
ABSTRACT
Particulate matter was sampled in Northern France during two summer and winter periods at both an urban background site (Douai, DO) and an industrialized coastal site (Grande-Synthe, GS). Ambient levels of particulate carbonaceous species and Polycyclic Aromatic Hydrocarbons (PAH) were measured by real-time measurements and via collection and analysis of offline filters (F). The comparison between online organic matter (OM) measured by an Aerosol Mass Spectrometer (AMS) and organic carbon (OC) determined by an offline thermal-optical method showed good linear trends in wintertime GS (r2 = 0.82 while only 0.50 in summer), and DO (r2 = 0.86 in summer and 0.92 in winter). However, significant differences were observed between analytical methods and sites with OCAMS/OCF ratios decreasing from 0.80 in DO during winter to ≈0.20 for GS in summer, suggesting that a large part of OM could be in the PM1–PM2.5 fraction. The simultaneous measurements of Black Carbon (BC) and Elemental Carbon (EC) concentrations in PM2.5 were also well correlated at both sites with r2 = 0.61–0.97 and slopes between 0.6 and 0.8. PAHs were analyzed in PM2.5 and also measured online by AMS in PM1. Their wintertime concentrations were highly correlated in DO (r2 = 0.98) and to a lesser degree in GS (r2 = 0.67). r2 values determined for comparison between online and offline parameters (OC and PAHs) in GS were lower than in DO, probably due to a more complex aerosol composition and a higher variability of the physical and chemical properties resulting from the coastal situation and diversity of emission sources in the vicinity of GS.
Copyright © 2018 American Association for Aerosol Research
EDITOR:
1. Introduction
The size and chemical composition of atmospheric particles lead to harmful effects on human health and impact both global climate and ecosystems (Kelly and Fussell Citation2012; IPCC and Stocker Citation2014). Atmospheric particulate matter (PM2.5) could be responsible for up to a 9.3-month decrease of lifetime expectancy in France (Amann et al. Citation2005). The determination of their chemical composition is in particular crucial to better understand their origins, their formation mechanisms, and the processes that can occur at their surface.
Yet, the complexity of atmospheric PM prevents the characterization of their entire chemical composition by a single technique. Offline methods are often subject to sampling artifacts, partly due to the presence of a large number of semi-volatile organic compounds (SVOCs) mixed with atmospheric oxidants (McMurry Citation2000) and their poor time resolution (several hours or days). Those methods are also labor intensive and time consuming due to sample treatment (collection, extraction) and analysis. Moreover, the chemical speciation of organic aerosol (OA) is limited by the wide variety of organic compounds presenting different properties (oxidation state, volatility, hygroscopicity). Gas or liquid chromatographs coupled to mass spectrometers are classically used to identify OA at a molecular scale (Mirivel et al. Citation2009, Citation2010, Citation2011; Pratt and Prather Citation2011). Those techniques have indeed improved the characterization of organic aerosol (OA) chemical composition with the identification of a few thousand organic compounds; however, a large part of OA still remains unknown (Goldstein and Galbally Citation2007). In recent years, real-time measuring instruments such as aerosol mass spectrometers (abbreviated “AMS” hereafter) have been developed leading to a global analysis which covers the entire mass of OA but without the possibility of chemical speciation (DeCarlo et al. Citation2006; Canagaratna et al. Citation2007). Among online techniques, highly time-resolved AMS with Quadrupole (Q-AMS) or Time-of-Flight (ToF-AMS and HR-ToF-AMS) analyzers (Canagaratna et al. Citation2007) provided new insights on the identification of emission sources and chemical transformations of atmospheric particles sampled throughout the world (Zhang et al. Citation2007; Jimenez et al. Citation2009; Riffault et al. Citation2015). Online mass spectrometry techniques used in aerosol applications have also been reported recently in several reviews (Aubriet and Carré Citation2010; Pratt and Prather Citation2012) highlighting the significant advances to better understand aerosol formation and aging and their chemical composition thanks to the higher sensitivity and fast response time of those techniques.
Another global analysis of OA consists of different thermal, optical, and thermal/optical carbon methods to determine the concentrations of Organic Carbon (OC), Elemental Carbon (EC), or Black Carbon (BC). For the organic fraction, one of the critical parameters is to estimate the ratio to convert the organic carbon mass (expressed in μgC m−3) into organic matter mass (μg m−3), which is essential for mass closure studies (Turpin and Lim Citation2001; Chan et al. Citation2010). BC concentrations are derived from absorption coefficient measurements at one or more wavelengths whereas EC is associated with thermal-optical analysis (Petzold et al. Citation2013). It must be noted that there is no reference method for either method. Usually, the link between BC mass concentrations, optical attenuations, and light absorption coefficients is determined from instrument inter-comparisons (Bae et al. Citation2007; Slowik et al. Citation2007; Snyder and Schauer Citation2007; Müller et al. Citation2011). However, the use of different methods leads to a large degree of uncertainty. Moreover, sampling artifacts can also affect attenuation measurements. This is why algorithms and empirical expressions have been developed to correct for the multiple scattering from the filter fibers and the collected aerosols, and the shadowing effect due to mass accumulation of impacted particles (Weingartner et al. Citation2003; Arnott et al. Citation2005; Schmid et al. Citation2006; Virkkula et al. Citation2007; Collaud Coen et al. Citation2010). It has been demonstrated that the uncorrected BC concentrations measured by aethalometers lead to an overestimation of absorption coefficients (Collaud Coen et al. Citation2010).
Like BC, PAHs are emitted from the incomplete combustion or pyrolysis of organic materials, mainly originating from several anthropogenic activities, such as traffic, industries, vehicle exhaust, coal, and wood combustion (Sharma et al. Citation2007; Ravindra et al. Citation2008). PAHs are ubiquitous pollutants present in both the gas and particulate phases in the atmosphere, and are particularly monitored due to their known toxic, carcinogenic, and/or mutagenic properties on human health (Billet et al. Citation2008). Their moderate vapor pressure favors their adsorption and condensation onto the surface of pre-existing particles to form an organic coating (Moosmüller et al. Citation2009). Many analytical methods have been developed for the quantitative detection of particle-phase PAHs (Liu et al. Citation2007) with chromatographic techniques being the most widespread ones for PAH collected on filters (Ma et al. Citation2010; Singh et al. Citation2011). A few real or near-real time analytical techniques such as AMS (Dzepina et al. Citation2007; Poulain et al. Citation2011), single-particle laser desorption/ionization (LDI) coupled with time-of-flight mass spectrometry (Zimmermann et al. Citation2003; Sodeman et al. Citation2005), or two-step laser mass spectrometry analysis (L2MS) with impaction sampling (Kalberer et al. Citation2004) have also been developed for PAH measurements.
In this work, we carried out an extensive comparison of carbonaceous species during two summer and two winter field campaigns at an urban background site and an industrialized coastal site located in Northern France. The results obtained from real-time measurements with an HR-ToF-AMS and offline analyses from 24 h filter samples were compared in an attempt to highlight the correlations or discrepancies between analytical methods. For the last decades, many studies in the literature have reported results obtained with one of those methods but to the best of our knowledge, the comparison of these techniques with very different time resolutions had not yet been carried out in a single study for the aforementioned chemical families associated with fine particulate matter, and for such a dataset obtained during two seasons and for two site typologies.
2. Methodology
2.1. Field campaign description
Atmospheric aerosols were sampled at two sites located in Northern France. One was an industrialized coastal site in Grande-Synthe (abbreviated “GS” hereafter) (51°00′N, 02°18′E and 9 m above sea level), located 6 km southwest of Dunkirk. This densely populated area is highly impacted by several major industries. Previous field experiments performed at this location indicate that GS is potentially subject to the influence of industrial pollutant emissions such as Volatile Organic Compounds (VOCs; Badol et al. Citation2008; Roukos et al. Citation2009, Citation2011; Xiang et al. Citation2012), heavy metals (Flament et al. Citation2008; Alleman et al. Citation2010; Choël et al. Citation2010; Marris et al. Citation2012), or PM (Rimetz-Planchon et al. Citation2008; Cazier et al. Citation2011). The other site was located near the city center of Douai (abbreviated “DO” hereafter) (50°22′N, 03°04′E and 24 m above sea level), close to the Scarpe River channel (about 50 m), and surrounded almost exclusively with residential areas and traffic roads, representing a typical urban background site. Previous studies from our group in DO have determined the chemical composition of toxic organic compounds like PAHs and their oxygenated and nitrated derivatives (O-PAHs and N-PAHs, respectively) as well as long-chain mono- and dicarboxylic acids, which have been quantified following PM2.5 and PM10 filter sampling (Mirivel et al. Citation2009, Citation2010, Citation2011) over short field campaigns.
The exact sampling periods in DO were 18 November to 10 December 2010 and 7 July to 8 August 2011 for the winter and summer field campaigns, respectively; and 30 May to 20 June 2011 (summer) and 28 January to 14 February 2012 (winter) for GS. The meteorological conditions encountered during the field campaigns demonstrated that the sampling periods are representative of warm and cold seasons (average temperatures of 1.9 ± 3.6 and 18.1 ± 3.4°C, respectively). Similar instrumentation was deployed during all field campaigns, except the Aethalometer which was not available for the winter field campaign in DO. The concentrations of particulate and gaseous species measured at both sites during the field campaigns have already been described in a companion paper (Crenn et al. Citation2017).
2.2. Offline analyses
In this study, 10 PAHs, i.e., two C16 (fluoranthene, FLA; pyrene, PYR), two C18 (benzo(a)anthracene, BaA; chrysene, CHR), three C20 (benzo(b)fluoranthene, BbF; benzo(k)fluoranthene, BkF; benzo(a)pyrene, BaP), and three C22 (dibenzo(a,h)anthracene, DahA; benzo(g,h,i)perylene, BghiP; and indeno(1,2,3-c,d)pyrene, IP), present in the particulate phase (Tasdemir and Esen Citation2007), were quantified. PM2.5 samples were also analyzed for EC and OC concentrations by an EC/OC analyzer (Model 6.2, Sunset Laboratory Inc., Tigard, OR, USA) using a TOT method and an NIOSH-derived temperature program (referred as “quartz.par” in the instrument) summarized in Table S1. More details on all the analytical procedures can be found in the supplemental information (SI).
2.3. Online analyses
An isokinetic flow sampled PM2.5 toward two online instruments installed in an air-conditioned room: (i) a dual-wavelength Aethalometer (Model AE-21, Magee Scientific, Berkeley, CA, USA) for BC measurements; (ii) a High-Resolution Time-of-Flight Aerosol Mass Spectrometer (HR-ToF-AMS; Aerodyne Research Inc., Billerica, MA, USA) measuring the real-time mass concentrations of non-refractory submicron aerosols (NR-PM1). The Aethalometer measured automatically every 10 min refractory BC (λ = 880 nm) and BCUV (λ = 370 nm) concentrations. More details are given in the SI.
A complete description of the HR-ToF-AMS principle can be found elsewhere (DeCarlo et al. Citation2006; Canagaratna et al. Citation2007) but it basically consists in sampling ambient particles through a critical orifice of 100 µm diameter at a flow rate of ∼80 mL min−1 focused into a narrow beam by an aerodynamic lens system (Zhang et al. Citation2002, Citation2004) that is transmitted into a vacuum chamber where particles are accelerated according to their aerodynamic diameter. Then, particle components are flash-vaporized by impaction at 600°C. The resultant vapor molecules are immediately ionized by electron impact at 70 eV. Positive ions are guided into the time-of-flight mass spectrometer in the V-mode (higher sensitivity but low mass resolution) or in the W-mode (less sensitivity but higher mass resolution) alternately, before being detected. AMS are now usually operated with a dryer upstream the inlet to avoid artifacts due to the presence of moisture in the air flow (Matthew et al. Citation2008). However, due to its unavailability during the first campaign aerosols were sampled without drying. To maintain consistency, subsequent campaigns were also performed without drying the aerosols before entering into the AMS. The possible impact of not drying aerosols during these campaigns, as well as the specific data treatment of the organic and PAH species, are discussed more extensively in the SI, Section S1.2.2. PAH concentrations measured by the AMS during both summer sampling periods were below the limit of quantification for the 4 min sampling time base used in this work. Therefore, only winter data are presented here.
3. Results and discussion
3.1. Comparison of PAH concentrations
The time series of the total PAH concentrations measured from daily PM2.5 filter analysis ([PAH]F) and by the AMS ([PAH]AMS) are shown in for the winter field campaigns at both sites. Results for [PAH]AMS considering PAHs from C16 to C34 and from C16 to C22 are both presented. [PAH]F and [PAH]AMS with the same carbon number for both sites are compared in . Despite the large difference in measurement time resolutions, when AMS data were averaged over 24 h, a strong similarity (with minor differences, discussed below) between the two PAH time series was observed irrespective of the site. The profiles of [PAH]AMS with the same carbon number as measured on filters were consistent in DO where [PAH]AMS C16-C34 logically overestimated the filter concentrations as discussed later in this section. Remarkably good correlations between PAH concentrations were observed with [PAH]F highly correlated with both [PAH]AMS C16-C34 (r2 = 0.97; n = 7) and [PAH]AMS C16-C22 (r2 = 0.98; slope = 0.66). The obtained results are consistent with a previous study which found a high correlation (r2= 0.97; slope = 0.77) between PAH concentrations measured by a Q-AMS and the sum of 18 PAHs identified in PM1 filter samples collected at a mid-level mountain site in Melpitz, Germany (Poulain et al. Citation2011).
Figure 1. (Top) Time series of winter PAH measurements by online AMS (averaged data; open triangles: sum of C16 to C34; solid triangles: sum of C16 to C22) and from daily PM2.5 filter samples collected in winter (solid circles: sum of C16 to C22 PAHs) in DO (n = 7) and GS (n = 9). (Bottom) Scatterplots comparing C16 to C22 PAH concentrations between both methods at both sites, together with the corresponding linear regression fits through the data (coefficients are given with a 1σ statistical uncertainty). One outlier was excluded from the GS dataset.
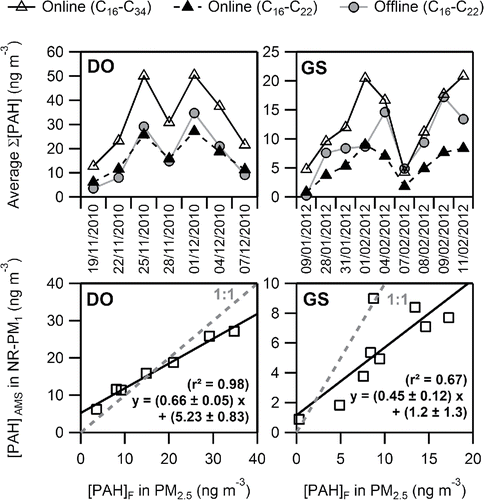
On the contrary in GS, [PAH]AMS C16-C34 were lower than [PAH]F regardless of the period and the discrepancy was even more pronounced with [PAH]AMS C16-C22. Total PAH concentrations were also well correlated but to a lesser degree than in DO with determination coefficients ranging from 0.62 between [PAH]F and [PAH]AMS C16-C34 (n = 9) up to 0.67 between [PAH]F and [PAH]AMS C16-C22. It is interesting to note that excellent correlations between individual PAH concentrations were observed at DO (r2 > 0.91). In GS, even when one outlier was excluded from the fits, the correlation coefficients were lower (r2∼0.7 for C16, C18, and C20 and r2 = 0.34 for C22) although the dispersion of points could be explained by the largely smaller concentrations observed at this site (see Figures S1 and S2 in SI).
Several reasons could explain the non-unity slopes observed, i.e., 0.66 ± 0.05 in DO and 0.45 ± 0.12 in GS:
| (i) | the HPLC analysis focuses on 10 PAHs specifically identified while AMS, based on mass-to-charge ratios can also measure isomers of these 10 PAHs, which could lead to an underestimation of the ambient PAH concentrations measured on filters. Nevertheless, the fact that a slight overestimation is rather observed in DO, and a strong one in GS tend to point out that the filter analyses capture most of the PAHs involved in the observed concentrations at least at the urban site; | ||||
| (ii) | due to longer sampling times, the collection of PAH on filters can be subject to sampling artifacts. The negative artifacts (desorption, reactivity) seem negligible because of the low concentrations of oxidants expected during the winter season (maximum hourly concentrations of ozone did not exceed 53 and 66 µg m−3 in DO and GS, respectively, during the campaigns) and the low temperatures measured prevented PAH desorption (Tavg = 1.9 and −1.0°C in DO and GS, respectively). Moreover, negative artifacts would lead to an underestimation of [PAH]F contrary to the trend observed. As for the positive adsorption artifact, organic compounds in the gas phase can be adsorbed on the surface of the collected particles or the filter fibers during sampling, which may lead to their overestimation in the particulate phase up to 50% (Turpin et al. Citation2000). However, high humidity levels, like those observed in this work (78% in DO and 77% in GS) may prevent the adsorption of hydrophobic organic compounds such as PAHs (Pankow et al. Citation1993; Storey et al. Citation1995) and no significant trend was observed between semi-volatile species like C16 and C18 PAHs and the non-volatile C20 and C22 (Figures S1 and S2); | ||||
| (iii) | the uncertainties on PAH identification by the AMS have been estimated at 35–38% (Dzepina et al. Citation2007), which encompass at least the overall discrepancy observed at the DO site. The combined uncertainty for offline, filter-based PAH measurements varied from 12 to 16%, depending upon the PAH; | ||||
| (iv) | finally, there may be a significant contribution of PM1-PM2.5 to the PM2.5 fraction in GS. During the winter campaign in DO, thanks to a PM1 analyzer available at a monitoring station close (∼800 m) to our sampling site, the contribution of PM1 and NR-PM1 to the PM2.5 total mass could be estimated at 94% and ∼70%, respectively, so the PM1-PM2.5 could be considered negligible. In GS, only PM2.5 mass concentrations were available with NR-PM1 representing only 52% of the fine particulate matter. This observation is however consistent with previous measurements in Dunkirk, where PM1/PM2.5 ratios of 35% and 53% have been determined under urban and industrial influences, respectively (Cazier et al. Citation2011). Back trajectory analysis also indicates that during summertime, a significant fraction of air masses was arriving from the sea. So, the contribution from refractory sea salts in the larger size fraction may have contributed to lower PM1/PM2.5 ratios during summer. PAHs emitted by urban and/or industrial combustion processes could moreover have been adsorbed on the surface of larger particles (like coarse-mode marine aerosols or dust emitted from the piles of ore present in the industrial area) and thus explain the larger discrepancy between both measurements. | ||||
The PAHAMS time series showed several short events from local sources, which could not be observed with the sampling times classically used for filter collection: the PAHAMS C16-C22 maximum concentration reached for instance 336 ng m−3 in GS (while the PAHAMS and PAHF mean concentrations over the filter sampling period were 5.2 ng m−3 and 9.4 ± 4.7 ng m−3, respectively) and 802 ng m−3 in DO (17 ng m−3 and 17 ± 12 ng m−3, respectively). These observations highlight the relevance for the deployment of field instruments with minute-timebase resolution for the identification of PAH sources as suggested by Poulain et al. (Citation2011). However, the low concentrations encountered during the summer sampling periods would constrain the user to increase the averaging acquisition time to reach lower detection limits.
3.2. Comparison of the organic fraction
The organic fraction was measured both online in NR-PM1 ([OM]AMS) and offline after PM2.5 collection on filter and OC determination ([OC]F). In DO, the average [OC]F values ranged from 2.23 ± 1.25 µgC m−3 (summer) to 5.79 ± 0.13 µgC m−3 (winter). The highest levels were also measured during the cold period in GS with an average concentration of 5.49 ± 0.43 µgC m−3 compared to 1.65 ± 0.22 µgC m−3 in summer, highlighting a clear seasonal variation at both sites but no significant differences between sites. Similar conclusions could be drawn from the average [OM]AMS values with 7.7 ± 5.7 µg m−3 observed in DO in winter and 1.6 ± 2.0 µg m−3 in summer, whereas in GS 6.8 ± 5.1 µg m−3 were measured in winter (and 0.40 ± 0.56 µg m−3 in summer).
The elemental analysis performed using high mass resolution data from the AMS (Aiken et al. Citation2007) allowed for the determination of the temporal evolution of the OMAMS/OCAMS ratio in winter (). On average, the wintertime OMAMS/OCAMS ratios were 1.63 in DO and 1.76 in GS. The results of the elemental analysis showed that organic aerosols in GS were more oxidized than in DO, and representative of relatively functionalized aerosols resulting from chemical ageing during their transport in the atmosphere. The ratio was stable during the winter campaign at both sites with a variation of 8.4% in GS and 6.6% in DO, respectively, suggesting similar emission sources for organics and/or similarity in the timescale and nature of their atmospheric processing. As mentioned in Section S1.3 of the SI, the summertime ratio was considered constant at 1.6 at both sites, consistent with both the HR analysis of V-mode data and the literature (Turpin and Lim Citation2001).
Figure 2. Time series of particulate organics observed in (left) DO and (right) GS in (top) winter and (bottom) summer. Each subpanel represents, from top to bottom, [OM]AMS measured in NR-PM1, OMAMS/OCAMS ratios determined from elemental analysis (only in winter), and averaged [OM]AMS (open triangles), [OC]AMS (solid triangles), and [OC]F (solid circles) measured in PM2.5.
![Figure 2. Time series of particulate organics observed in (left) DO and (right) GS in (top) winter and (bottom) summer. Each subpanel represents, from top to bottom, [OM]AMS measured in NR-PM1, OMAMS/OCAMS ratios determined from elemental analysis (only in winter), and averaged [OM]AMS (open triangles), [OC]AMS (solid triangles), and [OC]F (solid circles) measured in PM2.5.](/cms/asset/51cfc396-c820-4306-bb38-71474eaf3adc/uast_a_1403008_f0002_b.gif)
Measurements were compared by plotting OCAMS in NR-PM1 vs. OCF in PM2.5 and determining the slope for each campaign (Figure S3). Good linear trends were observed between OC real-time measurements from the HR-ToF-AMS and organic carbon determined by a thermal-optical method. The lowest determination coefficients were observed at the industrialized coastal site with values ranging from 0.50 in summer to 0.82 in winter and the highest values at the urban background site (r2 = 0.86 and 0.92 in winter and summer, respectively). However, significant OCAMS-to-OCF ratio differences were observed; the highest ratio (0.61) was measured in DO in winter when PM1 represented 94% of the total PM2.5 mass, and the lowest one (0.29) during the summer campaign in GS. In the absence of additional measurements to help interpret these observations, one can only provide some tentative explanations. A large part of OM could be in the PM1–2.5 fraction in summertime GS while the elevated wintertime ratios could indicate freshly emitted organics in the submicron fraction. The wintertime OMAMS/PM2.5 ratio of 0.24 in DO (0.23 in GS, respectively) was much higher than the summertime OMAMS/PM2.5 ratio of 0.08 in DO (0.04 in GS, respectively). These ratios probably point out some seasonal contribution of refractory and/or inorganic species in the PM1–2.5 fraction. In GS, the total mass of NR-PM1 was also dominated by inorganics, on average contributing around 70% for both seasons. Back trajectory analysis suggests that during summer, local and sea salt aerosols were contributing significantly at both sites while in winter aerosols were associated with long-range continental air masses coming from continental Europe. This could explain why inorganic and refractory species were dominating during summer.
also shows the time series of [OC]F, [OC]AMS and averaged [OM]AMS following similar trends regardless of the sampling sites and periods but differing significantly in term of concentration levels between the two analytical methods and the two size fractions. For the same reasons as already mentioned in Section 3.1, no significant losses (by desorption and/or reactivity) of SVOCs are expected. Even if reactivity would occur with atmospheric oxidants, initially it would not alter the number of carbons but only the oxidation state. Continuous atmospheric oxidation may lead to the fragmentation of organic molecules but due to low temperatures observed during the sampling periods, significant evaporative loss of organics was unlikely. A previous study also showed that OC concentrations measured during a summer field campaign at an urban site were not affected by a significant desorption artifact using a device with front and backup filters (Viana et al. Citation2006). By comparing between undenuded filters and denuded ones, they also estimated that the positive adsorption artifact could account for as much as 1/3 of the total OC measured on filter, which would reduce the discrepancy observed in our study. Nevertheless, they suggested that part of the difference could be due to the volatilization of semi-volatile species to re-establish the equilibrium with the gas phase. The slope differences can also be partly explained by the uncertainty on the EC/OC split which can significantly modify OC measurements, and is affected by the use of different protocols involving different temperature steps. Measurement precision ranges were estimated from 4 to 13% for OC and 6 to 21% for EC between ACE-Asia and IMPROVE protocols (Schauer et al. Citation2003). Differences of the order of 20% (Schmid et al. Citation2001) or 25% (Husain et al. Citation2007) have been observed between NIOSH and IMPROVE programs and have been estimated as quite consistent. Finally, the high values of ambient relative humidity combined with the sampling of non-dried aerosols into the AMS for these campaigns have possibly led to high water content in fine aerosol composition and an increase of the collection efficiency (Matthew et al. Citation2008), resulting in an underestimation of the OMAMS concentrations.
The values of OMAMS/OC for the DO and GS winter campaigns are given in Table S2 together with other studies performed around the world, where OC values are either coming from the AMS or from filter analysis by thermo-optical methods. Some of these studies used a cut-off diameter of 1 µm for filter collection to overcome the PM1–2.5 coarse fraction thus avoiding the possible difference in the oxidation state of organic aerosol between PM1 and PM2.5. Indeed, PM1 are generally associated with relatively fresh emissions, growing by condensation during their transport into the atmosphere to more oxidized organic matter. Despite this consideration and the differences of instrumentation and analytical methods between the studies, OMAMS/OCAMS and OMAMS/OCF values were in the same order of magnitude, with OMAMS/OCF 16% higher than OMAMS/OCAMS for both sites. Results for urban aerosols are comprised in the range 1.61 to 1.79 with highest values observed in winter. Back trajectory analysis indicates that aerosols were mostly coming from continental Europe during winter, while in summer, they were mostly regional and from the sea. Another possible reason of observed high OM/OC ratios during winter could be the high RH and foggy weather which usually provides a suitable environment for aqueous phase oxidation (Ervens et al. Citation2011). Calm conditions during winter may have also allowed more processing of the stagnant air masses. No significant OMAMS/OCAMS ratio differences were observed between our two sampling sites. However values determined at a rural site in Ontario, Canada (Chan et al. Citation2010) showed a higher variability as a function of the wind directions with values ranging from 1.58 to 2.08 probably due to different aging processes of air masses. The winter value in DO was very close to the summer OM/OC measurement found at an urban site in Marseilles, France (El Haddad et al. Citation2011). The winter ratio in GS was in excellent agreement with the one observed for rural aerosols sampled in the Pearl River Delta, China (Huang et al. Citation2011), where an average ratio of 1.77 was highly correlated with the O/C ratio (r2 = 0.98). This rural site was not influenced by local emissions, which suggests aerosols sampled in GS reflected relatively aged air masses with oxidized organic aerosols and/or underwent a very quick modification of their physical and chemical properties between the local emission sources and the receptor site.
3.3. Comparison of EC and BC measurements
presents the time series of Black Carbon concentrations (termed as [BC]AE) in PM2.5 samples monitored continuously by the Aethalometer and averaged [BC]AE calculated on the same time resolution as the concentrations of elemental carbon (termed as [EC]F) determined by the EC/OC analyses of PM2.5 collected on filters at both sites. [EC]F were relatively low regardless of the sampling sites and periods, from 0.46 ± 0.10 µgC m−3 (summer) to 0.80 ± 0.39 µgC m−3 (winter) in GS; and from 0.46 ± 0.18 µgC m−3 (summer) to 1.11 ± 0.10 µgC m−3 (winter) in DO. On average, [BC]AE were roughly lower than [EC]F with average values varying from 0.32 ± 0.28 µgC m−3 during the warm period in DO to 0.67 ± 0.57 µgC m−3 (winter) and a summer average of 0.29 ± 0.31 µgC m−3 in GS. Although [BC]AE and [EC]F were relatively similar on average, the [BC]AE time series showed several short-term events with values ranging up to 5.48 µgC m−3 in DO, 3.17 µgC m−3 and 6.60 µgC m−3 in GS in summer and winter, respectively.
Figure 3. (Top) Time series of [BC]AE and (bottom) averaged [BC]AE (black triangles) and [EC]F (gray circles) in PM2.5 sampled in (from left to right) DO in summer, GS during the winter and summer.
![Figure 3. (Top) Time series of [BC]AE and (bottom) averaged [BC]AE (black triangles) and [EC]F (gray circles) in PM2.5 sampled in (from left to right) DO in summer, GS during the winter and summer.](/cms/asset/7f58aa61-f8fc-4060-8db0-e6ab325385d9/uast_a_1403008_f0003_b.gif)
The comparison of [BC]AE vs. [EC]F is shown in Figure S4. BC and EC concentrations were highly correlated for the summer campaigns with good determination coefficients in GS (r2 = 0.81, n = 7) and DO (r2 = 0.97, n = 10) but to a lesser degree in winter (r2 = 0.64), where BC vs. EC measurements are pretty scattered. On average, the BC/EC ratios were lower than unity with values of 0.83 for DO and varying from 0.48 (summer) to 0.65 (winter) for GS. BC/EC ratios were then compared to values found in the literature (Table S3). The majority of these studies have reported good linear trends with r2 ranging up to 0.97 but poor correlations were also observed for rural/remote environments with r2 values below 0.5. The BC/EC ratio reported in the literature varied widely from 0.61 to 3.3 for sampling sites influenced by traffic, with a majority of values close to unity or with EC concentrations slightly higher than BC.
The use of different analytical techniques could be one of the reasons that contributed to an important measurement uncertainty. Part of the measurement uncertainties can come from the EC/OC analyzer as previously mentioned for OC measurements (see Section 3.2). Besides, two intercomparisons for Aethalometers showed a unit-to-unit variability of 30%, pointing out the lack of a generally accepted calibration standard and the need to measure regularly the spot area size and flow rate. The ratio differences could also result from the use of artifact correction algorithms.
In our work, the relative underestimation of the corrected [BC]AE comparatively to the measured [EC]F may be explained by the constant values given in the correction algorithm (Weingartner et al. Citation2003). Higher constant values of Cref, ranging from 2.9 to 4.3, have been determined following an intercomparison between five correction schemes applied to several datasets (Collaud Coen et al. Citation2010). Such an increase in Cref would cause a decrease of babs and [BC]AE (see Section S1.2.1 of the SI), whereas a lower value of the single scattering albedo (SSA or ω0) would increase babs and [BC]AE. Collaud Coen et al. (Citation2010) have shown a clear seasonal dependence of Cref with higher values during summer partly due to the dependence of SSA with Cref which highlights the need to estimate SSA values with a high time resolution during field campaigns. Indeed, the SSA has a great temporal variability with lower and noisier values in winter (Andrews et al. Citation2011). In addition, industrial plumes are likely to significantly alter the SSA values in GS by increasing the aerosol optical extinction. Thus, a better knowledge of the aerosol physical and chemical composition and aerosol light scattering measurements (Cref and SSA) are needed to improve the AE correction procedure. As shown in Table S3, slope differences are observed between seasons and sites for thermal and thermal-optical methods suggesting that the composition of the sampled aerosol and its physical and chemical properties as well as the local environment (urban, rural, or remote) also play an important role for instrument correlation (Liousse et al. Citation1993), especially for the determination of the specific attenuation cross-section, σATN. The chemical ageing process can change the chemical composition of BC aerosols by reactivity with other species present during their transport in the atmosphere to the sampling site. As far as this study is concerned, the poor winter correlation in GS might have been due to aerosol characteristics varying during strong sulfate events where acidic particles have been observed under a high relative humidity (Crenn et al. Citation2017). The presence of sulfur compounds emitted by the industrial sector can indeed lead to the formation of a sulfate shell around BC particles (Schnaiter et al. Citation2005), which increases particle size and can affect optical properties by increasing the σATN value. Jeong et al. (Citation2004) have concluded that [BC]AE might be overestimated due to the hygroscopic behavior of carbonaceous materials during sulfate haze events. This is also consistent with results which showed that a lower r2 might be caused by the large variability in optical properties of BC aerosols due its mixing state (Ahmed et al. Citation2009).
The variability of the slopes may also be attributed to the condensation of SVOCs onto particles, aerosol water content, or the presence of liquid organic particles, which could also affect the Cref value (Weingartner et al. Citation2003). As previously described, the near-UV wavelength of the AE-21 Aethalometer (λ = 370 nm) can be used to identify the presence of UV absorbing organic compounds such as aromatics even if values of (BCUV)AE are only qualitative. Overall, [BC]AE-to-(BCUV)AE varied from 0.87 (winter) to 1.07 (summer) in GS and was equal to 1.12 in DO in summer. No significant correlation was observed between [PAH]AMS and (BCUV)AE for the only winter campaign with both instruments (GS), indicating that PAHs were not the only chemical species absorbing in the UV. Aromatic VOCs have indeed largely been identified and quantified from measurements in the Dunkirk area (Roukos et al. Citation2009; Xiang et al. Citation2012). The presence of aromatic SVOCs—either directly emitted or as oxidation products of primary VOCs—have therefore to be expected as a potential influencing parameter which can explain (BCUV)AE levels. Further investigations will be necessary to determine the organic species responsible for the absorption in the near UV-region.
4. Conclusion
This extensive study has compared real-time measurements and chemical analyses of carbonaceous aerosols collected on filters for two seasons and two sites. The comparison showed significant correlations for the total PAH measurements with r2 between 0.67 and 0.98. However, the limits of detection obtained by chromatographic techniques were better than the AMS and thus useful in summer when the compounds were present at low concentrations in the atmosphere. Linear trends were also observed for organic carbon, regardless of the type of sampling site and season. The study of OM/OC ratios suggested that a large part of the organic fraction was in the PM1–2.5 size range except in DO during winter where 80% of the organic carbon was in the NR-PM1 fraction. The elemental analysis showed more highly oxidized organic aerosols at the industrial and coastal site during the cold period. Good data consistency was also obtained between thermal-optical EC and optical BC measurements with high correlations ranging from 0.64 to 0.97 but BC/EC ratios less than unity. Many parameters depending on aerosol composition and its physicochemical properties, various artifacts and uncertainties in the analytical techniques can explain the observed differences. This highlights the critical need to combine several types of methods to provide chemical speciation as well as high temporal resolution in order to explain specific pollution events and identify sources at sites highly influenced by various anthropogenic emissions.
UAST_1403008_Supplemental_File.zip
Download Zip (934.2 KB)Funding
IMT Lille Douai and PC2A acknowledge financial support from the CaPPA (Chemical and Physical Properties of the Atmosphere) project funded by the French National Research Agency (ANR) through the PIA (Programme d'Investissement d'Avenir) under contract “ANR-11-LABX-0005-01,” and two CPER projects funded by the French Ministry of Higher Education and Research, the CNRS and the European Regional Development Fund (ERDF): “Climibio,” and “IRENI” (additionally financed by the Communauté Urbaine de Dunkerque). V. Crenn gratefully thanks Armines and the Région Hauts-de-France for his PhD grant. A. Chakraborty acknowledges support from the “Climibio” project for his postdoctoral fellowship.
References
- Ahmed, T., Dutkiewicz, V. A., Shareef, A., Tuncel, G., Tuncel, S., and Husain, L. (2009). Measurement of Black Carbon (BC) by an Optical Method and a Thermal-Optical Method: Intercomparison for Four Sites. Atmos. Environ., 43:6305–6311. doi:10.1016/j.atmosenv.2009.09.031.
- Aiken, A. C., DeCarlo, P. F., and Jimenez, J. L. (2007). Elemental Analysis of Organic Species with Electron Ionization High-Resolution Mass Spectrometry. Anal. Chem., 79:8350–8358. doi:10.1021/ac071150w.
- Alleman, L. Y., Lamaison, L., Perdrix, E., Robache, A., and Galloo, J.-C. (2010). PM10 Metal Concentrations and Source Identification Using Positive Matrix Factorization and Wind Sectoring In A French Industrial Zone. Atmos. Res., 96:612–625.
- Amann, M., Bertok, I., Cofala, J., Gyarfas, F., Heyes, C., Klimont, Z., Schöpp, W., and Winiwarter, W. (2005). Baseline Scenarios for the Clean Air for Europe (CAFE) Programme. Report No. 1. International Institute for Applied Systems Analysis, Laxenburg, Austria.
- Andrews, E., Ogren, J. A., Bonasoni, P., Marinoni, A., Cuevas, E., Rodríguez, S., Sun, J. Y., Jaffe, D. A., Fischer, E. V., Baltensperger, U., Weingartner, E., Coen, M. C., Sharma, S., Macdonald, A. M., Leaitch, W. R., Lin, N.-H., Laj, P., Arsov, T., Kalapov, I., Jefferson, A., and Sheridan, P. (2011). Climatology of Aerosol Radiative Properties in the Free Troposphere. Atmos. Res., 102:365–393. doi:10.1016/j.atmosres.2011.08.017.
- Arnott, W. P., Hamasha, K., Moosmüller, H., Sheridan, P. J., and Ogren, J. A. (2005). Towards Aerosol Light-Absorption Measurements with a 7-Wavelength Aethalometer: Evaluation with a Photoacoustic Instrument and 3-Wavelength Nephelometer. Aerosol Sci. Technol., 39:17–29. doi:10.1080/027868290901972.
- Aubriet, F., and Carré, V. (2010). Potential of Laser Mass Spectrometry for the Analysis of Environmental Dust Particles—A Review. Anal. Chim. Acta., 659:34–54. doi:10.1016/j.aca.2009.11.047.
- Badol, C., Locoge, N., and Galloo, J.-C. (2008). Using a Source-Receptor Approach to Characterise VOC Behaviour in A French Urban Area Influenced by Industrial Emissions. Sci. Total Environ., 389:429–440. doi:10.1016/j.scitotenv.2007.09.002.
- Bae, M.-S., Hong, C.-S., Kim, Y. J., Han, J.-S., Moon, K.-J., Kondo, Y., Komazaki, Y., and Miyazaki, Y. (2007). Intercomparison of two Different Thermal-Optical Elemental Carbons and Optical Black Carbon During ABC-EAREX2005. Atmos. Environ., 41:2791–2803. doi:10.1016/j.atmosenv.2006.11.040.
- Billet, S., Abbas, I., Goff, J. L., Verdin, A., André, V., Lafargue, P.-E., Hachimi, A., Cazier, F., Sichel, F., Shirali, P., and Garçon, G. (2008). Genotoxic Potential of Polycyclic Aromatic Hydrocarbons-coated onto Airborne Particulate Matter (PM2.5) in Human Lung Epithelial A549 cells. Cancer Lett., 270:144–155. doi:10.1016/j.canlet.2008.04.044.
- Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R. (2007). Chemical and Microphysical Characterization of Ambient Aerosols With the Aerodyne Aerosol Mass Spectrometer. Mass Spectrom. Rev., 26:185–222. doi:10.1002/mas.20115.
- Cazier, F., Dewaele, D., Delbende, A., Nouali, H., Garçon, G., Verdin, A., Courcot, D., Bouhsina, S., and Shirali, P. (2011). Sampling Analysis and Characterization of Particles in the Atmosphere of Rural, Urban and Industrial Areas. Procedia Environ. Sci., 4:218–227.
- Chan, T. W., Huang, L., Leaitch, W. R., Sharma, S., Brook, J. R., Slowik, J. G., Abbatt, J. P. D., Brickell, P. C., Liggio, J., Li, S.-M., and Moosmüller, H. (2010). Observations of OM/OC and Specific Attenuation Coefficients (SAC) in Ambient fine PM at a Rural Site in Central Ontario, Canada. Atmos. Chem. Phys., 10:2393–2411.
- Choël, M., Deboudt, K., and Flament, P. (2010). Development of Time-Resolved Description of Aerosol Properties at the Particle Scale During an Episode of Industrial Pollution Plume. Water Air Soil Pollut., 209:93–107. doi:10.1007/s11270-009-0183-9.
- Collaud Coen, M., Weingartner, E., Apituley, A., Ceburnis, D., Fierz-Schmidhauser, R., Flentje, H., Henzing, J. S., Jennings, S. G., Moerman, M., Petzold, A., Schmid, O., and Baltensperger, U. (2010). Minimizing Light Absorption Measurement Artifacts of the Aethalometer: Evaluation of Five Correction Algorithms. Atmos. Meas. Tech., 3:457–474. doi:10.5194/amt-3-457-2010.
- Crenn, V., Fronval, I., Petitprez, D., and Riffault, V. (2017). Fine Particles Sampled at An Urban Background Site and An Industrialized Coastal Site in Northern France — Part 1: Seasonal Variations and Chemical Characterization. Sci. Total Environ., 578:203–218. doi:10.1016/j.scitotenv.2015.11.165.
- DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L. (2006). Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer. Anal. Chem., 78:8281–8289. doi:10.1021/ac061249n.
- Dzepina, K., Arey, J., Marr, L. C., Worsnop, D. R., Salcedo, D., Zhang, Q., Onasch, T. B., Molina, L. T., Molina, M. J., and Jimenez, J. L. (2007). Detection of Particle-Phase Polycyclic Aromatic Hydrocarbons In Mexico City Using An Aerosol Mass Spectrometer. Int. J. Mass Spectrom., 263:152–170. doi:10.1016/j.ijms.2007.01.010.
- El Haddad, I., Marchand, N., Wortham, H., Piot, C., Besombes, J.-L., Cozic, J., Chauvel, C., Armengaud, A., Robin, D., and Jaffrezo, J.-L. (2011). Primary Sources of PM2.5 Organic Aerosol in an Industrial Mediterranean city, Marseille. Atmos. Chem. Phys., 11:2039–2058. doi:10.5194/acp-11-2039-2011.
- Ervens, B., Turpin, B. J., and Weber, R. J. (2011). Secondary Organic Aerosol Formation in Cloud Droplets and Aqueous Particles (aqSOA): A Review of Laboratory, Field and Model Studies. Atmos. Chem. Phys., 11:11069–11102. doi:10.5194/acp-11-11069-2011.
- Flament, P., Mattielli, N., Aimoz, L., Choël, M., Deboudt, K., Jong, J., de Rimetz-Planchon, J., and Weis, D. (2008). Iron Isotopic Fractionation in Industrial Emissions and Urban Aerosols. Chemosphere. 73:1793–1798. doi:10.1016/j.chemosphere.2008.08.042.
- Goldstein, A. H., and Galbally, I. E. (2007). Known and Unexplored Organic Constituents in the Earth's Atmosphere. Environ. Sci. Technol., 41:1514–1521.
- Huang, X.-F., He, L.-Y., Hu, M., Canagaratna, M. R., Kroll, J. H., Ng, N. L., Zhang, Y.-H., Lin, Y., Xue, L., Sun, T.-L., Liu, X.-G., Shao, M., Jayne, J. T., and Worsnop, D. R. (2011). Characterization of Submicron Aerosols at A Rural Site in Pearl River Delta of China Using an Aerodyne High-Resolution Aerosol Mass Spectrometer. Atmos. Chem. Phys., 11:1865–1877. doi:10.5194/acp-11-1865-2011.
- Husain, L., Dutkiewicz, V. A., Khan, A. J., and Ghauri, B. M. (2007). Characterization of Carbonaceous Aerosols in Urban Air. Atmos. Environ., 41:6872–6883. doi:10.1016/j.atmosenv.2007.04.037.
- IPCC, Stocker, T. F. (Eds.) (2014). Climate Change 2013: The Physical Science Basis; Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge Univ. Press, New York, NY.
- Jeong, C.-H., Hopke, P. K., Kim, E., and Lee, D.-W. (2004). The comparison between thermal-optical transmittance elemental carbon and Aethalometer black carbon measured at multiple monitoring sites. Atmos. Environ., 38:5193–5204. doi:10.1016/j.atmosenv.2004.02.065.
- Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K. L., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R. (2009). Evolution of Organic Aerosols in the Atmosphere. Science, 326:1525–1529. doi:10.1126/science.1180353.
- Kalberer, M., Henne, S., Prevot, A. S. H., and Steinbacher, M. (2004). Vertical Transport and Degradation of Polycyclic Aromatic Hydrocarbons in an Alpine Valley. Atmos. Environ., 38:6447–6456. doi:10.1016/j.atmosenv.2004.06.021.
- Kelly, F. J., and Fussell, J. C. (2012). Size, Source and Chemical Composition as Determinants of Toxicity Attributable to Ambient Particulate Matter. Atmos. Environ., 60:504–526. doi:10.1016/j.atmosenv.2012.06.039.
- Liousse, C., Cachier, H., and Jennings, S. G. (1993). Optical and Thermal Measurements of Black Carbon Aerosol Content In Different Environments: Variation Of The Specific Attenuation Cross-Section, Sigma (σ). Atmos. Environ. Part A. Gen. Top., 27:1203–1211.
- Liu, L., Liu, Y., Lin, J.-M., Tang, N., Hayakawa, K., and Maeda, T. (2007). Development of Analytical Methods For Polycyclic Aromatic Hydrocarbons (PAHs) in Airborne Particulates: A Review. J. Environ. Sci., 19:1–11.
- Ma, W.-L., Li, Y.-F., Qi, H., Sun, D.-Z., Liu, L.-Y., and Wang, D.-G. (2010). Seasonal Variations of Sources of Polycyclic Aromatic Hydrocarbons (PAHs) to a Northeastern Urban City, China. Chemosphere, 79:441–447. doi:10.1016/j.chemosphere.2010.01.048.
- Marris, H., Deboudt, K., Augustin, P., Flament, P., Blond, F., Fiani, E., Fourmentin, M., and Delbarre, H. (2012). Fast Changes in Chemical Composition and Size Distribution of Fine Particles During the Near-Field Transport of Industrial Plumes. Sci. Total Environ., 427–428:126–138. doi:10.1016/j.scitotenv.2012.03.068.
- Matthew, B. M., Middlebrook, A. M., and Onasch, T. B. (2008). Collection Efficiencies in an Aerodyne Aerosol Mass Spectrometer as a Function of Particle Phase for Laboratory Generated Aerosols. Aerosol Sci. Technol., 42:884–898. doi:10.1080/02786820802356797.
- McMurry, P. H. (2000). A Review of Atmospheric Aerosol Measurements. Atmos. Environ., 34:1959–1999.
- Mirivel, G., Riffault, V., and Galloo, J.-C. (2009). Development and Validation of An Ultra-High-Performance Liquid Chromatography Coupled to Time-Of-Flight Mass Spectrometry Method To Quantify Benzoic Acid and Long-Chain Monocarboxylic Acids (C12-C28) in Atmospheric Aerosols. J. Chrom. A., 1216:6481–6489. doi:10.1016/j.chroma.2009.07.041.
- Mirivel, G., Riffault, V., and Galloo, J.-C. (2010). Simultaneous Determination by Ultra-Performance Liquid Chromatography–Atmospheric Pressure Chemical Ionization Time-of-Flight Mass Spectrometry of Nitrated and Oxygenated PAHs Found in Air and Soot Particles. Anal. Bioanal. Chem., 397:243–256. doi:10.1007/s00216-009-3416-2.
- Mirivel, G., Riffault, V., and Galloo, J.-C. (2011). Analysis of Phthalic, Isophthalic and Long-Chain (C4-C12) Dicarboxylic Acids in Atmospheric Aerosols by UPLC/ESI/ToF-MS. Anal. Methods, 3:1172–1179. doi:10.1039/C0AY00677G.
- Moosmüller, H., Chakrabarty, R. K., and Arnott, W. P. (2009). Aerosol Light Absorption and its Measurement: A Review. J. Quant. Spectrosc. Ra. Transfer., 110:844–878. doi:10.1016/j.jqsrt.2009.02.035.
- Müller, T., Henzing, J. S., de Leeuw, G., Wiedensohler, A., Alastuey, A., Angelov, H., Bizjak, M., Collaud Coen, M., Engström, J. E., Gruening, C., Hillamo, R., Hoffer, A., Imre, K., Ivanow, P., Jennings, G., Sun, J. Y., Kalivitis, N., Karlsson, H., Komppula, M., Laj, P., Li, S.-M., Lunder, C., Marinoni, A., Martins dos Santos, S., Moerman, M., Nowak, A., Ogren, J. A., Petzold, A., Pichon, J. M., Rodriquez, S., Sharma, S., Sheridan, P. J., Teinilä, K., Tuch, T., Viana, M., Virkkula, A., Weingartner, E., Wilhelm, R., and Wang, Y. Q. (2011). Characterization and Intercomparison of Aerosol Absorption Photometers: Result of Two Intercomparison Workshops. Atmos. Meas. Tech., 4:245–268. doi:10.5194/amt-4-245-2011.
- Pankow, J. F., Storey, J. M. E., and Yamasaki, H. (1993). Effects of Relative Humidity on Gas/Particle Partitioning of Semivolatile Organic Compounds to Urban Particulate Matter. Environ. Sci. Technol., 27:2220–2226. doi:10.1021/es00047a032.
- Petzold, A., Ogren, J. A., Fiebig, M., Laj, P., Li, S.-M., Baltensperger, U., Holzer-Popp, T., Kinne, S., Pappalardo, G., Sugimoto, N., Wehrli, C., Wiedensohler, A., and Zhang, X.-Y. (2013). Recommendations for Reporting “Black Carbon” Measurements. Atmos. Chem. Phys., 13:8365–8379. doi:10.5194/acp-13-8365-2013.
- Poulain, L., Iinuma, Y., Müller, K., Birmili, W., Weinhold, K., Brüggemann, E., Gnauk, T., Hausmann, A., Löschau, G., Wiedensohler, A., and Herrmann, H. (2011). Diurnal Variations of Ambient Particulate Wood Burning Emissions and Their Contribution to the Concentration of Polycyclic Aromatic Hydrocarbons (PAHs) in Seiffen, Germany. Atmos. Chem. Phys., 11:12697–12713. doi:10.5194/acp-11-12697-2011.
- Pratt, K. A., and Prather, K. A. (2011). Mass Spectrometry of Atmospheric Aerosols—Recent Developments and Applications. Part I: Off-Line Mass Spectrometry Techniques. Mass Spectrom. Rev., 31:1–16. doi:10.1002/mas.20322.
- Pratt, K. A., and Prather, K. A. (2012). Mass Spectrometry of Atmospheric Aerosols—Recent Developments and Applications. Part II: On-Line Mass Spectrometry Techniques. Mass Spectrom. Rev., 31:17–48. doi:10.1002/mas.20330.
- Ravindra, K., Sokhi, R., and Vangrieken, R. (2008). Atmospheric Polycyclic Aromatic Hydrocarbons: Source Attribution, Emission Factors and Regulation. Atmos. Environ., 42:2895–2921. doi:10.1016/j.atmosenv.2007.12.010.
- Riffault, V., Arndt, J., Marris, H., Mbengue, S., Setyan, A., Alleman, L. Y., Deboudt, K., Flament, P., Augustin, P., Delbarre, H., and Wenger, J. (2015). Fine and Ultrafine Particles in the Vicinity of Industrial Activities: A Review. Crit. Rev. Environ. Sci. Technol., 45:2305–2356. doi:10.1080/10643389.2015.1025636.
- Rimetz-Planchon, J., Perdrix, E., Sobanska, S., and Brémard, C. (2008). PM10 Air Quality Variations in an Urbanized and Industrialized Harbor. Atmos. Environ., 42:7274–7283. doi:10.1016/j.atmosenv.2008.07.005.
- Roukos, J., Locoge, N., Sacco, P., and Plaisance, H. (2011). Radial Diffusive Samplers for Determination of 8-h Concentration of BTEX, Acetone, Ethanol and Ozone in Ambient Air During a Sea Breeze Event. Atmos. Environ., 45:755–763.
- Roukos, J., Riffault, V., Locoge, N., and Plaisance, H. (2009). VOC in an Urban and Industrial Harbor on the French North Sea Coast During Two Contrasted Meteorological Situations. Environ. Pollut., 157:3001–3009. doi:10.1016/j.envpol.2009.05.059.
- Schauer, J. J., Mader, B. T., DeMinter, J. T., Heidemann, G., Bae, M. S., Seinfeld, J. H., Flagan, R. C., Cary, R. A., Smith, D., Huebert, B. J., Bertram, T., Howell, S., Kline, J. T., Quinn, P., Bates, T., Turpin, B., Lim, H. J., Yu, J. Z., Yang, H., and Keywood, M. D. (2003). ACE-Asia Intercomparison of a Thermal-Optical Method for the Determination of Particle-Phase Organic and Elemental Carbon. Environ. Sci. Technol., 37:993–1001. doi:10.1021/es020622f.
- Schmid, H., Laskus, L., Jürgen Abraham, H., Baltensperger, U., Lavanchy, V., Bizjak, M., Burba, P., Cachier, H., Crow, D., Chow, J., Gnauk, T., Even, A., ten Brink, H., Giesen, K.-P., Hitzenberger, R., Hueglin, C., Maenhaut, W., Pio, C., Carvalho, A., Putaud, J.-P., Toom-Sauntry, D., and Puxbaum, H. (2001). Results of the “Carbon Conference” International Aerosol Carbon Round Robin Test Stage I. Atmos. Environ., 35:2111–2121. doi:10.1016/S1352-2310(00)00493-3.
- Schmid, O., Artaxo, P., Arnott, W. P., Chand, D., Gatti, L. V., Frank, G. P., Hoffer, A., Schnaiter, M., and Andreae, T. W. (2006). Spectral Light Absorption By Ambient Aerosols Influenced By Biomass Burning in the Amazon Basin. I: Comparison and Field Calibration of Absorption Measurement Techniques. Atmos. Chem. Phys., 6:3443–3462.
- Schnaiter, M., Linke, C., Möhler, O., Naumann, K.-H., Saathoff, H., Wagner, R., Schurath, U., and Wehner, B. (2005). Absorption Amplification of Black Carbon Internally Mixed With Secondary Organic Aerosol. J. Geophys. Res., 110, n/a–n/a. doi:10.1029/2005JD006046.
- Sharma, H., Jain, V. K., and Khan, Z. H. (2007). Characterization and Source Identification of Polycyclic Aromatic Hydrocarbons (PAHs) in the Urban Environment of Delhi. Chemosphere, 66:302–310. doi:10.1016/j.chemosphere.2006.05.003.
- Singh, D. P., Gadi, R., and Mandal, T. K. (2011). Characterization of Particulate-Bound Polycyclic Aromatic Hydrocarbons and Trace Metals Composition of Urban Air in Delhi, India. Atmos. Environ., 45:7653–7663. doi:10.1016/j.atmosenv.2011.02.058.
- Slowik, J. G., Cross, E. S., Han, J.-H., Davidovits, P., Onasch, T. B., Jayne, J. T., Williams, L. R., Canagaratna, M. R., Worsnop, D. R., Chakrabarty, R. K., Moosmüller, H., Arnott, W. P., Schwarz, J. P., Gao, R.-S., Fahey, D. W., Kok, G. L., and Petzold, A. (2007). An Inter-Comparison of Instruments Measuring Black Carbon Content of Soot Particles. Aerosol Sci. Technol., 41:295–314. doi:10.1080/02786820701197078.
- Snyder, D. C., and Schauer, J. J. (2007). An Inter-Comparison of Two Black Carbon Aerosol Instruments and a Semi-Continuous Elemental Carbon Instrument in the Urban Environment. Aerosol Sci. Technol., 41:463–474. doi:10.1080/02786820701222819.
- Sodeman, D. A., Toner, S. M., and Prather, K. A. (2005). Determination of Single Particle Mass Spectral Signatures from Light-Duty Vehicle Emissions. Environ. Sci. Technol., 39:4569–4580. doi:10.1021/es0489947.
- Storey, J. M. E., Luo, W., Isabelle, L. M., and Pankow, J. F. (1995). Gas/Solid Partitioning of Semivolatile Organic Compounds to Model Atmospheric Solid Surfaces as a Function of Relative Humidity. Clean Quartz. Environ. Sci. Technol., 29:2420–2428. doi:10.1021/es00009a039.
- Tasdemir, Y., and Esen, F. (2007). Urban air PAHs: Concentrations, temporal changes and gas/particle partitioning at a traffic site in Turkey. Atmos. Res., 84:1–12. doi:10.1016/j.atmosres.2006.04.003.
- Turpin, B. J., and Lim, H.-J. (2001). Species Contributions to PM2.5 Mass Concentrations: Revisiting Common Assumptions for Estimating Organic Mass. Aerosol Sci. Technol., 35:602–610. doi:10.1080/02786820119445.
- Turpin, B. J., Saxena, P., and Andrews, E. (2000). Measuring and Simulating Particulate Organics in the Atmosphere: Problems and Prospects. Atmos. Environ., 34:2983–3013. doi:10.1016/S1352-2310(99)00501-4.
- Viana, M., Chi, X., Maenhaut, W., Cafmeyer, J., Querol, X., Alastuey, A., Mikuška, P., and Večeřa, Z. (2006). Influence of Sampling Artefacts on Measured PM, OC, and EC Levels in Carbonaceous Aerosols in an Urban Area. Aerosol Sci. Technol., 40:107–117. doi:10.1080/02786820500484388.
- Virkkula, A., Mäkelä, T., Hillamo, R., Yli-Tuomi, T., Hirsikko, A., Hämeri, K., and Koponen, I. K. (2007). A Simple Procedure for Correcting Loading Effects of Aethalometer Data. J. Air Waste Manag. Assoc., 57:1214–1222. doi:10.3155/1047-3289.57.10.1214.
- Weingartner, E., Saathoff, H., Schnaiter, M., Streit, N., Bitnar, B., and Baltensperger, U. (2003). Absorption of Light by Soot Particles: Determination of the Absorption Coefficient by Means of Aethalometers. J. Aerosol Sci., 34:1445–1463. doi:10.1016/S0021-8502(03)00359-8.
- Xiang, Y., Delbarre, H., Sauvage, S., Léonardis, T., Fourmentin, M., Augustin, P., and Locoge, N. (2012). Development of a Methodology Examining the Behaviours of VOCs Source Apportionment With Micro-Meteorology Analysis in an Urban and Industrial Area. Environ. Pollut., 162:15–28. doi:10.1016/j.envpol.2011.10.012.
- Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R. (2007). Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenically-Influenced Northern Hemisphere Midlatitudes. Geophys. Res. Lett., 34:L13801. doi:10.1029/2007gl029979.
- Zhang, X., Smith, K. A., Worsnop, D. R., Jimenez, J., Jayne, J. T., and Kolb, C. E. (2002). A Numerical Characterization of Particle Beam Collimation by an Aerodynamic Lens-Nozzle System: Part I. An Individual Lens or Nozzle. Aerosol Sci. Technol., 36:617–631. doi:10.1080/02786820252883856.
- Zhang, X., Smith, K. A., Worsnop, D. R., Jimenez, J. L., Jayne, J. T., Kolb, C. E., Morris, J., and Davidovits, P. (2004). Numerical Characterization of Particle Beam Collimation: Part II Integrated Aerodynamic-Lens–Nozzle System. Aerosol Sci. Technol., 38:619–638. doi:10.1080/02786820490479833.
- Zimmermann, R., Ferge, T., Gälli, M., and Karlsson, R. (2003). Application of Single-Particle Laser Desorption/Ionization Time-Of-Flight Mass Spectrometry For Detection of Polycyclic Aromatic Hydrocarbons From Soot Particles Originating From An Industrial Combustion Process. Rapid Commun. Mass Spectrom., 17:851–859. doi:10.1002/rcm.979.