4 (0.4) in the SV, while other HySOx+/SO+ ratios showed diverse trends. This substantially low abundance of SO3+ has also been observed in SV-AMS study for hydroxymethanesulfonate (HMS), which is a tracer of aqueous-phase chemistry (Gilardoni et al. Citation2016). The differences of HySOx+/SO+ fragmentation patterns have also been observed between organosulfate and ammonium sulfate in the laboratory (Huang et al. Citation2015). Although lower HySOx+/SO+ ratios were observed for both sulfones and (NH4)2SO4 in the CV due to additional thermal decomposition, the differences of SO3+/SO+ ratios between organic sulfone (∼10−6 to 10−5) and (NH4)2SO4 (10−2) are still discernable in the CV. However, for typical mass concentrations of ambient aerosols, those differences in the CV may be more limited by signal-to-noise levels, similar to organic/inorganic nitrate separation in the CV.
3. Results and discussion
3.1. Fragmentation patterns
3.1.1. Sulfate
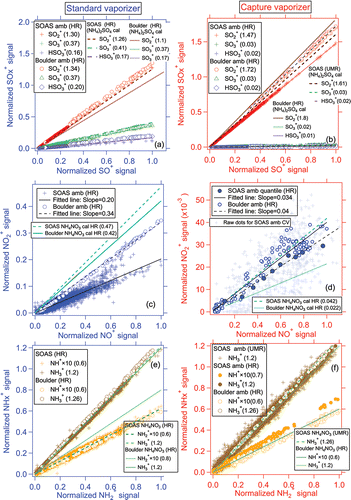
The ambient fragmentation patterns of sulfate for both vaporizers in the Boulder and SOAS studies are shown in . The ambient HySOx+/SO+ ratios for both vaporizers were consistent with those from pure (NH4)2SO4 calibrations (Hu et al. Citation2016b; Xu et al. Citation2017), supporting the dominance of (NH4)2SO4 over organic sulfur species in both campaigns. The CV exhibited slightly higher SO2+/SO+ ratios (1.47–1.72) than the SV (1.26–1.34), while lower SO3+/SO+ and HSO3+/SO+ ratios (<0.03) were observed in the CV than SV (0.2–0.4). The quantification of UMR SO3+ and HSO3+ are more easily influenced by organic aerosols than UMR SO2+ and SO+ due to the much larger contributions of organic ions at those m/z, as shown in Figure S2 (C5H4O+ and C6H8+ at m/z 80; C5H5O+ and C6H9+ at m/z 81). Although those contributions are approximately subtracted with the fragmentation table approach (Allan et al. Citation2004b) that method can have errors when the organic fractions of the UMR signals are large. Thus, in ambient plumes with high OA concentrations and relative high OA/SO4 ratios such as biomass burning plumes, UMR sulfate in the SV may have higher errors (Milic et al. Citation2016). UMR quantification of sulfate with the CV could more easily be based entirely on SO+ and SO2+ (by modifying the fragmentation table), eliminating this error.
Figure 4. Scatterplots of major ion ratios in the SV vs. the CV (a vs. b) for sulfate, (c vs. d) for nitrate, and (e vs. f) for ammonium during the SOAS and Boulder ambient studies. The ratios of fragment ions from pure (NH4)2SO4 and NH4NO3 particles during SOAS calibration experiments are also shown. For clearer comparison, all signals were normalized to the maximum signal of each species on the x-axis during each study. Note that the ambient HR nitrate, sulfate, and ammonium shown from the CV in SOAS are from the 3 day long HR dataset (see methods section). The UMR data for CV are from C-ToF. All SV data are HR. All lines were determined using orthogonal distance regression with fits forced through zero. Regression slopes are given in the figure legends.
No significant variations in time of the sulfate fragmentation pattern were observed in both vaporizers during the SOAS study. Organosulfate groups (−OSO3) from species derived from isoprene epoxydiols oxidation consistently accounted for a very small fraction (∼5%) of measured total sulfate signal in AMS (Hettiyadura et al. Citation2015; Hu et al. Citation2015; Liao et al. Citation2015; Riva et al. Citation2016). This is consistent with the small variation of the sulfate fragmentation pattern in SOAS. Additional investigation of organosulfate detection with the CV is recommended.
3.1.2. Nitrate
The ratios of fragment ions of NO2+/NO+ from the SOAS and Boulder studies are shown in . Generally, ambient NO2+/NO+ ratios for the SV (0.2 and 0.34, respectively) were higher than for the CV (0.03–0.04), which is consistent with the results for pure NH4NO3 on both vaporizers (Hu et al. Citation2016b). Much lower NO2+/NO+ ratios were seen in the ambient SOAS study for the SV dataset than the ratios from pure NH4NO3 particles during calibrations (), indicating a substantial RONO2 contribution to the ambient nitrate of SOAS study. By applying the RONO2 method of Fry et al. (Citation2013) to SOAS, ∼80% of ambient submicron nitrate was estimated to be RONO2, which agreed reasonably well with RONO2 measured with some independent methods (Ayres et al. Citation2015; Lee et al. Citation2016). During SOAS, the NO2+/NO+ (HR) ratios for the CV (0.03) were lower than for pure NH4NO3 (0.04, ), which is qualitatively consistent with the substantial presence of RONO2 during this study. However, NH4NO3 and RONO2 cannot reliably be discriminated using the Fry et al. method with the CV data due to the very low ambient nitrate mass concentrations (40 ± 20 ng m−3), which is lower than the estimated limit of detection for this method (100 ng m−3) discussed in Section 2.4. The NO2+/NO+ ratio for ambient nitrate in the CV in SOAS (0.03, , when ∼80% of the nitrate is organic) is higher than that obtained from the pure organic nitrate (∼0.0045) in the chamber studies (). Possible reasons for this difference include, (Equation1[1] ) different instrument conditions, (2) different type of organic nitrates between chamber and ambient studies, (3) organic interference at m/z 30 and m/z 46 in the UMR ambient dataset causing a larger quantification uncertainty of those ratios, or (4) other small unknown effects that become important at low signal levels.
3.1.3. Ammonium
Fragment ions from ammonium are thought to arise mainly from the direct ionization of gas-phase NH3 formed from thermal decomposition of the salts (Hu et al. Citation2016b). In both the SV and CV, the fragmentation pattern of ammonium in the ambient SOAS datasets show ratios of NH3+/NH2+ (∼1.2–1.3) and NH+/NH2+ (∼0.06) that are consistent with ratios from pure NH4NO3 calibration particles measured during the campaign (). For SOAS with varied nitrate/sulfate mass ratio in the NO3-OFR (Figure S3), constant NH3+/NH2+ ratios were observed, consistent with previous results (Hu et al. Citation2016b; Jimenez et al. Citation2016; Xu et al. Citation2017).
3.2. Quantification of CE
The comparisons of ambient total and inorganic species mass concentrations for both vaporizers are shown in . show the regression slopes and correlation coefficients (R) of those comparisons from the different studies. A composition-dependent CE was applied to the SV-AMS data (Middlebrook et al. Citation2012). The time dependence of the estimated CE for both the ambient and NO3-OFR datasets of the SV-AMS during SOAS are shown in Figure S4. For the ambient data, most of the CE values were between 0.5 and 0.6 with periodic increases up to 0.7–0.8. All datasets using the CV were analyzed using a CE of 1. In the following discussion, we summarize and discuss the comparison results by species.
Figure 5. Regression slopes and correlation coefficients (R) of total submicron aerosol mass concentration between AMS and SMPS in ambient and NO3-OFR datasets during SOAS study. The same parameters are shown for comparisons of the main chemical species between SV-AMS and CV-AMS, and PILS-IC vs. CV-AMS in different studies. This plot is the summary plot of . An uncertainty band of 44% is shown, which is calculated based on the quantification uncertainties of AMS and SMPS. The total uncertainty for the AMS vs. PILS-IC comparison is similar.
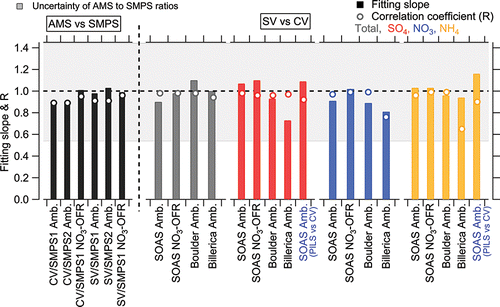
Figure 6. Ambient time series and scatterplots of ambient (a) total, (b) sulfate, (c) nitrate, (d) ammonium, and (e) chloride between the SV and CV during the SOAS study. Two SMPS datasets and one PILS-IC dataset from the SOAS field study are also shown here to compare to total and speciated inorganic AMS mass. All black solid lines were determined using orthogonal distance regression with fits forced through zero intercept and in all figures shown below. Regression slopes are given in the figure legends. The gray solid lines and in all figures shown below are the orthogonal distance regression lines without intercept constraining to be zero. The values from both regression methods are listed in Table S2.
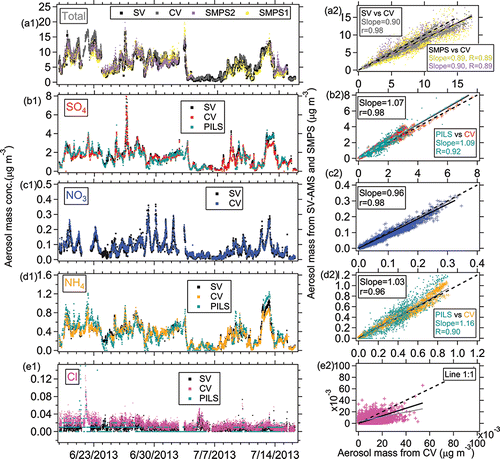
Figure 7. Time series and scatterplots of (a) total, (b) sulfate, (c) nitrate, (d) ammonium, and (e) chloride for the SV and CV after NO3-OFR during the SOAS study. SMPS data coupled with the NO3-OFR in the SOAS field study are also shown.
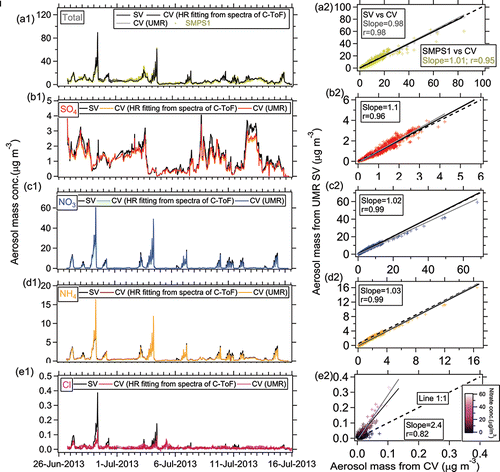
Figure 8. Ambient time series and scatterplots of (a) total, (b) sulfate, (c) nitrate, (d) ammonium, and (e) chloride for the SV and CV in the Boulder study.
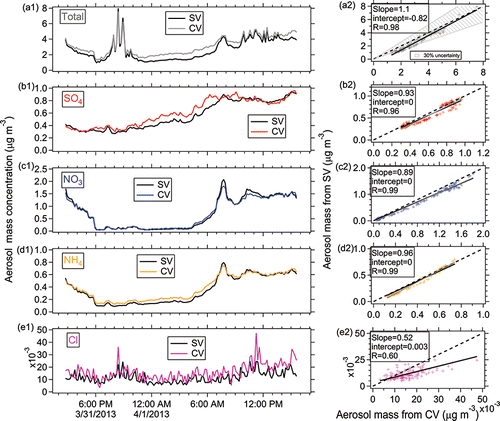
Figure 9. Ambient time series and scatterplots of (a) total, (b) sulfate, (c) nitrate, and (d) ammonium for the SV and CV in the Billerica study.
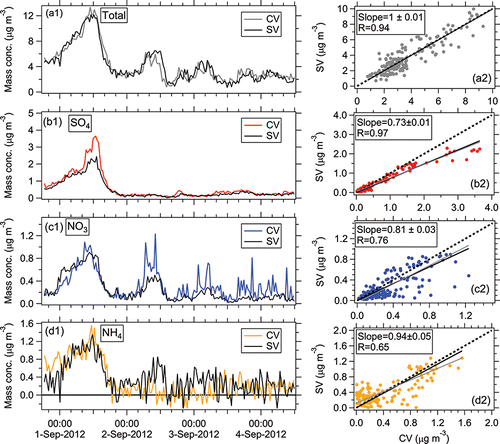
Table 2. Summary of orthogonal distance regression slopes (with fits forced through zero interceptFootnote*) and correlation coefficients (R) of total and inorganic species measured by AMS or ACSM between the SV and CV and PILS-IC vs. CV in different field studies. Those results are also illustrated in .
Table 3. Summary of orthogonal distance regression slopes (with fits forced through zero intercept) and correlation coefficients (R) for SMPS vs. AMS for total aerosol mass. Those results are also illustrated in the .
3.2.1. Total NR-PM1 aerosol
Good agreement of total aerosol mass concentrations between two SMPS and CV-AMS was also observed in SOAS, with regression slopes between 0.89 and 1.0 and ( and ). The uncertainty (accuracy) of the AMS to SMPS ratio was estimated to be 44% by propagating the uncertainties of AMS quantification (36%) and SMPS total mass estimation (28% in Section 2.3). The consistency of total aerosol mass concentrations measured between SMPS and CV-AMS suggests the CV is capable of quantifying non-refractory sub-micron ambient aerosols with negligible particle bounce losses. The CE of ambient aerosol in the CV is roughly equivalent to 1. More ambient comparisons between SV and CV, in conjunction with SMPS, in areas with externally mixed aerosol are recommended.
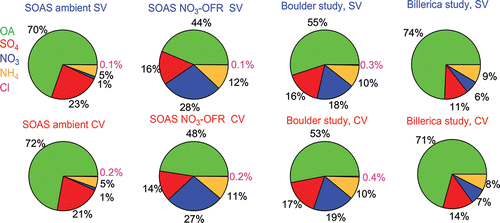
Good correlation of the total NR-PM1 concentrations between SV and CV was also found for the SOAS studies (R > 0.94, and ). The total mass concentrations of aerosols detected by the CV (CE = 1) were within 12% of those measured by the SV when the composition-dependent CE was used. During the three ambient studies, the main chemical component of non-refractory submicron aerosol was OA (>50%, as shown in ). Therefore, the good correlation of total aerosols between the SV and CV is also indicative of good agreement of OA in those studies. The small intercept of total aerosols in the comparison for the Boulder study () was mainly caused by the enhanced signals of OA ions below m/z 44 in the CV. The Boulder study is the only study where a significant intercept was observed. The campaign-average chemical compositions of ambient aerosols measured by the SV and CV were very similar (), indicating that aerosol composition measurements with the CV can be accurate when ammonium, sulfate, and chloride RIE are measured. A detailed analysis of the comparisons for OA composition and quantification will be presented in a future publication.
Figure 10. Average chemical compositions of non-refractory submicron aerosols during SOAS, Boulder, and Billerica studies for the SV and CV. Note that the average chemical composition in SOAS shown here is for the period 19 June–16 July 2013.
3.2.2. Sulfate
In all studies, the time series of sulfate from the SV and CV all tracked well, with all Pearson's R values above 0.96. The regression slopes (0.93–1.1) between the SV and CV for the SOAS and Boulder studies were within 10% of the 1:1 line (). Good agreement of sulfate with another independent instrument, particle-into-liquid sampler coupled to ion chromatography (PILS-IC, Guo et al. Citation2015) vs. CV during SOAS study is also observed (slope = 1.09 and R = 0.92), as shown in . In the Billerica study (), the lower regression slope (0.73) between two ACSMs may be due to the uncertainty of default sulfate RIE (1.2) applied in the SV that was not calibrated prior to the study. A sulfate RIE of 1 for the SV-ACSM (which is within the range of calibrated RIEs observed in previous studies) would result in a comparable agreement to the AMS datasets (Crenn et al. Citation2015). Laboratory-generated pure (NH4)2SO4 showed a CE below 1 in the CV, ∼0.8 (Hu et al. Citation2016b) and 0.9 (Xu et al. Citation2017) at normal vaporizer temperatures (500–600°C). However, the good agreement of ambient sulfate for the CV and SV in different studies suggested that the CE of ambient sulfate from the CV was ∼1. Similarly, the CE of ambient sulfate when using the SV (>0.5) is also usually higher than that of pure (NH4)2SO4 (0.2–0.4) due to less bounce (Matthew et al. Citation2008). The better detection of sulfate in the ambient aerosols than pure sulfate species is likely related with its internal mixing with nitrate and OA, which reduces the bounce fraction (Canagaratna et al. Citation2007). Xu et al. (Citation2017) also showed an increased sulfate CE with higher fractions of NH4NO3 in the NH4NO3 and (NH4)2SO4 mixtures for the CV in the laboratory studies.
Besides the comparison of sulfate difference signal (open minus close signal) discussed above, it is also interested to compare the sulfate background detection. It can help understanding if there is slow desorption for sulfate in the CV due to its longer residence time than SV. The time series of particle beam open (ambient + background) and closed (background) sulfate signals in SOAS are shown in Figure S5. Consistent with laboratory experiments (Hu et al. Citation2016b), much lower ambient sulfate background was found for the CV than for the SV. Experiments have been conducted in the laboratory to investigate the lifetime of sulfate signal decay by maintaining the particle beam close/open time intervals at several minutes (Hu et al. Citation2016b). The lower background for ambient sulfate in the CV is mainly caused by the faster sulfate signal decay in the CV (τ < 2 s) than that in SV (τ ∼ 13 s) at vaporizer temperatures around 500–600°C. The interval of MS mode switching during ambient measurement in both AMS was around 4–6 s. Systematically higher closed/open ratios were also observed in the ambient SV datasets (∼0.17 vs. 0.05–0.12 in MS mode in laboratory studies) than those for the CV (∼0.04 in the ambient dataset and ∼0.01 in the laboratory studies, Hu et al. Citation2016b).
3.2.3. Nitrate
The time series of HR nitrate mass concentration between the two vaporizers agreed well with each other with a narrow range in slopes of 0.89–1.02 (correlation coefficient R > 0.97, ) for the SOAS (including the NO3-OFR measurement) and Boulder studies. A very good linear correlation for nitrate between the two vaporizers was observed over a large range of concentrations (0–60 µg m−3, ). The good correlation indicates the CE of ambient nitrate is also approximately unity in the CV. Note that nitrate signals for all studies shown here are the total nitrate signal without separating organic nitrate and inorganic nitrate.
Larger deviations in the comparisons of NO3 mass concentrations (slope = 0.81) were observed for the UMR Billerica dataset (both UMR ACSM) studies when the default fragmentation table was used (). A similar regression slope (0.82) was also found in the mass comparison of UMR nitrate (default fragmentation table applied) between both vaporizers in SOAS, as shown in Figure S9. The high-resolution fitting showed that at m/z 30 and 46 where most nitrate signal is detected, there are other organic ions (e.g., m/z 30: CH2O+ and CH4N+; m/z 46: CH2O2+), as shown in Figure S6. This larger slope difference could be due to changing interferences of organic ions to mass spectra obtained from SV and CV slightly differently that are not captured with default fragmentation table.
CH2O+ was found to be a prominent ion at m/z 30 in the SOAS study, which has also been observed in other biogenically influenced areas with low nitrate concentrations, such as in the Amazon region (de Sá et al. Citation2016). In the presence of these organic interferences, the total nitrate mass concentrations calculated for the SV in SOAS, if only UMR analysis with the default fragmentation table were applied, would result in an overestimation by a factor of ∼2.3 compared to using HR analysis (Figure S10).
These overestimations suggest that the default UMR fragmentation table, at least for studies dominated by biogenic emissions (e.g., southeast US or Amazon), will require updated corrections regardless of vaporizer configuration. Based on results from the SOAS study, modified organic interference subtractions in the fragmentation table are shown in Table S1. By using the revised fragmentation tables, much better agreement of UMR and HR nitrate mass concentrations was found for both vaporizers (slope = 0.8–1) than when using the default UMR fragmentation table (∼2.3–2.7). This modified table is calculated only based on SOAS study. More detailed examination on AMS quantification for UMR inorganic nitrate from other areas with strong biogenic emissions and other types of emissions are recommended.
3.2.4. Ammonium
Ammonium also showed good agreement between the SV and CV with a regression slope ranging between 0.94 and 1.03 with R > 0.96 in the HR and C-ToF datasets and R = 0.65 in the UMR datasets of the ACSM comparison ( and ). Good agreement of ammonium between CV and PILS-IC during SOAS study are also found (slope = 1.16; R = 0.90, ). The lower correlation coefficient of ammonium in the Billerica ambient dataset is probably due to low signal-to-noise ratio, due in part to the noise left after subtracting average interferences of air fragments in the ACSM (e.g., m/z 16 O+ to NH2+ or m/z 17 OH+ to NH3+), the lower sensitivity of the ACSM compared to the AMS, and the relatively low ammonium concentrations ().
3.2.5. Chloride and chloride artifacts
Due to the extremely low concentrations of chloride in ambient air of these field studies (<0.1 µg m−3), it is difficult to make any firm conclusions on the quantification of ambient chloride in the SV vs. CV in this study. However, a reasonable linear correlation for chloride was still found between the two vaporizers (e.g., R = 0.6 in Boulder study). The deployment of the CV sampling ambient chloride should be further explored in areas with higher submicron chloride mass concentrations.
Extra chloride signals (maximum 0.28 µg m−3 and 0.1 µg m−3 in SV and CV, respectively) were observed when sampling NO3-OFR output for both vaporizers compared to ambient chloride. The regression slope between the SV and CV in the NO3-OFR (2.4) was systematically higher than from the corresponding ambient dataset (0.7, vs. ). No known source of chloride aerosol was present in the NO3-OFR. We hypothesize that this enhanced chloride signal was caused by surface chemistry on the vaporizer. As Drewnick et al. (Citation2015) and Hu et al. (Citation2016b) have shown, chloride salts (e.g., NaCl or NH4Cl, or HCl or other species formed from their decomposition) appear to be very sticky on the vaporizer surface and/or ionization chamber walls, and thus some chloride residue may always be present on the vaporizer. Thermal decomposition products from nitrate on the SV include gas-phase HNO3(g), NO2(g), or NO(g). HNO3(g) may react with chloride on the vaporizer analogously to the atmospheric heterogenous reaction, e.g., NaCl(s) + HNO3(g) → HCl(g) + NaNO3(s) (Harkel Citation1997). The gas-phase HCl(g) would then lead to extra chloride ion signal. Since this possible artifact signal is quite small relative to nitrate, this effect would be most apparent when sampling high NH4NO3 mass concentrations (e.g., 10–60 µg m−3 in NO3-OFR). Good linear correlations between nitrate and chloride signals in the SV and CV both support this possible mechanism ().
Figure 11. Scatterplots between nitrate equivalent mass concentration (RIE = 1) of chloride and nitrate detected by AMS with the SV and CV measuring outflow of the NO3-OFR during the SOAS study.
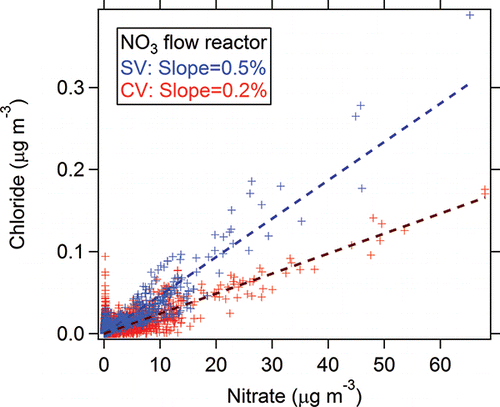
The linear regression slope in between chloride and nitrate (nitrate equivalent mass concentrations, both assuming RIE = 1) suggests the enhancement of chloride in the CV (0.2% chloride/nitrate) is substantially lower than in the SV (∼0.5% chloride/nitrate). The lower enhancement of chloride in the CV is probably because (Equation1[1] ) the main thermal decomposition product of nitrate in the CV is gas-phase NO(g) with less gas-phase HNO3(g) formed (Hu et al. Citation2016b); (2) potentially lower chloride residual in the CV because of shorter sampling history of the CV than the SV used in this study.
In addition to the SOAS OFR results, we have also observed increased chloride signals when sampling pure organic acid particles with both vaporizers (0.002–0.02% per nitrate mass, as shown in Figure S12). This indicates that artifact chloride signal might occur for acids other than HNO3(g). However, we cannot rule out the possibility of chloride contamination in the organic-containing solution given its very small magnitude. The chloride enhancement was found to be negligible when sampling pure (NH4)2SO4 particles in both vaporizers. AMS users should consider performing an RIE calibration for chloride, using ammonium chloride, before field and laboratory studies, although it may also result in an increased intensity of the interference discussed in this section.
3.3. Evaluation of composition-dependent CE
The SOAS NO3-OFR dataset showed a large variation of NH4NO3 fraction in its output (1%–80%), which provided a good opportunity to examine the validity of the composition-dependent CE parametrization of Middlebrook et al. (Citation2012). AMS/SMPS mass ratios as a function of NH4NO3 fraction in total aerosol are shown in . A clear NH4NO3-fraction-dependent CE was observed in the SV, which is similar to the SV results of Middlebrook et al. (Citation2012). However, AMS/SMPS mass ratios (∼1) in the CV did not show a dependence with varying NH4NO3 fraction, suggesting complete collection of ambient aerosol ().
Figure 12. Ratio of AMS to SMPS mass concentrations as a function of the measured ammonium nitrate mass fraction, for both the SV and CV. The CE parameterization of Middlebrook et al. (Citation2012) is also shown. The error bars are the standard deviation of the averaged decile values.
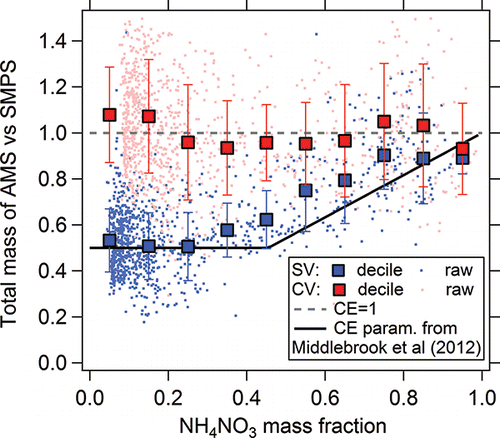
To further explore this effect, a constant CE of 0.55 was applied to the SV-data (Figure S13). This value was determined as the average of the dynamic composition-dependent CE used in Figure S4. Systematically higher nitrate and ammonium concentrations (a factor of 1.5) are observed in the SV with constant CE compared to the CV (Figure S13). The high regression slopes were mainly biased by the overestimated concentration when NH4NO3 fractions were high. Ambient organic nitrates typically only dominate total nitrate at lower ambient nitrate concentration (<0.5 µg m−3; Day et al. Citationin preparation). Thus, even if organic nitrates at high concentrations led to a similar increase of CE as NH4NO3 in Middlebrook et al. (Citation2012) (which has not been shown to our knowledge), their low ambient fractions will generally not result in an increased CE. For sulfate species, most of the high sulfate mass concentrations occurred during time periods with low NH4NO3 fractions. Thus, the regression slope of sulfate with constant CE applied was similar to that with composition-dependent CE used. This comparison provides strong support for the application of the composition-dependent CE for accurate quantification with SV in areas with variable (and relatively high: >50%) NH4NO3 fractions.
3.4. AMS size distribution measurements
For monodisperse inorganic standard species, the CV showed additional broadening of the size distribution measurements, compared to the SV (Hu et al. Citation2016b; Xu et al. Citation2017). This is consistent with the delayed vaporization due to multiple bounces and longer thermal decomposition timescales within CV. In ambient air, aerosol size distributions are usually relatively broad (Seinfeld and Pandis Citation1998). The capability of the CV to characterize typical ambient accumulation-mode size distributions is examined in this section.
Particle time-of-flight (PToF) and size distributions of ambient total and inorganic species from both vaporizers during the Boulder study are shown in . An apparent PToF shift (∼100 µs) in the CV was observed compared to that in the SV. This lag due to longer residence time in the CV is consistent with the PToF comparison from laboratory-generated standard species. The measured peak in the CV is up to 30% wider than in the SV (), consistent with the broadening effect observed for monodisperse particles of inorganic species (a factor of ∼1.8–5.5; Hu et al. Citation2016b).
Figure 13. (a) Measured particle time-of-flight distributions for ambient aerosols using the SV and CV in the Boulder study. (b) Comparison of the calibrated size distributions. The sizing with the CV was calibrated with pure ammonium sulfate particles. The relationships between particle velocity and size are shown in .
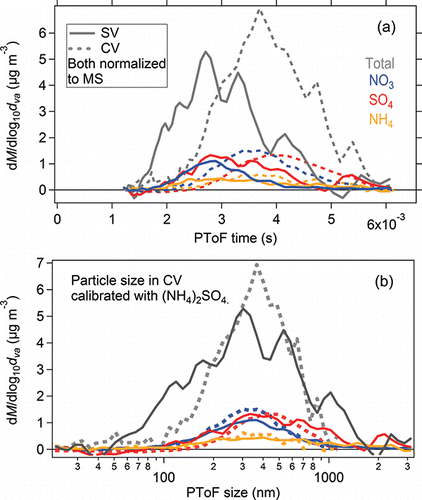
Table 4. Summary of peak widths of ambient aerosol PToF and size distributions in the Boulder study. FWHM method was used to calculate peaks widths here. The corresponding PToF and size distributions are shown in .
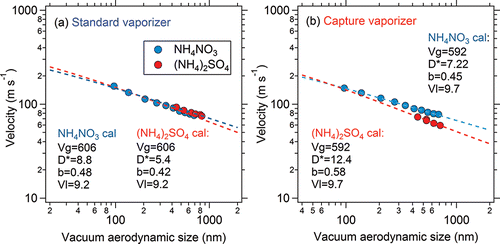
Calibrations of PToF size distributions based on monodisperse NH4NO3 and (NH4)2SO4 particles were carried out for both the SV and CV. The PToF size calibration curves from both vaporizers are shown in . In the SV, the PToF velocity calibration curve from NH4NO3 was consistent with that from (NH4)2SO4, while the PToF size calibration curves from NH4NO3 and (NH4)2SO4 in the CV were quite different because of the longer average residence time of (NH4)2SO4 than NH4NO3. Most (NH4)2SO4 signal in the SV is detected with limited broadening compared to NH4NO3 because the signal is dominated by the particles that do not bounce (Robinson et al. Citation2016). In contrast, (NH4)2SO4 particles in the CV may have several opportunities to vaporize as it rattles around in the vaporizer cavity. This explanation is consistent with the large broadening of pure, monodisperse (NH4)2SO4 size distributions (∼ a factor of 5.5 for 250 nm particles) shown in Hu et al. (Citation2016b).
Figure 14. Particle velocity vs. vacuum aerodynamic diameter of ammonium nitrate and ammonium sulfate in the (a) SV and (b) CV. The gas velocity within the aerodynamic lens (Vl), calculated from the lens flow rate and pressure, and the gas velocity upon exiting the nozzle (Vg) estimated by the measured arrival time of airbeam signal, were constrained in the fitting. Other parameters were obtained by curve fitting (according to Jayne et al. Citation2000; Allan et al. Citation2003a).
By using the NH4NO3 size calibrations to calculate the CV size distributions of ambient aerosols, a large shift of about 250 nm was observed compared to the SV (Figure S14). When the calibration curve from (NH4)2SO4 is applied, the updated size distributions from the CV is more consistent with the SV distributions (). This indicates that for typical ambient particles, mainly composed of OA and sulfate, most of the particles have vaporization/detection timescales closer to pure (NH4)2SO4 in the CV. We note that the CV size calibrations were performed here using the peaks of the AMS PToF signals from monodisperse particles. For SV the recommended calibration method is to use the arrival time plus ½ of the chopper opening time, rather than the peak. However, the latter method is not desirable for the CV due to the larger and more variable broadening observed.
The good agreement of the updated size distribution between the SV and CV supports that CV-AMS particle sizing is still useful for ambient measurements. The peak width of the size distributions using a calibration from the (NH4)2SO4 calibration curve (415 ± 74 nm) was close to that in the SV (513 ± 49 nm, ). The slightly narrower peak width of total aerosol for the CV here is not necessarily indicative of better sizing resolution detection but rather due to uncertainties in the pure (NH4)2SO4 sizing calibration and its application to ambient data, as (NH4)2SO4 data show both a large delay as well as broadening of the peaks, which may be greater than for mixed-composition ambient particles. We recommend using (NH4)2SO4 calibrations when analyzing data from sulfate/organic dominated ambient particles. Further systematic characterization of CV size distribution detection is recommended.
4. Conclusions
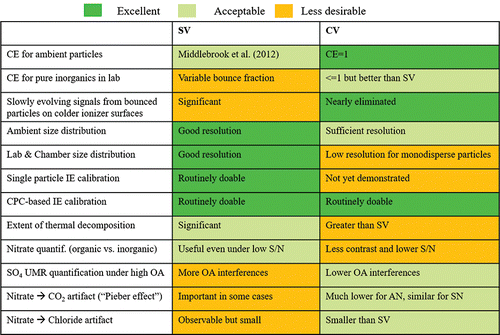
In this study, we have examined the performance of the AMS equipped with a CV in regards to chemical quantification, characterization, and sizing of ambient inorganic and total aerosols with four field datasets. AMSs with SV and CV installed were calibrated and sampled in parallel. A summary of the advantages and disadvantages of the SV and CV for AMS/ACSM measurements, based on the results in this study and the previous laboratory study (Hu et al. Citation2016b), is shown in .
Figure 15. Summary of the advantages and disadvantages of SV and CV usage in AMS/ACSM measurement based on this field study and Hu et al. (Citation2016b) laboratory study. Note that some effects (such as Pieber and chloride artifacts) need characterization in other instruments and studies.
The overall results showed good agreement of mass concentration of total and speciated inorganic composition detected between the SV and CV, and also SMPS or PILS-IC vs. CV-AMS (<16% deviation, R = 0.89–0.99 of HR comparisons for major inorganic species). For UMR datasets, good agreement and correlations of total and inorganic aerosols were also found (slopes are within 0.73–1.1, R > 0.65). The field studies indicate that CE ∼ 1 for ambient aerosol in the CV. The comparison results also support that the composition-dependent CE due to high nitrate fractions for ambient aerosol measurements when using an SV works well, and its robustness was demonstrated for a study with rapidly varying NH4NO3 fractions.
Compared to the SV, fragmentation patterns of nitrate and sulfate in the CV were altered due to additional thermal decomposition processes, with a shift in the ion distributions to smaller ions. The fragmentation pattern of ammonium showed little difference because the gas-phase NH3(g) from NH4NO3 and (NH4)2SO4 thermal decomposition was not influenced by additional thermal decomposition since it appears to be evaporated and ionized directly. We investigated the fragmentation patterns of organic vs. inorganic nitrate for both vaporizers. The differences in NO2+/NO+ ratios between organic and inorganic species observed in the SV can still be observed in the CV. This indicates that the distinguishing chemical information from those ions is still present with the CV. However, with background ambient aerosol concentrations, the quantification of the variations of those ratios may sometimes be limited by the signal-to-noise level in the CV due to the much lower concentrations of the larger ions. HySOx+/SO+ ratios between organic sulfone vs. inorganic (NH4)2SO4 showed similar phenomenology as nitrate in the SV and CV.
In biogenic areas, UMR nitrate mass is overestimated when using default fragmentation table (∼factor of 2–3 in SOAS) for both vaporizers, due to underestimation of the organic interferences. Updates to the fragmentation tables for both vaporizers are proposed in this study. Since the CV might be installed extensively in the commercial ToF-ACSM and Q-ACSM with PM2.5 lens, attention needs to be paid to the organic/air interferences in UMR inorganic quantification when inorganic concentration is lower compared to OA. A small artifact chloride signal can be observed when high NO3 (or organic acid) mass concentrations are sampled into the AMS with the SV or CV, presumably due to the reaction of gas-phase HNO3 released from thermal decomposition with residual chloride on the vaporizer. This effect is similar to the recently reported enhancement of CO2+ signal when sampling nitrate into AMS due to particle–vaporizer interactions (Pieber et al. Citation2016). For the levels observed here, this effect will always be negligible and much smaller than the measurement uncertainties for AMS total mass. However, it could be a non-negligible and even dominant interference for the measured AMS chloride when the sampled air contains high nitrate and low chloride levels, as demonstrated with an NO3-OFR dataset in this study. However, less chloride enhancement (0.2% per nitrate mass) was observed in the CV than in the SV (0.5% per nitrate mass), so in this case the CV simplifies quantification. Further study of this artifact is recommended.
Size distributions can be measured with the CV-AMS, with similar results to the SV-AMS. However, it is important to calibrate the size measurement with monodisperse particles. Using (NH4)2SO4 for size calibrations when analyzing data from sulfate/organic dominated ambient particles is suggested. Further study of this mode and calibration with mixed particles is recommended.
UAST_1296104_Supplemental_File.zip
Download Zip (2 MB)Acknowledgments
We sincerely thank Hyungu Kang, Jason Schroder, Brett Palm, and Amber Ortega of CU-Boulder and Suzane S. de Sá of Harvard University for experimental support in specific datasets and Hongyu Guo and Rodney Weber from Georgia Tech for SOAS PILS-IC data support.
Funding
This study was partially supported by NSF AGS-1243354 and AGS-1360834, EPRI 10004734, NASA NNX15AT96G, DOE (BER/ASR/SBIR) DE-SC0011105, DE-SC0016559, and DE-SC0001673, and NOAA NA13OAR4310063.
Related Research Data
References
- Aiken, A. C., Decarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Sun, Y., Zhang, Q., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Onasch, T. B., Alfarra, M. R., Prevot, A. S. H., Dommen, J., Duplissy, J., Metzger, A., Baltensperger, U., and Jimenez, J. L. (2008). O/C and OM/OC Ratios of Primary, Secondary, and Ambient Organic Aerosols with High-Resolution Time-of-Flight Aerosol Mass Spectrometry. Environ. Sci. Technol., 42:4478–4485. DOI:10.1021/Es703009q.
- Allan, J. D., Alfarra, M. R., Bower, K. N., Williams, P. I., Gallagher, M. W., Jimenez, J. L., McDonald, A. G., Nemitz, E., Canagaratna, M. R., Jayne, J. T., Coe, H., and Worsnop, D. R. (2003a). Quantitative Sampling Using an Aerodyne Aerosol Mass Spectrometer - 2. Measurements of Fine Particulate Chemical Composition in two U.K. Cities. J. Geophys. Res.-Atmos., 108(D3):4091. DOI:10.1029/2002jd002359.
- Allan, J. D., Bower, K. N., Coe, H., Boudries, H., Jayne, J. T., Canagaratna, M. R., Millet, D. B., Goldstein, A. H., Quinn, P. K., Weber, R. J., and Worsnop, D. R. (2004a). Submicron Aerosol Composition at Trinidad Head, California, during ITCT 2K2: Its Relationship With Gas Phase Volatile Organic Carbon and Assessment of Instrument Performance. J. Geophys. Res.-Atmos., 109:D23S24. DOI:10.1029/2003jd004208.
- Allan, J. D., Delia, A. E., Coe, H., Bower, K. N., Alfarra, M. R., Jimenez, J. L., Middlebrook, A. M., Drewnick, F., Onasch, T. B., Canagaratna, M. R., Jayne, J. T., and Worsnop, D. R. (2004b). A Generalised Method for the Extraction of Chemically Resolved Mass Spectra From Aerodyne Aerosol Mass Spectrometer Data. J. Aerosol Sci., 35:909–922. DOI:10.1016/j.jaerosci.2004.02.007.
- Allan, J. D., Jimenez, J. L., Williams, P. I., Alfarra, M. R., Bower, K. N., Jayne, J. T., Coe, H., and Worsnop, D. R. (2003b). Quantitative Sampling Using an Aerodyne Aerosol Mass Spectrometer: 1. Techniques of Data Interpretation and Error Analysis. J. Geophys. Res.-Atmos., 108:4091. DOI:10.1029/2003jd001607.
- Ayres, B. R., Allen, H. M., Draper, D. C., Brown, S. S., Wild, R. J., Jimenez, J. L., Day, D. A., Campuzano-Jost, P., Hu, W., de Gouw, J., Koss, A., Cohen, R. C., Duffey, K. C., Romer, P., Baumann, K., Edgerton, E., Takahama, S., Thornton, J. A., Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Wennberg, P. O., Nguyen, T. B., Teng, A., Goldstein, A. H., Olson, K., and Fry, J. L. (2015). Organic Nitrate Aerosol Formation via NO3 + biogenic Volatile Organic Compounds in the Southeastern United States. Atmos. Chem. Phys., 15:13377–13392. DOI:10.5194/acp-15-13377-2015.
- Bahreini, R., Ervens, B., Middlebrook, A. M., Warneke, C., de Gouw, J. A., DeCarlo, P. F., Jimenez, J. L., Brock, C. A., Neuman, J. A., Ryerson, T. B., Stark, H., Atlas, E., Brioude, J., Fried, A., Holloway, J. S., Peischl, J., Richter, D., Walega, J., Weibring, P., Wollny, A. G., and Fehsenfeld, F. C. (2009). Organic Aerosol Formation in Urban and Industrial Plumes near Houston and Dallas, Texas. J. Geophys. Res.: Atmos., 114:D00F16. DOI:10.1029/2008jd011493.
- Berresheim, H., Tanner, D. J., and Eisele, F. L. (1993). Method for Real-Time Detection of Dimethyl Sulfone in Ambient Air. Anal. Chem., 65:3168–3170. DOI:10.1021/ac00069a037.
- Brown, S. S., and Stutz, J. (2012). Nighttime Radical Observations and Chemistry. Chem. Soc. Rev., 41:6405–6447.
- Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R. (2007). Chemical and Microphysical Characterization of Ambient Aerosols With the Aerodyne Aerosol Mass Spectrometer. Mass Spectrom. Rev., 26:185–222. DOI:10.1002/Mas.20115.
- Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R. (2015). Elemental Ratio Measurements of Organic Compounds Using Aerosol Mass Spectrometry: Characterization, Improved Calibration, and Implications. Atmos. Chem. Phys., 15:253–272. DOI:10.5194/acp-15-253-2015.
- Carlton, A. M., de Gouw, J., Jimenez, J. L., Ambrose, J. L., Brown, S., Baker, K. R., Brock, C. A., Cohen, R. C., Edgerton, S., Farkas, C., Farmer, D., Goldstein, A. H., Gratz, L., Guenther, A., Hunt, S., Jaeglé, L., Jaffe, D. A., Mak, J., McClure, C., Nenes, A., Nguyen, T. K. V., Pierce, J. R., Selin, N., Shah, V., Shaw, S., Shepson, P. B., Song, S., Stutz, J., Surratt, J., Turpin, B. J., Warneke, C., Washenfelder, R. A., Wennberg, P. O., X., Z. (2017). The Southeast Atmosphere Studies (SAS): Coordinated Investigation and Discovery to Answer Critical Questions About Fundamental Atmospheric Processes. Bull. Am. Met. Soc. In review.
- Crenn, V., Sciare, J., Croteau, P. L., Verlhac, S., Fröhlich, R., Belis, C. A., Aas, W., Äijälä, M., Alastuey, A., Artiñano, B., Baisnée, D., Bonnaire, N., Bressi, M., Canagaratna, M., Canonaco, F., Carbone, C., Cavalli, F., Coz, E., Cubison, M. J., Esser-Gietl, J. K., Green, D. C., Gros, V., Heikkinen, L., Herrmann, H., Lunder, C., Minguillón, M. C., Močnik, G., O'Dowd, C. D., Ovadnevaite, J., Petit, J. E., Petralia, E., Poulain, L., Priestman, M., Riffault, V., Ripoll, A., Sarda-Estève, R., Slowik, J. G., Setyan, A., Wiedensohler, A., Baltensperger, U., Prévôt, A. S. H., Jayne, J. T., and Favez, O. (2015). ACTRIS ACSM intercomparison – Part 1: Reproducibility of Concentration and Fragment Results from 13 Individual Quadrupole Aerosol Chemical Speciation Monitors (Q-ACSM) and Consistency with Co-Located Instruments. Atmos. Meas. Tech., 8:5063–5087. DOI:10.5194/amt-8-5063-2015.
- Day, D. A., Campuzano-Jost, P., Palm, B. B., Hu, W., Nault, B. A., Wooldridge, P. J., Cohen, R. C., Docherty, K. S., Huffman, J. A., and Jimenez, J. L. (in preparation). Evaluation of Methods for Quantification of Bulk Particle-Phase Organic Nitrates Using Real-Time Aerosol Mass Spectrometry.
- DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L. (2006). Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer. Anal. Chem., 78:8281–8289. DOI:10.1021/Ac061249n.
- DeCarlo, P. F., Dunlea, E. J., Kimmel, J. R., Aiken, A. C., Sueper, D., Crounse, J., Wennberg, P. O., Emmons, L., Shinozuka, Y., Clarke, A., Zhou, J., Tomlinson, J., Collins, D. R., Knapp, D., Weinheimer, A. J., Montzka, D. D., Campos, T., and Jimenez, J. L. (2008). Fast Airborne Aerosol Size and Chemistry Measurements Above Mexico City and Central Mexico During the MILAGRO Campaign. Atmos. Chem. Phys., 8:4027–4048.
- de Sá, S. S., Palm, B. B., Campuzano-Jost, P., Day, D. A., Hu, W., Newburn, M. K., Brito, J., Liu, Y., Isaacman-VanWertz, G., Yee, L. D., Goldstein, A. H., Artaxo, P., Souza, R., Manzi, A., Jimenez, J. L., Alexander, M. L., and Martin, S. T. (2016). Mass Spectral Observations of Fine Aerosol Particles and Production of SOM at an Anthropogenically Influenced Site During GoAmazon2014 Wet Season. Atmos. Chem. Phys. Discuss., 1–58. DOI:10.5194/acp-2016-1020
- Docherty, K. S., Jaoui, M., Corse, E., Jimenez, J. L., Offenberg, J. H., Lewandowski, M., and Kleindienst, T. E. (2012). Collection Efficiency of the Aerosol Mass Spectrometer for Chamber-Generated Secondary Organic Aerosols. Aerosol Sci. Tech., 47:294–309. DOI:10.1080/02786826.2012.752572.
- Drewnick, F., Diesch, J. M., Faber, P., and Borrmann, S. (2015). Aerosol Mass Spectrometry: Particle–Vaporizer Interactions and Their Consequences for the Measurements. Atmos. Meas. Tech., 8:3811–3830. DOI:10.5194/amt-8-3811-2015.
- Drewnick, F., Hings, S. S., DeCarlo, P., Jayne, J. T., Gonin, M., Fuhrer, K., Weimer, S., Jimenez, J. L., Demerjian, K. L., Borrmann, S., and Worsnop, D. R. (2005). A New Time-of-Flight Aerosol Mass Spectrometer (TOF-AMS) - Instrument Description and First Field Deployment. Aerosol Sci. Technol., 39:637–658. DOI:10.1080/02786820500182040.
- Eatough, D. J., and Hansen, L. D. (1984). Carbonaceous Particles in the Atmosphere 1983 Bis-hydroxymethyl Sulfone: A Major Aerosol Product of Atmospheric Reactions of SO2(g). Sci. Total Environ., 36:319–328.
- Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L. (2010). Response of an Aerosol Mass Spectrometer to Organonitrates and Organosulfates and Implications for Atmospheric Chemistry. Proc. Natl. Acad. Sci. USA, 107:6670–6675. DOI:10.1073/pnas.0912340107.
- Friedel, R. A., Shultz, J. L., and Sharkey, A. G. (1959). Mass Spectrum of Nitric Acid. Anal. Chem., 31:1128–1128. DOI:10.1021/ac60150a615.
- Fry, J. L., Draper, D. C., Zarzana, K. J., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Brown, S. S., Cohen, R. C., Kaser, L., Hansel, A., Cappellin, L., Karl, T., Hodzic Roux, A., Turnipseed, A., Cantrell, C., Lefer, B. L., and Grossberg, N. (2013). Observations of Gas- and Aerosol-Phase Organic Nitrates at BEACHON-RoMBAS 2011. Atmos. Chem. Phys., 13:8585–8605. DOI:10.5194/acp-13-8585-2013.
- Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Brauers, T., Brown, S. S., Dorn, H. P., Dubé, W. P., Fuchs, H., Mensah, A., Rohrer, F., Tillmann, R., Wahner, A., Wooldridge, P. J., and Cohen, R. C. (2011). SOA From Limonene: Role of NO3 in Its Generation and Degradation. Atmos. Chem. Phys., 11:3879–3894. DOI:10.5194/acp-11-3879-2011.
- Gilardoni, S., Massoli, P., Paglione, M., Giulianelli, L., Carbone, C., Rinaldi, M., Decesari, S., Sandrini, S., Costabile, F., Gobbi, G. P., Pietrogrande, M. C., Visentin, M., Scotto, F., Fuzzi, S., and Facchini, M. C. (2016). Direct Observation of Aqueous Secondary Organic Aerosol From Biomass-Burning Emissions. Proc. Natl. Acad. Sci., 113:10013–10018. DOI:10.1073/pnas.1602212113.
- Guo, H., Xu, L., Bougiatioti, A., Cerully, K. M., Capps, S. L., Hite Jr, J. R., Carlton, A. G., Lee, S. H., Bergin, M. H., Ng, N. L., Nenes, A., and Weber, R. J. (2015). Fine-Particle Water and pH in the Southeastern United States. Atmos. Chem. Phys., 15:5211–5228. DOI:10.5194/acp-15-5211-2015.
- Harkel, M. J. T. (1997). The Effects of Particle-size Distribution and Chloride Depletion of Sea-Salt Aerosols on Estimating Atmospheric Deposition at a Coastal Site. Atmos. Environ., 31:417–427.
- Haynes, W. (2015). CRC Handbook of Chemistry and Physics. 96th ed. CRC Press, Boca Raton, FL.
- Hettiyadura, A. P. S., Stone, E. A., Kundu, S., Baker, Z., Geddes, E., Richards, K., and Humphry, T. (2015). Determination of Atmospheric Organosulfates using HILIC Chromatography with MS Detection. Atmos. Meas. Tech., 8:2347–2358. DOI:10.5194/amt-8-2347-2015.
- Hogrefe, O., Drewnick, F., Lala, G. G., Schwab, J. J., and Demerjian, K. L. (2004). Development, Operation and Applications of an Aerosol Generation, Calibration and Research Facility. Aerosol Sci. Technol., 38:196–214. DOI:10.1080/02786820390229516.
- Hu, W., Palm, B. B., Day, D. A., Campuzano-Jost, P., Krechmer, J. E., Peng, Z., de Sá, S. S., Martin, S. T., Alexander, M. L., Baumann, K., Hacker, L., Kiendler-Scharr, A., Koss, A. R., de Gouw, J. A., Goldstein, A. H., Seco, R., Sjostedt, S. J., Park, J. H., Guenther, A. B., Kim, S., Canonaco, F., Prévôt, A. S. H., Brune, W. H., and Jimenez, J. L. (2016a). Volatility and Lifetime Against OH Heterogeneous Reaction of Ambient Isoprene-Epoxydiols-Derived Secondary Organic Aerosol (IEPOX-SOA). Atmos. Chem. Phys., 16:11563–11580. DOI:10.5194/acp-16-11563-2016.
- Hu, W. W., Campuzano-Jost, P., Day, D. A., Croteau, P. L., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L. (2016b). Evaluation of the New Capture Vaporizer for Aerosol Mass Spectrometers (AMS) Through Laboratory Studies of Inorganic Species. Atmos. Meas. Tech. Discuss., 2016:1–55. DOI:10.5194/amt-2016-337.
- Hu, W. W., Campuzano-Jost, P., Palm, B. B., Day, D. A., Ortega, A. M., Hayes, P. L., Krechmer, J. E., Chen, Q., Kuwata, M., Liu, Y. J., de Sá, S. S., McKinney, K., Martin, S. T., Hu, M., Budisulistiorini, S. H., Riva, M., Surratt, J. D., St. Clair, J. M., Isaacman-Van Wertz, G., Yee, L. D., Goldstein, A. H., Carbone, S., Brito, J., Artaxo, P., de Gouw, J. A., Koss, A., Wisthaler, A., Mikoviny, T., Karl, T., Kaser, L., Jud, W., Hansel, A., Docherty, K. S., Alexander, M. L., Robinson, N. H., Coe, H., Allan, J. D., Canagaratna, M. R., Paulot, F., and Jimenez, J. L. (2015). Characterization of a Real-time Tracer for Isoprene Epoxydiols-Derived Secondary Organic Aerosol (IEPOX-SOA) From Aerosol Mass Spectrometer Measurements. Atmos. Chem. Phys., 15:11807–11833. DOI:10.5194/acp-15-11807-2015.
- Huang, D. D., Li, Y. J., Lee, B. P., and Chan, C. K. (2015). Analysis of Organic Sulfur Compounds in Atmospheric Aerosols at the HKUST Supersite in Hong Kong Using HR-ToF-AMS. Environ. Sci. Technol., 49:3672–3679. DOI:10.1021/es5056269.
- Huffman, J. A., Jayne, J. T., Drewnick, F., Aiken, A. C., Onasch, T., Worsnop, D. R., and Jimenez, J. L. (2005). Design, Modeling, Optimization, and Experimental Tests of a Particle Beam Width Probe for the Aerodyne Aerosol Mass Spectrometer. Aerosol Sci. Technol., 39:1143–1163. DOI:10.1080/02786820500423782.
- Jayne, J. T., Leard, D. C., Zhang, X. F., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R. (2000). Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles. Aerosol Sci. Technol., 33:49–70.
- Jayne, J. T., and Worsnop, D. R. (2016). Particle Capture Device. US Patent. US 20150040689 A1, Aerodyne Research, Inc.
- Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R. (2009). Evolution of Organic Aerosols in the Atmosphere. Science, 326:1525–1529. DOI:10.1126/science.1180353.
- Jimenez, J. L., Canagaratna, M. R., Drewnick, F., Allan, J. D., Alfarra, M. R., Middlebrook, A. M., Slowik, J. G., Zhang, Q., Coe, H., Jayne, J. T., and Worsnop, D. R. (2016). Comment on “The Effects of Molecular Weight and Thermal Decomposition on the Sensitivity of a Thermal Desorption Aerosol Mass Spectrometer”. Aerosol Sci. Technol., 50:i–xv. DOI:10.1080/02786826.2016.1205728.
- Jimenez, J. L., Jayne, J. T., Shi, Q., Kolb, C. E., Worsnop, D. R., Yourshaw, I., Seinfeld, J. H., Flagan, R. C., Zhang, X. F., Smith, K. A., Morris, J. W., and Davidovits, P. (2003). Ambient Aerosol Sampling Using the Aerodyne Aerosol Mass Spectrometer. J. Geophys. Res.-Atmos., 108:8425. DOI:10.1029/2001jd001213.
- Kuwata, M., Zorn, S. R., and Martin, S. T. (2012). Using Elemental Ratios to Predict the Density of Organic Material Composed of Carbon, Hydrogen, and Oxygen. Environ. Sci. Technol., 46:787–794. DOI:10.1021/es202525q.
- Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A. (2016). Highly Functionalized Organic Nitrates in the Southeast United States: Contribution to Secondary Organic Aerosol and Reactive Nitrogen Budgets. Proc. Natl. Acad. Sci., 1516–1521. DOI:10.1073/pnas.1508108113.
- Liao, J., Froyd, K. D., Murphy, D. M., Keutsch, F. N., Yu, G., Wennberg, P. O., St. Clair, J. M., Crounse, J. D., Wisthaler, A., Mikoviny, T., Jimenez, J. L., Campuzano Jost, P., Day, D. A., Hu, W., Ryerson, T. B., Pollack, I. B., Peischl, J., Anderson, B. E., Ziemba, L. D., Blake, D. R., Meinardi, S., and Diskin, G. (2015). Airborne Measurements of Organosulfates Over the Continental US. J. Geophys. Res.: Atmos., 120:2990–3005. DOI:10.1002/2014jd022378.
- Linstrom, P. J., and Mallard, W. G. (2016). NIST Chemistry WebBook, NIST Standard Reference Database Number 69. Available at http://webbook.nist.gov/chemistry.
- Matthew, B. M., Middlebrook, A. M., and Onasch, T. B. (2008). Collection Efficiencies in an Aerodyne Aerosol Mass Spectrometer as a Function of Particle Phase for Laboratory Generated Aerosols. Aerosol Sci. Technol., 42:884–898. DOI:10.1080/02786820802356797.
- Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R. (2012). Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data. Aerosol Sci. Technol., 46:258–271. DOI:10.1080/02786826.2011.620041.
- Milic, A., Mallet, M. D., Cravigan, L. T., Alroe, J., Ristovski, Z. D., Selleck, P., Lawson, S. J., Ward, J., Desservettaz, M. J., Paton-Walsh, C., Williams, L. R., Keywood, M. D., and Miljevic, B. (2016). Aging of Aerosols Emitted from Biomass Burning in Northern Australia. Atmos. Chem. Phys. Discuss., 2016:1–24. DOI:10.5194/acp-2016-730.
- Ng, N. L., Jayne, J. T., Herndon, S. C., Trimborn, A., Canagaratna, M. R., Croteau, P. L., Onasch, T. B., Sueper, D., Worsnop, D. R., Zhang, Q., and Sun, Y. L. (2011). An Aerosol Chemical Speciation Monitor (ACSM) for Routine Monitoring of the Composition and Mass Concentrations of Ambient Aerosol. Aerosol Sci. Technol., 45:780–794. DOI:10.1080/02786826.2011.560211.
- Palm, B. B., Campuzano-Jost, P., Day, D. A., Ortega, A. M., Fry, J. L., Brown, S. S., Zarzana, K., Dube, W., Wagner, N., Draper, D., Kaser, L., Jud, W., Karl, T., Hansel, A., and Jimenez, J. L. (2017). Secondary Organic Aerosol Formation From in situ OH, O3, and NO3 Oxidation of Ambient Air in an Oxidation Flow Reactor. Atmos. Chem. Phys. Discuss., 2017:1–46. DOI:10.5194/acp-2016-1080.
- Peck, J., Gonzalez, L. A., Williams, L. R., Xu, W., Croteau, P. L., Timko, M. T., Jayne, J. T., Worsnop, D. R., Miake-Lye, R. C., and Smith, K. A. (2016). Development of an Aerosol Mass Spectrometer Lens System for PM2.5. Aerosol Sci. Technol., 50:781–789. DOI:10.1080/02786826.2016.1190444.
- Pieber, S. M., El Haddad, I., Slowik, J. G., Canagaratna, M. R., Jayne, J. T., Platt, S. M., Bozzetti, C., Daellenbach, K. R., Fröhlich, R., Vlachou, A., Klein, F., Dommen, J., Miljevic, B., Jiménez, J. L., Worsnop, D. R., Baltensperger, U., and Prévôt, A. S. H. (2016). Inorganic Salt Interference on CO2+ in Aerodyne AMS and ACSM Organic Aerosol Composition Studies. Environ. Sci. Technol., 50: 10494–10503. DOI:10.1021/acs.est.6b01035
- Quinn, P. K., Bates, T. S., Coffman, D., Onasch, T. B., Worsnop, D., Baynard, T., de Gouw, J. A., Goldan, P. D., Kuster, W. C., Williams, E., Roberts, J. M., Lerner, B., Stohl, A., Pettersson, A., and Lovejoy, E. R. (2006). Impacts of Sources and Aging on Submicrometer Aerosol Properties in the Marine Boundary Layer Across the Gulf of Maine. J. Geophys. Res.-Atmos., 111:D23S36. DOI:10.1029/2006JD007582.
- Riva, M., Budisulistiorini, S. H., Zhang, Z., Gold, A., and Surratt, J. D. (2016). Chemical Characterization of Secondary Organic Aerosol Constituents from Isoprene Ozonolysis in the Presence of Acidic Aerosol. Atmos. Environ., 130:5–13.
- Robinson, E. S., Onasch, T. B., Worsnop, D., and Donahue, N. M. (2016). Collection Efficiency of a-Pinene Secondary Organic Aerosol Particles Explored via Light Scattering Single Particle Aerosol Mass Spectrometry. Atmos. Meas. Tech. Discuss., 2016:1–29. DOI:10.5194/amt-2016-271.
- Rollins, A. W., Kiendler-Scharr, A., Fry, J. L., Brauers, T., Brown, S. S., Dorn, H. P., Dubé, W. P., Fuchs, H., Mensah, A., Mentel, T. F., Rohrer, F., Tillmann, R., Wegener, R., Wooldridge, P. J., and Cohen, R. C. (2009). Isoprene Oxidation by Nitrate Radical: Alkyl Nitrate and Secondary Organic Aerosol Yields. Atmos. Chem. Phys., 9:6685–6703. DOI:10.5194/acp-9-6685-2009.
- Seinfeld, J. H., and Pandis, S. N. (1998). Atmospheric Chemistry and Physics: From Air Pollution to Climate Change. John Wiley, New York.
- Xu, L., Suresh, S., Guo, H., Weber, R. J., and Ng, N. L. (2015). Aerosol Characterization Over the Southeastern United States Using High-Resolution Aerosol Mass Spectrometry: Spatial and Seasonal Variation of Aerosol Composition and Sources with a Focus on Organic Nitrates. Atmos. Chem. Phys., 15:7307–7336. DOI:10.5194/acp-15-7307-2015.
- Xu, W., Croteau, P., Williams, L., Canagaratna, M., Onasch, T., Cross, E., Zhang, X., Robinson, W., Worsnop, D., and Jayne, J. (2017). Laboratory Characterization of an Aerosol Chemical Speciation Monitor with PM2.5 Measurement Capability. Aerosol Sci. Technol., 51:69–83. 10.1080/02786826.2016.1241859.