
ABSTRACT
In the Nano Aerosol Mass Spectrometer (NAMS), particles are irradiated with a high energy laser pulse to produce a plasma that quantitatively disintegrates each particle into positively charged atomic ions. Previous work with this method used electrodynamic focusing and trapping of particles 30 nm dia. and below. In the current work, an aerodynamic focusing inlet was used to study particles between 40 and 150 nm dia. The distribution of atomic ion charge states was found to be particle size dependent, shifting toward lower charges with increasing size. This shift also affected the calibration by which elemental composition was determined from atomic ion signal intensities. Size independent calibration could be achieved by restricting the analysis to particles that gave more than 90% of the total signal intensity as multiply charged ions. This approach worked best for particles smaller than about 100 nm dia. since most spectra met this criterion. For the nanoparticles studied, the elemental mole fractions of Group I and II metals, halogens, and low atomic mass nonmetals could be determined within 10% or less of the expected value when the mole fraction was at the 1% level or greater. Some transition and heavy metals could not be quantified, while others could. Quantification appeared to be dependent on the ability of the element to be vaporized. Elements with high melting and boiling points gave particle mass spectra similar to those obtained by laser desorption ionization—mostly singly charged ions with relative intensities strongly biased toward atoms with low ionization energies.
Copyright © 2017 American Association for Aerosol Research
EDITOR:
Introduction
Atmospheric aerosols have a significant influence on health and climate but many questions remain regarding their chemical composition, source, fate, and the magnitude of their environmental impact (Pöschl Citation2005; Solomon et al. Citation2007). Ultrafine aerosols (defined as airborne particles <100 nm in diameter) represent a particular analytical challenge owing to their small mass and heterogeneous composition that can change quickly with time (Kumar et al. Citation2016; Bzdek and Johnston Citation2010). A variety of online and offline methods have been developed and deployed for ultrafine particle analysis (Bzdek and Johnston Citation2010; Bzdek et al. Citation2012a). One such method developed by our group is the Nano Aerosol Mass Spectrometer (NAMS) (Wang and Johnston Citation2006), which uses a laser induced plasma ionization (LIPI) source (Reents and Schabel Citation2001; Mahadevan et al. Citation2002) to perform quantitative elemental analysis of size-selectively trapped particles in the 10–30 nm size range.
Field measurements with NAMS show that ambient nanoparticles around 20 nm in diameter are composed mostly of carbon, nitrogen, oxygen, silicon and sulfur (Bzdek et al. Citation2012a; Zordan et al. Citation2008; Klems et al. Citation2010, Citation2011; Bzdek et al. Citation2011; Pennington et al. Citation2012; Klems et al. Citation2012; Bzdek et al. Citation2012b, Citation2013; Pennington et al. Citation2006). Notably, metals were not detected in these studies, even though metals have been reported in ambient measurements of particles 50 nm in diameter and greater using laser desorption ionization (LDI) (Reinard et al. Citation2007; Rhoads et al. Citation2003; Phares et al. Citation2003; Lake et al. Citation2003; Bein et al. Citation2005). LDI is very sensitive toward some metals with the ability for detection at the parts-per-thousand level or lower (Pekney et al. Citation2006). In contrast, LIPI is sensitive to a wide range of elements including metals (Lee et al. Citation2006; Zhou et al. Citation2006) but at higher concentrations (Klems and Johnston Citation2013). Therefore, it is plausible that the lack of metals in the NAMS measurements most likely reflects their absence from small, ambient nanoparticles above a level of about 1 mole percent.
Elemental analysis by LIPI on the NAMS platform has been studied with respect to ion formation mechanism (Klems and Johnston Citation2013) and calibration/quantification procedures and performance (Zordan et al. Citation2010). These studies necessarily have focused on particles 30 nm in diameter and below that consist of nonmetals. In the work reported here, these studies are expanded to include larger particle sizes and a wider range of elements found in bulk ambient particulate matter. To accomplish these goals, the NAMS particle inlet was reconstructed with an aerodynamic lens system that permitted larger size particles to be efficiently transmitted into the ion source region. Particle size and composition effects on the mass spectra are discussed with regard to the ion formation mechanism and the ability of LIPI to perform quantitative elemental analysis.
Experimental methods
shows a schematic of the NAMS modified for analysis of particles between 40 and 150 nm in diameter. Particles enter the mass spectrometer through an aerodynamic lens assembly (Liu et al. Citation1995a,b) and differential pumping stages that are mounted orthogonal to the time-of-flight mass analyzer. The aerodynamic lens focuses particles into a tight beam that is intercepted by a focused, pulsed laser beam inside the mass analyzer source region at an operating pressure of 10−5 Torr. This arrangement is similar to that used previously by our group to perform LDI of particles in the same size range (Kane et al. Citation2001). In these experiments, the lower particle size limit (40 nm) was determined by the focusing ability of the aerodynamic lens assembly. The upper size limit (150 nm) was determined by limitations associated with ion formation by LIPI, as will be discussed later.
Figure 1. NAMS platform for analyzing particles between 40 and 150 nm in diameter.
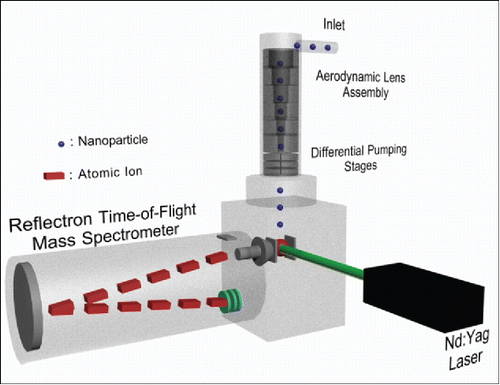
LIPI was performed with a Nd:YAG laser (Model CFR-400, Quantel, Bozeman, MT, USA) operating at 532 nm, 5 Hz repetition rate, and 230 mJ/pulse (9 ns nominal pulse width) focused to an effective spot size of about 0.1 mm dia. in the source region by a 71 mm focal length lens. When a particle was in the laser spot when the laser fired, a plasma was produced which vaporized the particle to give positively charged atomic ions. Ions were subsequently analyzed with a reflecting time-of-flight mass analyzer (Jordan TOF Products, Inc., Grass Valley, CA, USA). The resulting ion current vs. time profile was digitized, and the integrated signal intensities of individual mass-to-charge species were recorded. The relative signal intensity of each element was obtained by summing the intensities of all isotopic peaks present at various atomic charge states of that element (typically +1 to +4) and dividing by the total summed intensity of all elements. For isobaric ions (e.g., C+3 and O+4, O+2 and S+4), the signal intensities of the individual atomic species were deconvoluted using sucrose and HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) as calibrants (Zordan et al. Citation2010). Briefly, using the m/z 4 peak as an example, an average spectrum was obtained for the sucrose calibrant. The area ratio of the isobaric peak (C+3/O+4) to a neighboring charge state (O+3) was fit with an exponent that was iteratively varied until the total calculated elemental ratio matched the expected ratio (0.917 O/C ratio for sucrose). This fitted parameter was then applied to future spectra to determine the portions of the m/z 4 peak to assign to O+4 and C+3. A similar approach was then used with the HEPES calibrant in order to assign the m/z 8 peak to O+2 and S+4.
At least 100 particle spectra were recorded for each experiment, which was sufficient to assess the accuracy and precision of elemental analysis (Klems and Johnston Citation2013). In the text that follows, we discuss particle mass spectra in the context of single particles. In these experiments, the maximum hit rate was one particle spectrum per 100 laser shots, which corresponded to a probability that at most 1 in 100 particle mass spectra arose from two particles randomly in the laser beam at the same time.
Polydisperse test aerosols were produced with a commercial atomizer (TOPAS ATM226, Dresden, Germany) using solute concentrations of 5mM. Use of the same solute concentration for different test compounds caused the aerosol size distributions to be similar for different composition aerosols, which is an important feature of this work as will be discussed later. Depending on composition, the aerosol size distributions generated under these conditions exhibited mode diameters between about 40 and 70 nm and geometric standard deviations of 1.5–1.6, as measured with a Scanning Mobility Particle Sizer (SMPS, TSI Inc., Shoreview, MN, USA). Monodisperse aerosols were produced by sending the polydisperse aerosol through a differential mobility analyzer (TSI, Inc.).
Test compounds used in this study to generate aerosols were LiCl, NaCl, KCl, MgCl2, CaCl2, NaF, NaBr, CsI, Cr(NO3)3, Fe(NO3)3, AgNO3, Al(NO3)3, Hg(NO3)2, Cd(NO3)2, Pb(NO3)2, MnCl2, TiO2, ZnO, Na2HPO4, Na2SeO4, diamond nanoparticles, polydimethylsiloxane (PDMS), sucrose (C12H22O11) and HEPES (C8H18N2O4S). All compounds were dissolved or suspended in 18.2 MΩ, Milli-Q filtered water (Millipore Corporation, Billerica, MA, USA), with the exception of PDMS which was suspended in ethanol. For insoluble compounds, atomization produced aerosol with mobility diameters in the same size range as the soluble compounds. All chemicals were obtained from Sigma-Aldrich Co. (St. Louis, MO, USA) and were ACS grade when available. These test compounds spanned the range of elements commonly detected in ambient particles by LIPI or LDI.
Safety considerations
Aerosols, especially those containing heavy metals and/or manganese, are inhalation hazards. To mitigate the risk of exposure, the atomizer was placed in a fume hood and a single length of copper tubing was used to carry the aerosol to the instrumentation. Excess flow from the aerosol generator was split off and sent through a series of filter cartridges (McMaster-Carr) before being vented into the fume hood. The entire apparatus was checked for leaks prior to each experiment.
Results and discussion
Size dependence of single particle mass spectra
The LIPI mass spectrum of a polydisperse sucrose aerosol is shown in Figure S1. This spectrum is the average of 100 single particle mass spectra and because the aerosol analyzed was polydisperse (64 nm mode diameter), this average represents the contribution from many particle sizes. When compared to our previously published spectra of sucrose particles in the 10–20 nm size range (Zordan et al. Citation2008; Bzdek et al. Citation2011; Wang et al. Citation2006), the larger particles analyzed in Figure S1 gave a substantial number of singly charged atomic ions, whereas the smaller particles analyzed previously gave only multiply charged ions.
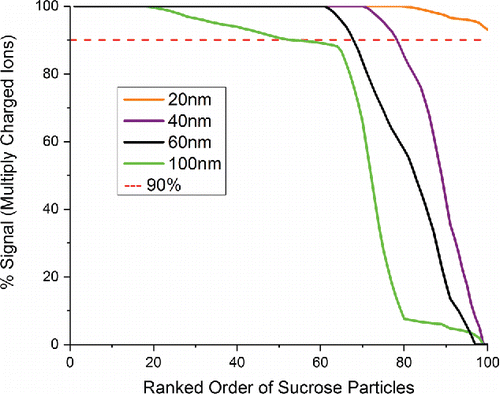
The difference in atomic charge state distribution between small and large particles was studied further by examining particle-to-particle variations of the LIPI mass spectra as a function of particle size. In this experiment, 100 single particle spectra were obtained for each of four monodisperse sucrose samples using a differential mobility analyzer to select particle diameters of 20, 40, 60, and 100 nm. The 20 nm dataset was obtained by sending particles through the original electrodynamic inlet, while the 40, 60 and 100 nm datasets were obtained by sending particles through the aerodynamic inlet. Although two different inlets were employed, the ion source configuration and voltages were the same for each dataset.
summarizes the charge state distributions on a particle-by-particle basis for the four particle sizes. This figure was constructed by calculating the percent of total signal intensity encompassed by multiply charged atomic ions for each particle and then plotting these data from highest percent intensity on the left to lowest percent intensity on the right. For the 20 nm dataset, most particles gave 100% of the signal intensity as multiply charged ions. Singly charged ions were observed in about 10% of the particle spectra, but their combined intensity was low - never exceeding 10% of the total intensity. As the particle size increased, a greater fraction of the particles gave singly charged ions and in some cases these ions dominated the total signal intensity. A striking feature of the plots in is the abrupt change in signal intensity. Most particles in each dataset had either >90% or <10% of the signal intensity as multiply charged ions. Very few had intermediate percentages. A similar dropoff is shown for HEPES in Figure S2.
Figure 2. Summary of sucrose charge state distributions in single particle mass spectra from four monodisperse aerosol datasets, 100 spectra each. For each mass spectrum, the percentage of total signal intensity attributed to multiply charged atomic ions was calculated, and the data were sorted from highest percentage on the left to lowest percentage on the right.
Previously, we reported and discussed systematic variations in the charge state distributions of 20 nm diameter particles composed of either HEPES or NaCl (Klems and Johnston Citation2013). In this work, it was found that the signal intensities of all +2 atomic ions in the spectra were positively correlated with each other, while at the same time they were anti-correlated with the signal intensities of +3 and +4 ions. These correlations were explained on the basis of plasma energetics. When a particle was irradiated, either a “hot” or “cold” plasma could be generated depending on variations of the laser pulse energy and particle location in the laser beam. Hotter plasmas are defined as those giving higher proportions of +3 and +4 ions which require a substantial amount of energy in order to be formed, while colder plasmas are defined as those giving higher proportions of +2 ions which require a smaller amount of energy in order to be formed. Similar charge state correlations are illustrated in Figure S3 for polydisperse sucrose aerosol. Particles showing a high signal intensity for C+1 also show a high intensity for O+1. Particles showing a high signal intensity for C+2 also show a high intensity for O+2. Particles showing a high signal intensity for C+3/4 also show a high signal intensity for O+3/4.
The particle size dependent data in can be interpreted in a similar way. Hotter plasmas give mostly multiply charged ions, while colder plasmas give mostly singly charged ions. Because it takes more energy to convert a large particle into atomic ions than a small particle, the fraction of particles exhibiting a cold plasma spectrum increases as the particle size increases. In fact, for particle sizes above about 150 nm, hardly any spectra at all are recorded with our apparatus that contain multiply charged atomic ions. As discussed below, the inability to produce multiply charged ions causes the effective upper particle size limit of NAMS for quantification to be in the 100–150 nm size range when quantification is based on hot spectra alone.
Quantification of elemental composition as a function of particle size
The particle size dependence of LIPI mass spectra has important consequences for quantification. The ability of LIPI to quantify elemental composition is based on the premise that complete ionization is achieved, in other words particles are quantitatively converted to atoms and the atoms are quantitatively ionized. In the complete ionization limit, the combined signal intensities of atomic ions of a given element relative to the total signal intensity from all ions gives a quantitative measure of the mole fraction of that element in the particle. However, this relationship can be disrupted if either (1) some of the particle is not completely vaporized to atoms or (2) some of the vaporized atoms are not ionized. For incomplete vaporization, a composition bias can arise if some components, e.g., refractory materials, prove harder to vaporize then others. For incomplete ionization, a composition bias can arise from collisions between neutral and ionic species in the plume, which favor charge migration to the lowest ionization energy species. In the discussion below, we refer to this phenomenon as quenching. Quenching is unlikely to be a major factor when multiply charged ions dominate the mass spectrum, since these species are so energetic that any collisions at all with un-ionized species will lead to their loss.
Quantitative elemental analysis as a function of particle size was examined using a 100-particle dataset from a polydisperse sucrose aerosol as the calibration aerosol and then applying this calibration to 100-particle datasets of the monodisperse aerosols. Calibration was performed in the manner reported previously (Wang and Johnston Citation2006; Zordan et al. Citation2010). As shows, when all 100 spectra in each of the datasets were used for calibration and analysis (hot and cold spectra), the measured oxygen-to-carbon (O/C) elemental ratio did not match the expected value. Additionally, the deviation between measured and expected values increased as the monodisperse particle diameter diverged from the mode diameter of the polydisperse calibration set (∼65 nm). In contrast, when only the hot spectra from the calibration and analysis datasets were used, correct O/C ratios were measured for all particle sizes. (In this experiment, hot was defined as multiply charged ions comprising ≥90% of the total signal intensity, while cold was defined as <90% of the total signal intensity.) The results in show that quenching, as defined by the predominance of singly charged atomic ions in the mass spectrum, can substantially affect quantitative measurements, but its impact can be minimized or eliminated by restricting measurements to hot spectra. It should be noted that the approach of using a hot subset of spectra for quantitative analysis has been reported previously (Reents and Schabel Citation2001), though that study used a different criterion where >10% of the total signal from a particle mass spectrum had to arise from multiply charged ions. The fact that size-independent calibration can be achieved by restricting the analysis to hot spectra suggests that quenching is the main cause of size-dependent changes in the mass spectra.
Table 1. Measured O/C ratios for monodisperse sucrose aerosols using two different quantification methods. Measurement uncertainties are reported as 1 standard deviation. (Expected O/C ratio is 0.917).
Calibration using both hot and cold spectra can be achieved if the calibration and sample aerosols have the same size distribution. This principle is suggested by , which shows closest agreement between measured and expected mole ratios when the diameter of the size-selected aerosol matches the mode diameter of the polydisperse aerosol. It is evaluated more rigorously in and , where the same atomizer with the same approximate solute concentration was used to generate different composition aerosols having similar size distributions. In this experiment, polydisperse aerosols of sucrose and HEPES were used for calibration as described previously (Zordan et al. Citation2010), and then applied to elemental analysis of polydisperse aerosols having of varying amounts of HEPES and cesium iodide.
Figure 3. Average mass spectrum of 100 particles from a polydisperse aerosol composed of a 2:1 molar ratio HEPES and cesium iodide. Measurement uncertainties are reported as 1 standard deviation. Atomic ions from the various elements are marked by the bars above the spectrum.
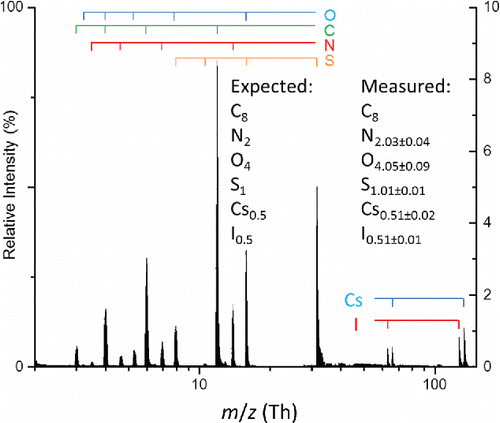
Table 2. Expected and measured elemental mole ratios from aerosols composed of HEPES and cesium iodide.
The LIPI mass spectrum from a 2:1 molar ratio of HEPES:CsI averaged over 100 single particles is shown in . Expected and measured elemental mole ratios for aerosols having this and other molar ratios of HEPES and cesium iodide are given in . It should be emphasized that all spectra in these datasets, whether hot or cold, were used for calibration and analysis, and that the size distributions of all aerosols matched each other closely. In general, the expected and measured elemental mole fractions in match within 10% of each other. The only exceptions are the Cs and I mole fractions for the 9.5:1 HEPES:CsI aerosol, where the expected and measured values deviate by slightly more than 10%, which is attributable to the relatively large standard deviations of this measurement. It should be noted that for this sample, the expected mole fractions for Cs and I are less than 0.7%.
Proceeding in a similar manner, quantitative measurements of elemental compositions were obtained for polydisperse aerosols containing a variety of combinations of HEPES and inorganic salts. These include Group 1 (Li, Na, K) and Group 2 (Mg, Ca) metals, several other low atomic weight metals (Al, Mn, Cu, Zn), halogens (F, Cl, Br), and low atomic weight nonmetals (C, N, O, Si, P, S, Se). A full list of compounds examined along with their calculated relative elemental composition is available in Table S1.
Problem elements for quantification
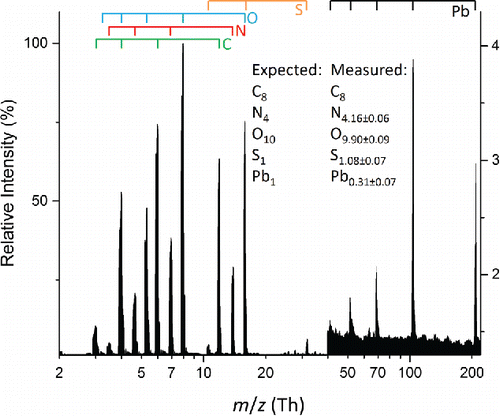
Several elements proved difficult to measure accurately and can be divided into two separate groups. The first includes silver, cadmium, mercury, and lead which all showed decreased sensitivity relative to other elements in the particle. An example is shown in for a polydisperse aerosol consisting of an equimolar mixture of lead nitrate and HEPES. The expected and measured elemental mole ratios match for C, N, O and S but not Pb. The agreement between expected and measured values is especially notable for nitrogen and oxygen which have contributions from multiple chemical species. Lead, on the other hand, is underrepresented in the spectrum, with a measured mole fraction a little more than half of the expected mole fraction. Underrepresentation of lead by the same approximate amount is also observed for an aerosol consisting of lead nitrate alone (Figure S4).
Figure 4. Average mass spectrum of 100 particles from a polydisperse aerosol composed of a 1:1 molar ratio lead(II) nitrate and HEPES. Measurement uncertainties are reported as 1 standard deviation. Atomic ions from the various elements are marked by the bars above the spectrum.
A similar underrepresentation was observed for polydisperse aerosols containing silver, cadmium and mercury both for the salts alone and in mixtures with HEPES (Table S2). While low atomic weight nonmetals were quantified properly (whether from a single or multiple chemical species, e.g., nitrate and HEPES), the metals were systematically underrepresented by a factor of 3 to 15 depending upon the specific aerosol examined.
Quenching does not seem to be the reason for underrepresentation of these elements, since the proportion of singly vs. multiply charged ions in the mass spectra were no different than in spectra from the HEPES-CsI aerosols discussed previously where accurate analysis was achieved. A different possibility to consider is a substantial m/z dependence on ion transmission in the time-of-flight mass analyzer. However, a systematic ion transmission dependence is inconsistent with the ability to quantify Cs and I but not Ag, Cd, Hg and Pb. Inspection of shows that the magnitude of the underrepresentation is element specific rather than particle composition specific. In other words, the magnitude of the underrepresentation for a given element is approximately the same for both metal nitrate and HEPES-metal nitrate composition particles. This behavior suggests that underrepresentation arises from incomplete vaporization of the element, which would result in a lower than expected atomic ion signal intensity independent of the other elements present in the particle.
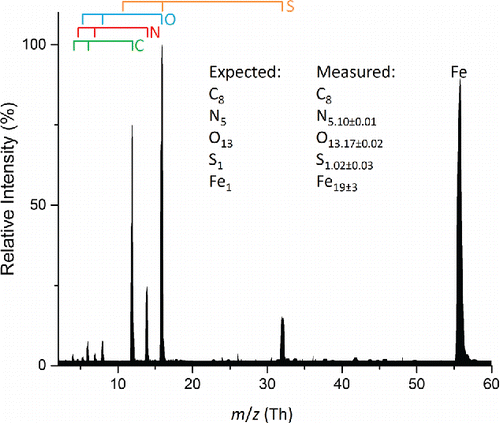
The second group of problem elements includes titanium, chromium, and iron. An example is shown in for a polydisperse aerosol consisting of an equimolar mixture of iron(III) nitrate and HEPES. Relative to and , the charge state distributions in are shifted strongly toward singly charged ions and the measured Fe mole fraction is overrepresented by more than an order of magnitude. Interestingly, the measured mole fractions of other elements (C, N, O, S) match the expected values, and two of these (N, O) have sources from multiple chemical species. The mass spectrum of polydisperse aerosol composed of iron(III) nitrate is similar – overrepresentation of iron in this case by a factor of 100, while the measured mole fractions of nitrogen and oxygen match the expected values (Figure S5). Mass spectra of polydisperse aerosols composed of chromium(III) nitrate and titanium dioxide showed similar features: signal intensity dominated by singly charged ions with the metal in each case overrepresented relative to the other elements present (Table S2).
Figure 5. Average mass spectrum of 100 particles from a polydisperse aerosol composed of a 1:1 molar ratio iron(III) nitrate and HEPES. Measurement uncertainties are reported as 1 standard deviation. Atomic ions from the various elements are marked by the bars above the spectrum.
Zhou et al (Zhou et al. Citation2007) have suggested that plasma formation by LIPI is a two-step process. First, the particle begins to heat and neutrals start to evaporate. Second, the energy absorbed by the particle reaches its cohesive energy threshold and a plasma is formed. Using this model as a guide, the behavior of problem elements can be understood. Particles containing the underrepresented metals (Ag, Cd, Hg, Pb) reach the cohesive energy threshold, forming a plasma that generates multiply charged ions. However, only a portion of the underrepresented metal is vaporized, so its ion signal intensity is lower than expected. Particles containing overrepresented metals (Ti, Fe, Cr) do not reach the cohesive energy threshold, so only a small portion of each particle is vaporized. The overrepresentation of these metals and the lack of multiply charged ions are the consequence of ion-neutral collisions in the plume, which favors charge migration to the lowest ionization energy atoms, which happen to be these elements.
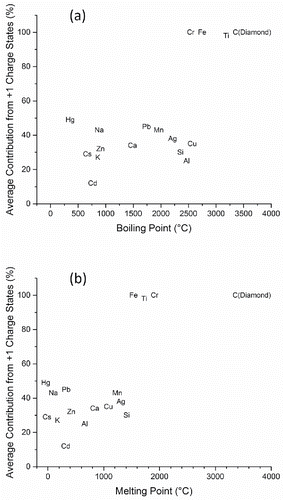
Using melting and boiling points (Lide Citation1991) as proxies for the cohesive energy threshold, the correlation between detection of singly charged ions and energy required for particle vaporization is illustrated in . This figure indicates that charge state distributions in the mass spectra correlate strongly with both melting and boiling points. Elements that induce nearly 100% of the signal intensity in singly charged states (Ti, Fe, Cr) uniformly have the highest melting and boiling points. Notably, manganese which has much lower melting and boiling points than the two neighboring elements on the Periodic Table (Cr and Fe), also has mass spectra dominated by multiply charged ions. To explore this correlation more thoroughly, aerosols generated from carbon in the form of diamond nanoparticles (melting point 3547°C) were generated and the mass spectra were compared with that from sucrose. While sucrose aerosol gave mostly multiply charged atomic ions, diamond gave only C+1.
Figure 6. Plot of percentage of total signal intensity attributed to singly charged ions in the average mass spectrum of aerosol containing a specific element vs. (a) boiling point and (b) melting point of that element.
Problem elements for quantification, whether under- or over- represented, can be traced to the difficulty of completely vaporizing a particle as opposed to other instrumental influences. To confirm that key instrumental parameters are not the source of over/under representation, we performed additional experiments to elucidate the effects of gas pressure and electric field on ion formation. For these experiments we chose two particle compositions, one exhibiting underrepresentation of the metal (lead nitrate) and the other overrepresentation of the metal (iron nitrate). First, the background pressure in the ion source was varied between 10−5 and 10−4 torr by adding argon. (Higher pressures could not be examined since the background pressure in the mass analyzer and detector regions became too large.) No effect on the mass spectra was observed, which is not surprising since few collisions would be expected. Furthermore, Ar+ was not observed in the mass spectra suggesting that it did not cause quenching. Second, the electric field gradient in the source region was varied. Again, no effect was observed other than the loss of mass resolving power and a reduction of the total signal intensity. These results confirm that quantification problems arise primarily from laser-particle interaction rather than ion source characteristics.
Conclusions
In this study, we have expanded use of the Nano Aerosol Mass Spectrometer (NAMS) to larger size particles and a wider range of particle compositions. Quantitative elemental analysis by NAMS is based on use of a laser induced plasma ionization (LIPI) source to achieve the “complete ionization limit,” meaning that the laser pulse induces formation of a plasma that quantitatively converts the particle into singly and multiply charged atomic ions. Achieving the complete ionization limit creates challenges for analyzing a wide range of particle sizes and compositions. With respect to particle size, a systematic change was observed in which smaller particles tended to produce hotter plasmas, i.e., mass spectra dominated by multiply charged atomic ions, while larger particles tended to produce colder plasmas, i.e., mass spectra dominated by singly charged atomic ions. Since quantification is dependent on the distribution of charge states produced, calibration procedures must take into account this particle size dependence. Two approaches were examined. First, the calibration and analytical datasets were restricted to hot mass spectra only, which had the advantage of creating a particle size-independent calibration, but also the disadvantage of excluding many mass spectra especially for particles above 100 nm. Second, the size distributions of the calibration and analytical datasets were matched, which had the advantage of using all particle mass spectra in the analysis but the disadvantage that changes in the particle size distribution during an experiment could lead to an incorrect calibration.
The experimental arrangement in used to analyze large (40–150 nm) particle diameters relies on the random probability that a particle passing through the source region at a high speed is in the laser beam when the laser fires. The probability that a particle is intercepted by the laser pulse is much lower than in the original NAMS configuration for particles in the 10–30 nm size range that are electrodynamically trapped inside a small volume. As a result, the configuration for analyzing 40–150 nm diameter particles is 10–100 times (depending on particle size and the calibration method used) less efficient for a given particle number concentration than the configuration for analyzing 10–30 nm diameter particles. For this reason, the large particle configuration for NAMS is best suited for laboratory experiments where particle concentrations are high and/or long analysis times (time required to obtain a statistically significant number of particle spectra) are possible. This configuration is less useful for ambient measurements if short time resolution is required. Future work would benefit from developing improved ways to efficiently deliver ultrafine particles over a broad size range into the laser beam.
This study also considered a wider range of particle compositions and found that some elements could be quantitatively determined by LIPI but other elements could not. Incomplete vaporization of an element can lead to its systematic underrepresentation in the mass spectra. In extreme cases, vaporization is so incomplete that the plume becomes dominated by ion-neutral collisions. These collisions quench formation of multiply charged ions and cause elements having low ionization energies to be overrepresented in the mass spectra.
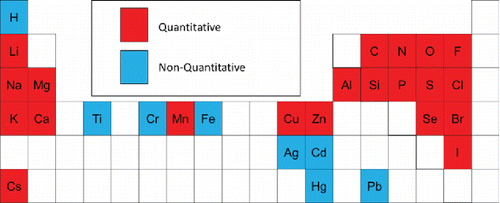
Elements examined for analysis by LIPI are summarized in . Note that hydrogen was not included in this study, but has been discussed previously (Wang et al. Citation2006). All elements studied to date can be detected at the 1 mole percent level or greater and most can be quantified. However, as discussed above, some transition and heavy metals can be detected at this level but not quantified. In general, LIPI quantitatively measures the relative elemental composition of nanoparticles containing Group I and II metals, halogens, and low atomic mass nonmetals. Detection at the 1 mole percent level or greater means that trace analysis at, e.g., the part-per-thousand level or less is not possible with LIPI, though other methods such as LDI may be applicable.
Figure 7. Elements for which quantitative analysis is/is not possible by LIPI under the conditions used in this study.
While the particles studied in this work focused mainly on atmospherically relevant elements, future experiments with additional particle compositions may provide further insight into the ionization mechanism. The ability to detect and/or quantify most elements relevant to ambient nanoparticles makes NAMS in general and LIPI in particular a valuable tool for studying aerosol processes.
UAST_1335390_Supplementary_File.zip
Download Zip (222.4 KB)Acknowledgments
The authors thank Svilen Bobev for helpful discussions concerning cohesive energy thresholds.
Funding
This research was supported by National Science Foundation grant number CHE 1408455.
Related Research Data
References
- Bein, K. J., Zhao, Y., Wexler, A. S., and Johnston, M. V. (2005). Speciation of Size-Resolved Individual Ultrafine Particles in Pittsburgh, Pennsylvania. J. Geophys. Res.: Atmospheres, 110:D07S05.
- Bzdek, B. R., Horan, A. J., Pennington, M. R., DePalma, J. W., Zhao, J., Jen, C. N., Hanson, D. R., Smith, J. N., McMurry, P. H., and Johnston, M. V. (2013). Quantitative and Time-Resolved Nanoparticle Composition Measurements During New Particle Formation. Faraday Discuss., 165:25–43.
- Bzdek, B. R., and Johnston, M. V. (2010). New Particle Formation and Growth in the Troposphere. Anal. Chem., 82:7871–7878.
- Bzdek, B. R., Pennington, M. R., and Johnston, M. V. (2012a). Single Particle Chemical Analysis of Ambient Ultrafine Aerosol: A Review. J. Aerosol Sci., 52:109–120.
- Bzdek, B. R., Zordan, C. A., Luther, G. W., and Johnston, M. V. (2011). Nanoparticle Chemical Composition During New Particle Formation. Aerosol Sci. Technol., 45:1041–1048.
- Bzdek, B. R., Zordan, C. A., Pennington, M. R., Luther, G. W., and Johnston, M. V. (2012b). Quantitative Assessment of the Sulfuric Acid Contribution to New Particle Growth. Environ. Sci. Technol.,46:4365–4373.
- Kane, D. B., Oktem, B., and Johnston, M. V. (2001). Nanoparticle Detection by Aerosol Mass Spectrometry. Aerosol Sci. Technol., 34:520–527.
- Klems, J., and Johnston, M. (2013). Origin and Impact of Particle-to-Particle Variations in Composition Measurements with the Nano-Aerosol Mass Spectrometer. Anal. Bioanalytical Chem., 405:6995–7003.
- Klems, J. P., Pennington, M. R., Zordan, C. A., and Johnston, M. V. (2010). Ultrafine Particles Near a Roadway Intersection: Origin and Apportionment of Fast Changes in Concentration. Environ. Sci. Technol., 44:7903–7907.
- Klems, J. P., Pennington, M. R., Zordan, C. A., McFadden, L., and Johnston, M. V. (2011). Apportionment of Motor Vehicle Emissions from Fast Changes in Number Concentration and Chemical Composition of Ultrafine Particles Near a Roadway Intersection. Environ. Sci. Technol., 45:5637–5643.
- Klems, J. P., Zordan, C. A., Pennington, M. R., and Johnston, M. V. (2012). Chemical Composition of Ambient Nanoparticles on a Particle-by-Particle Basis. Anal. Chem., 84:2253–2259.
- Kumar, P., Wiedensohler, A., Birmili, W., Quincey, P., and Hallquist, M. (2016). Ultrafine Particles Pollution and Measurements, in Comprehensive Analytical Chemistry, Elsevier, Cambridge, MA, pp. 369–390.
- Lake, D. A., Tolocka, M. P., Johnston, M. V., and Wexler, A. S. (2003). Mass Spectrometry of Individual Particles between 50 and 750 nm in Diameter at the Baltimore Supersite. Environ. Sci. Technol., 37:3268–3274.
- Lee, D., Miller, A., Kittelson, D., and Zachariah, M. R. (2006). Characterization of metal-bearing diesel nanoparticles using single-particle mass spectrometry. J. Aerosol Sci., 37:88–110.
- Lide, D. R. (1991). CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL.
- Liu, P., Ziemann, P. J., Kittelson, D. B., and McMurry, P. H. (1995a). Generating Particle Beams of Controlled Dimensions and Divergence 1. Theory of Particle Motion in Aerodynamic Lenses and Nozzle Expansions. Aerosol Sci. Technol., 22:293–313.
- Liu, P., Ziemann, P. J., Kittelson, D. B., and McMurry, P. H. (1995b). Generating Particle Beams of Controlled Dimensions and Divergence 2. Experimental Evaluation of Particle Motion in Aerodynamic Lenses and Nozzle Expansions. Aerosol Sci. Technol., 22:314–324.
- Mahadevan, R., Lee, D., Sakurai, H., and Zachariah, M. R. (2002). Measurement of Condensed-Phase Reaction Kinetics in the Aerosol Phase Using Single Particle Mass Spectrometry. J. Phys. Chem. A, 106:11083–11092.
- Pekney, N. J., Davidson, C. I., Bein, K. J., Wexler, A. S., and Johnston, M. V. (2006). Identification of Sources of Atmospheric PM at the Pittsburgh Supersite, Part I: Single Particle Analysis and Filter-Based Positive Matrix Factorization. Atmos. Environ., 40, Supplement 2:411–423.
- Pennington, M. R., Bzdek, B. R., DePalma, J. W., Smith, J. N., Kortelainen, A. M., Hildebrandt Ruiz, L., Petäjä, T., Kulmala, M., Worsnop, D. R., and Johnston, M. V. (2006). Identification and Quantification of Particle Growth Channels During New Particle Formation. Atmospheric Chem. Phys., 13:10215–10225.
- Pennington, M. R., Klems, J. P., Bzdek, B. R., and Johnston, M. V. (2012). Nanoparticle Chemical Composition and Diurnal Dependence at the CalNex Los Angeles Ground Site. J. Geophys. Res.: Atmospheres, 117:D00V10.
- Phares, D. J., Rhoads, K. P., Johnston, M. V., and Wexler, A. S. (2003). Size-Resolved Ultrafine Particle Composition Analysis 2. Houston. J. Geophys. Res.: Atmospheres, 108:8420.
- Pöschl, U. (2005). Atmospheric Aerosols: Composition, Transformation, Climate and Health Effects. Angew. Chem.-Int. Ed., 44:7520–7540.
- Reents, W. D., and Schabel, M. J. (2001). Measurement of Individual Particle Atomic Composition by Aerosol Mass Spectrometry. Anal. Chem., 73:5403–5414.
- Reinard, M. S., Adou, K., Martini, J. M., and Johnston, M. V. (2007). Source Characterization and Identification by Real-Time Single Particle Mass Spectrometry. Atmos. Environ., 41:9397–9409.
- Rhoads, K. P., Phares, D. J., Wexler, A. S., and Johnston, M. V. (2003). Size-Resolved Ultrafine Particle Composition Analysis 1. Atlanta. J. Geophys. Res.: Atmospheres, 108:8418.
- Solomon, S. Q. D., Manning, M., Chen, Z., Marquis, M., Averyt, K. B., Tignor, M., Miller, H. L. (2007). Climate Change 2007: The Physical Science Basis Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. Intergovernmental Panel on Climate Change:996.
- Wang, S., and Johnston, M. V. (2006). Airborne Nanoparticle Characterization with a Digital Ion Trap–Reflectron Time of Flight Mass Spectrometer. Int. J. Mass Spectrom., 258:50–57.
- Wang, S., Zordan, C. A., and Johnston, M. V. (2006). Chemical Characterization of Individual, Airborne Sub-10-nm Particles and Molecules. Anal. Chem., 78:1750–1754.
- Zhou, L., Park, K., Milchberg, H. M., and Zachariah, M. R. (2007). Understanding the Interaction of an Intense Laser Pulse with Nanoparticles: Application to the Quantification of Single Particle Mass Spectrometry. Aerosol Sci. Technol., 41:818–827.
- Zhou, L., Rai, A., and Zachariah, M. R. (2006). Component and Morphology Biases on Quantifying the Composition of Nanoparticles Using Single-Particle Mass Spectrometry. Int. J. Mass Spectrom., 258:104–112.
- Zordan, C. A., Pennington, M. R., and Johnston, M. V. (2010). Elemental Composition of Nanoparticles with the Nano Aerosol Mass Spectrometer. Anal. Chem., 82:8034–8038.
- Zordan, C. A., Wang, S., and Johnston, M. V. (2008). Time-Resolved Chemical Composition of Individual Nanoparticles in Urban Air. Environ. Sci. Technol., 42:6631–6636.