
Abstract
Nanoparticle formation and growth driven by acid-base chemistry was investigated by introducing gas-phase sulfuric acid (H2SO4) with ammonia (NH3) or dimethylamine (DMA) into a flow tube reactor. A thermal desorption chemical Ionization mass spectrometer was used to measure the size-resolved chemical composition of H2SO4-DMA and H2SO4- NH3 nanoparticles formed under dry conditions and at 60% relative humidity. In contrast with predictions for bulk aqueous systems, nanoparticles showed a strong size-dependent composition gradient and did not always reach a fully neutralized state in excess of gas-phase base. Smaller particles were more acidic, with an acid:base ratio of 0.7 ± 0.1 and 1.3 ± 0.3 for 8.6 and 9.5 nm H2SO4-DMA particles formed under dry and humid conditions, respectively, and 3.1 ± 0.6 and 3.4 ± 0.3 for 7.5 nm H2SO4-NH3 particles formed under dry and humid conditions, respectively. The acidity of particles generally decreased as particles grew. H2SO4-DMA particles became fully neutralized as they grew to 14 nm, but H2SO4-NH3 particles at 12 nm were still acidic and were never observed to reach bulk sample thermodynamic equilibrium for the experimental conditions in this study. Thermodynamic modeling demonstrated that the observed trends can be reproduced by modifying acid dissociation constants to minimize acid-base chemistry, which may be caused by steric or mixing effects, and by considering volatilization of the neutral base.
Copyright © 2018 American Association for Aerosol Research
EDITOR:
Introduction
Particle nucleation from gaseous precursors represents a significant source of aerosols in the atmosphere (Finlayson-Pitts and Pitts Jr Citation2000; Kulmala et al. Citation2004; Seinfeld and Pandis Citation2006; Zhang et al. 2012). Newly formed particles can continue growing in the atmosphere to ∼100 nm, at which size, they may act as cloud condensation nuclei and thereby impact cloud properties and lifetimes with implications for the global radiative energy balance (Kerminen et al. Citation2005; Spracklen et al. Citation2008; Kuang et al. Citation2009; Merikanto et al. Citation2009). Nucleation and subsequent growth are collectively known as new particle formation (NPF). The species involved in NPF, and the underlying mechanisms, have been the subjects of many studies over the past two decades. Sulfuric acid (H2SO4) is well recognized to be critical in many NPF events (Weber et al. Citation1996, Citation1997; McMurry et al. Citation2000; Weber et al. Citation2001; Kulmala et al. Citation2006; Sipilä et al. Citation2010). Amines and ammonia (NH3) can bond strongly with H2SO4, and greatly enhance NPF (Ball et al. Citation1999; Korhonen et al. Citation1999; Yu Citation2006; Kurtén et al. Citation2008; Benson et al. Citation2009; Berndt et al. Citation2010; Erupe et al. Citation2011; Kirkby et al. Citation2011; Zollner et al. Citation2012; Almeida et al. Citation2013; Glasoe et al. Citation2015). Recent studies suggest that organic acids such as methanesulfonic acid (Kreidenweis et al. Citation1989; Wyslouzil et al. Citation1991; Dawson et al. Citation2012; Chen et al. Citation2016), and highly oxidized, low-volatility organic compounds (Donahue et al. Citation2013; Schobesberger et al. Citation2013; Bianchi et al. Citation2014; Ehn et al. Citation2014; Riccobono et al. Citation2014; Jokinen et al. Citation2015; Bianchi et al. Citation2016; Tröstl et al. Citation2016) also play an important role in NPF.
While both amines and NH3 can enhance NPF in the presence of H2SO4, laboratory studies and theoretical calculations have shown that amines are more effective compared to NH3 due to their higher basicity (Kurtén et al. Citation2008; Barsanti et al. Citation2009; Berndt et al. Citation2010; Erupe et al. Citation2011; Yu et al. Citation2012; Zollner et al. Citation2012; Almeida et al. Citation2013; Glasoe et al. Citation2015). Field studies have also reported the presence of aminium salts in ambient particles (Facchini et al. Citation2008; Pratt et al. Citation2009; Sorooshian et al. Citation2009; Smith et al. Citation2010), and the strong correlation between NPF and the presence of amines (Freshour et al. Citation2014; Jen et al. Citation2014; Hemmilä et al. Citation2018). Displacement of NH3 with amines in H2SO4-NH3 clusters and particles has been observed in laboratory studies (Bzdek et al. Citation2010; Qiu et al. Citation2011; Liu et al. Citation2012), with a close-to-collision-limited rate for small clusters (Bzdek et al. Citation2010). Given that the concentration of NH3 in the atmosphere is at least one order of magnitude higher than that of amines (Ge et al. Citation2011), both amines and NH3 are expected to be important in NPF, depending on the proximity of their emission sources. Water vapor has also been reported to be critical during the initial steps of NPF (Berndt et al. Citation2010; Erupe et al. Citation2011; Loukonen et al. Citation2010; Zollner et al. Citation2012; Henschel et al. Citation2014; Yu et al. Citation2017).
After clusters formed in the H2SO4-base system, they can continue to grow by the formation of particle-phase ionic (and thus non-volatile) dissociated acids and bases, condensation of H2SO4 due to its very low saturation vapor pressure, and coagulation with existing clusters and/or particles (Kurtén et al. Citation2008; Chen et al. Citation2012; Keskinen et al. Citation2013; Lawler et al. Citation2016; Lehtipalo et al. Citation2016). Although many studies have focused on NPF in the H2SO4-base system, only a few studies focused on the chemical composition of those newly formed clusters and particles due to the challenges associated with the determination of chemical composition of clusters and nanometer-sized particles. Measurements of molecular clusters that have fewer than 20 molecules in the H2SO4-base system (corresponding to a particle diameter of about 2 nm) made at the Cosmics Leaving OUtdoor Droplets (CLOUD) chamber at CERN show a formation process involving stepwise addition of H2SO4 and base, typically in a 1:1 ratio (Kirkby et al. Citation2011; Almeida et al. Citation2013; Schobesberger et al. Citation2013; Bianchi et al. Citation2014; Kurten et al. Citation2014; Schobesberger et al. Citation2015). Strong acid-base interactions in these molecular clusters drive cluster growth during the early stages of NPF. However, in a recent study at the CLOUD7 campaign, hygroscopicity measurements of H2SO4-base particles formed in excess of dimethylamine (DMA) or NH3 show that particles as small as 10 nm are much more hygroscopic than particles at 15 nm, suggesting that the smaller particles are more acidic in the acid-base system (Kim et al. Citation2016). The acidity of newly formed particles decreases as they grow (Kim et al. Citation2016). This is further confirmed by direct measurements of the chemical composition of those nanoparticles formed in the same experiments using Thermal Desorption Chemical Ionization Mass Spectrometry (TDCIMS) (Lawler et al. Citation2016). The formation of highly acidic particles in excess of base is not consistent with the theory for bulk aqueous systems, which predicts that bulk sample thermodynamic equilibrium between H2SO4 and base should be reached (Lehtipalo et al. Citation2016). These findings demonstrate that uncertainties remain in understanding particle growth in the H2SO4-base system.
In this study, we present chemical composition measurements of newly formed particles in the diameter range of 8–21 nm in the H2SO4-base system. We specifically focus on size-resolved acid:base ratios of newly formed H2SO4-base nanoparticles. The effect of relative humidity (RH) on nanoparticle composition is also explored. Thermodynamic modeling studies were performed to provide possible explanations for the experimental observations. The work provides valuable insights into the growth of particles after nucleation from gaseous precursors.
Experiment
Experimental setup
Particles were formed by mixing gaseous H2SO4 and DMA or NH3 in purified air using a flow tube reactor fabricated from borosilicate glass as shown in . The flow tube measured 7.6 cm in diameter and 100 cm in length, with a total volume of 4.8 L. Three inlets were located at the upstream end for separately introducing reactants: gas-phase H2SO4, DMA or NH3, and dry or humidified air. The inlets were perforated to enable rapid mixing of reactant gases in the upstream region. Particles were collected at the downstream end of the flow tube for analysis. The residence time of gases in the flow tube was ∼1 min.
Figure 1. Schematic of the flow tube reactor.
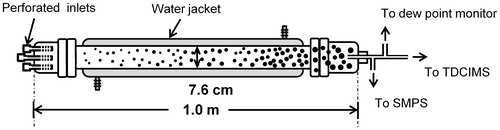
Gas flows were controlled and monitored with mass flow controllers. Gas-phase H2SO4 was generated by passing a 1 sLpm flow of gaseous N2, generated from the headspace of a liquid N2 dewar, through a 30 cm long glass saturator filled with 5 ml of 96% liquid H2SO4 (Fisher Scientific). The saturator was water jacketed to control the H2SO4 temperature at 30oC. Concentrations of H2SO4 in the flow tube were calculated by assuming that gas-phase H2SO4 reached its saturation vapor pressure before exiting the saturator. This assumption is supported by several experimental and theoretical studies (Brus et al. Citation2010; Herrmann et al. Citation2010; Panta et al. Citation2012; Zollner et al. Citation2012; Neitola et al. Citation2015). Our apparatus is identical to that described in Zollner et al. (Citation2012), who measured the [H2SO4] as it exited a 30 °C saturator as a function of carrier gas flow rate. They found that concentration varied linearly with flow rate over the range of 0.5–1.5 sLpm, and concluded that saturation conditions apply over this flow range. Neitola et al. (Citation2015) employed a lower carrier gas flow (0.5 sLpm) and directly measured [H2SO4] over a broader range of temperatures (0–40 °C), also concluding that the carrier gas is saturated with sulfuric acid under these conditions. Gas-phase NH3 and DMA were generated with two permeation tube systems. Each consisted of a mass flow controller that passed 0.1 sLpm of N2 through a glass tube holding a permeation tube (VICI AG International) containing liquid NH3 (Aldrich, 99.99%) or DMA (Aldrich, 99%). The permeation tubes were both kept at 30 °C to ensure constant permeation rates. The NH3 and DMA flows were directed into an active dilution system that mixed the sample with purified air to achieve the desired concentration (Freshour et al. Citation2014). Concentrations of DMA and NH3 were determined from the dilution factor and permeation rates, the latter of which were gravimetrically calibrated over 3–6 months. Purified air (2.9 sLpm) was generated from the compressed dry air with a zero air generator (Aadco, model 737-12). Water vapor was introduced into the flow tube by passing the dry purified air through a flask containing 60 °C nanopure water (>18 MΩ) followed by a saturator. The saturator was water jacketed and maintained at a constant temperature to control RH in the flow tube. A dew point monitor (General Eastern, model M3) was placed downstream of the flow tube to measure RH.
For the H2SO4 + DMA experiments, initial concentrations of H2SO4 and DMA were estimated to be 2.5 × 1010 and 8.9 × 1010 cm−3, respectively, under both dry and humid conditions. For the H2SO4 + NH3 experiments, initial concentrations of H2SO4 and NH3 were estimated to be 1.3 × 1010 and 2.5 × 1012 cm−3, respectively, under both dry and humid conditions. The initial gas phase acid:base ratio was below bulk sample thermodynamic equilibrium values of 1:2 (i.e., excess base was always present) in both H2SO4 + DMA and H2SO4 + NH3 experiments. The total flow rate through the flow tube was 4 sLpm, resulting in an average residence time of 72 s. It should be noted that additional wall losses occur within the flow tube for both H2SO4 (most likely, irreversible) and base compounds (possibly irreversible due to reactive uptake by adsorbed H2SO4). Based on a prior study that employed a flow tube with a similar diameter but twice the length and twice the flow rate, with similar residence time (Brus et al. Citation2010), we estimate total wall loss of H2SO4 to be 50–66% of the initial concentration over the entire length of the flow tube.
The size distribution of particles emerging from the flow tube was measured continually with a scanning mobility particle sizer (SMPS) consisting of a nano-differential mobility analyzer (nano-DMA; model 3085, TSI, Inc.) and a butanol-based ultrafine condensation particle counter (CPC; model 3025, TSI, Inc.). The minimum detectable diameter of the CPC, as reported by the manufacturer for 50% detection efficiency, is ∼2.5 nm.
Nanoparticle composition measurements
A TDCIMS was used to measure the size-resolved chemical composition of newly formed particles. The instrument has been previously described in detail (Voisin et al. Citation2003; Smith et al. Citation2004; Lawler et al. Citation2014). Briefly, aerosol particles were sampled continually from the flow tube reactor at 1.5 Lpm. Particles in the sampled flow were charged by a unipolar charger (UPC) (Chen and Pui Citation1999; McMurry et al. Citation2009). The charged particles were then size-selected by a nano-DMA and collected by electrostatic deposition onto a Pt filament. The collection potential of the filament was set to 4000 VDC. The collection time ranged from 30–120 min, depending on the particle concentration and collection efficiency. After collection, the Pt filament was translated into the ionization region of the mass spectrometer, where the filament was resistively heated under atmospheric pressure from room temperature to ∼550 °C to desorb and/or decompose particle-phase species. The volatilized molecules were ionized by chemical ionization and detected using a high-resolution time-of-flight mass spectrometer (HTOF; Tofwerk AG). The heating of the Pt filament was programmed to be at constant ambient temperature for the first 10 s, ramp up to ∼550 °C for the next 40 s, maintain at ∼550 °C for 15 s, and then return to ambient temperature for 5 s. The timespan of the heating cycle was 70 s for each collected sample. The filament and the ionization region were continually purged with N2 to minimize contamination. Instrument background was assessed after each collection by performing the same procedure but without applying a collection potential to the Pt filament. The TDCIMS can operate in either positive or negative ion mode, and only one polarity can be monitored for each collection. Base-related species were detected in positive ion mode and H2SO4-related species in negative ion mode. Chemical ionization reagent ions were generated directly in the ion source using a 210Po source. Trace amounts of H2O and O2 in the N2 flow formed (H2O)nH+ and (H2O)nO2−, which served as reagent ions in positive and negative ion modes (with n = 1–3), respectively. All ion signals reported were corrected for the background and normalized to reagent ion abundance. The TDCIMS data are presented as sums of background-corrected, detected ions integrated over the whole desorption period for each collected sample.
The TDCIMS was periodically calibrated using laboratory-generated, 20 nm diameter dimethylaminium sulfate (DMAS) and ammonium sulfate (AS) particles. The DMAS and AS particles were generated by atomizing dilute DMAS and AS solutions, respectively, using a constant output atomizer (model 3076; TSI, Inc.). The DMAS solution was made by stoichiometrically mixing DMA (Aldrich, 40 wt. % in water) and H2SO4 (Fisher Scientific, 96.2%) in nanopure water. The AS solution was made by dissolving ammonium sulfate (Alfa Aesar, 99%) powder in nanopure water. Acid:base ratios for particles generated during the flow tube experiments were determined by comparing the measured ion ratios with those of atomized DMAS and AS particles. For the atomized particles, we assign TDCIMS-measured acid:base ratio to the ratio corresponding to fully neutralized salts (0.5). We have confirmed that 20 nm diameter AS particles were close to fully neutralized with an acid:base ratio of ∼0.60 ± 0.05 by performing ion chromatography on particle samples that were atomized and then collected on aluminum foil disks using a sequential spot sampler (model SSS110, Aerosol Devices, Inc.).
Model description
A series of calculations were performed to provide a mechanistic explanation for the TDCIMS-based observations. Neutral and ionized fractions of acid and base at thermodynamic equilibrium were estimated by numerically solving a system of non-linear equations including: mass balance, electroneutrality, and acid dissociation (Ka). Activity coefficients of both the ionic and neutral species were neglected. While consideration of activity coefficients would change the absolute pH values and ratios presented, it would not change the conclusions regarding the likely mechanisms responsible for the observed acid:base ratios. Further, realistic consideration of activity coefficients requires knowledge of the dielectric constant of the solvent, which is arguably unknown in these particles. A bulk aqueous solution was assumed for the base case calculations, thus pKa values for H2SO4, NH3, and DMA in water were used (Barsanti et al. (Citation2009) and references therein). For the H2SO4-NH3 base case, a comparison of our results with those obtained using the online Extended Aerosol Inorganics model (“CitationE-AIM Model”; Clegg et al. Citation1998; Wexler and Clegg Citation2002), the latter of which includes activity coefficient corrections, demonstrates <1% difference in calculated moles of the major ions (SO42−, NH4+). The activity coefficients of the neutral species were close to unity in the aqueous solution, while the activity coefficients of the ionic species were <1. Sensitivity cases were also explored for both H2SO4-NH3 and H2SO4-DMA, in which the pKa values and acid:base ratios were varied to represent deviations in acid dissociation equilibria from a bulk aqueous solution.
Results and discussion
Size distributions of H2SO4-DMA and H2SO4-NH3 nanoparticles
shows size distributions of H2SO4-DMA () and H2SO4-NH3 () particles generated in the flow tube. The reactions of H2SO4 with DMA or NH3 effectively formed particles under both dry conditions and at 60% RH. Particles were formed in the diameter range of 4–40 nm with a geometric mean mobility diameter of 13.5 nm in the H2SO4-DMA system, and in the diameter range of 3–18 nm with a geometric mean mobility diameter of 8.5 nm in the H2SO4-NH3 system. Compared to the dry conditions, the presence of water vapor enhances particle number concentration by a factor of 1.3 and 2.1 for the H2SO4-DMA and H2SO4-NH3 systems, respectively, suggesting that water plays an important role in particle nucleation. Although H2SO4-base particles are highly hygroscopic, the diameters of particles generated under humid conditions do not show significant increases compared with those formed under dry conditions. The similarity in the measured particle size distributions under dry and humid conditions most likely represents the competition of gas-phase precursors between nucleation and nanoparticle growth. Higher levels of H2SO4 and base participating in nucleation under humid conditions resulted in fewer precursors available in the flow tube for nanoparticle growth.
Figure 2. Size distributions of particles measured by SMPS from the reactions of (a) 2.5 × 1010 cm−3 H2SO4 with 8.9 × 1010 cm−3 DMA, and (b) 2.5 × 1010 cm−3 H2SO4 with 1.3 × 1012 cm−3 NH3 under dry conditions and at 60% RH.
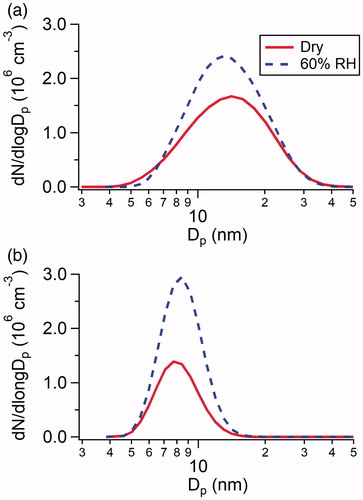
The enhancing effect of water on nucleation has previously been observed in laboratory experiments in the H2SO4-base systems (Berndt et al. Citation2010; Erupe et al. Citation2011; Zollner et al. Citation2012; Yu et al. Citation2017). Theoretical calculations showed that some clusters in the H2SO4-NH3 and H2SO4-DMA systems are stabilized by water, and could be more important in cluster growth pathways (Loukonen et al. Citation2010; Henschel et al. Citation2014). However, an inhibiting effect of water vapor on NPF has been postulated to explain field observations (Sihto et al. Citation2006; Laaksonen et al. Citation2008), presumably caused by other indirect effects. For example, combining field measurements and aerosol dynamics model simulations, Hamed et al. (Citation2011) attributed the inhibiting effect of water on NPF to the fact that solar radiation, which drives photochemistry and leads to the H2SO4 formation, usually peaks at noon when RH is low.
The formation and growth of particles in the flow tube were very stable (Figure S1). This enabled us to continually collect particles to explore their size-resolved chemical composition over a long period of time. The results of these size-resolved composition measurements are presented next.
Ions detected by TDCIMS and their desorption profiles
Particles collected onto the Pt filament started to desorb as the temperature of the filament was ramped from ambient temperature to ∼550 °C. shows desorption profiles of ions in negative and positive ion modes during typical heating cycles for particle samples collected from the H2SO4-DMA ( and ) and H2SO4-NH3 ( and ) systems. For both systems, ions including SO3−, HSO4−, and SO5− appeared in the negative ion mode as the temperature of the Pt filament increased. Ion intensities increased with time due to the increasing temperature of the Pt filament, and then decreased due to the sample depletion. All of the detected negative ions show multiple peaks throughout the thermal evolution of the collected samples. The HSO4− ions first appeared at a lower temperature, and this early peak is likely attributable to sulfuric acid. Detected HSO4− ions later in the desorption arise from a combination of H2SO4 re-volatilization from the ion source walls and further decomposition of salts on the wire, the latter of which is described in detail by Kiyoura and Urano (Citation1970). Overall, HSO4− was a minor component of the late desorption products. The SO3− ion dominated the later generated ions as the Pt filament was ramped to a higher temperature, followed by SO5− formed by the reaction of SO3 with the reagent ion O2−. Later generated ions of SO3− and SO5− showed similar desorption patterns, and they are attributed to the thermal decomposition of sulfate salts. Desorption profiles of atomized DMAS and AS particles in the calibration experiments are shown in Figure S2 for comparison. Ions including SO3−, SO5−, and HSO4− were observed in the negative ion mode in the calibration TDCIMS spectra. Their desorption profiles are similar to particles generated in the flow tube.
Figure 3. Typical desorption profiles of ions formed by heating H2SO4-DMA (a, c) and H2SO4-NH3 (b, d) particles collected on the Pt filament. The top panels show ions collected in the negative ion mode. The lower panels show ions collected in the positive ion mode. The dashed lines show the evolution of the filament temperature.
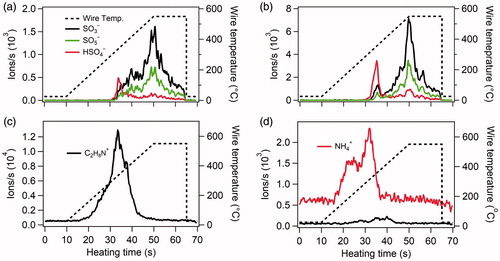
Dimethylammonium (C2H6NH2+ or DMAH+) and ammonium (NH4+) ions were detected as the main ions in the positive ion mode in the H2SO4-DMA () and H2SO4-NH3 () systems, respectively. An elevated level of NH4+ was observed even before heating the Pt filament, suggesting that NH4+ started to desorb from particle samples immediately after the Pt filament was moved into the ionization region, whose surfaces were kept at 60 °C to reduce partitioning of semi-volatile compounds. While NH4+ was observed before heating the Pt filament for particles formed in the H2SO4-NH3 system, atomized AS particles used for calibration showed a similar trend (Figure S2). Since both experiment and calibration analyses were conducted under identical operating conditions, we omitted NH4+ ions produced prior to the heating of the Pt filament in our calculation of total ion signal.
Trace amounts of DMAH+ were observed as a contaminant in H2SO4-NH3 particles and did not disappear even after the TDCIMS inlet system was thoroughly cleaned. Atomized AS particles (Figure S2b) showed no significant DMAH+, indicating that the contaminant was not from the TDCIMS inlet system, but from gas-phase DMA residue adsorbed on walls of the flow tube from earlier H2SO4-DMA experiments. The DMAH+ signal was ∼10% of the NH4+ signal and did not show a clear dependence on particle size. Given that the TDCIMS has higher sensitivity to DMAH+ than to NH4+ (Figure S2c), the lower intensity of the DMAH+ signal indicates that the presence of this ion does not significantly impact the interpretation of our measurements.
Acid:base ratio of H2SO4-NH3 and H2SO4-DMA nanoparticles
shows size-resolved acid:base ratio of H2SO4-DMA particles formed under dry conditions and at 60% RH in excess of DMA. The acid:base ratio of H2SO4-DMA particles at 8.6 nm formed under dry conditions is 0.7 ± 0.1; all ratios presented here are averages and the uncertainties are represented by the standard deviation obtained from repeated experiments. The ratio decreases as the size increases, reaching a minimum of ∼0.2 at 10.6 nm, and then increases again with increasing particle size until the neutralized state is reached (acid:base ratio = 0.5). An acid:base ratio of 0.2 suggests that excess DMAH+ was present in the 10.6 nm H2SO4-DMA particles. The acid:base ratio for 20 nm particles was 0.5 ± 0.1, indicating that particles became fully neutralized once they grew to 20 nm under dry conditions. Particles formed at 60% RH show a different trend compared to those formed under dry conditions. Smaller particles at 9.5 nm were acidic, showing an acid:base ratio of 1.3 ± 0.3. The ratio rapidly decreases with size, and reaches ∼0.5 at 12 nm. Particles larger than 12 nm were fully neutralized by DMA.
Figure 4. Size resolved acid:base ratio of newly formed particles in the H2SO4-DMA system under dry conditions and at 60% RH. The concentrations of H2SO4 and DMA introduced into the flow tube reactor were 2.5 × 1010 and 8.9 × 1010 cm−3, respectively, for both the dry conditions and at 60% RH. The error bars represent standard deviations of at least three repeated measurements. The horizontal black line represents the acid:base ratio of fully neutralized H2SO4-DMA particles.
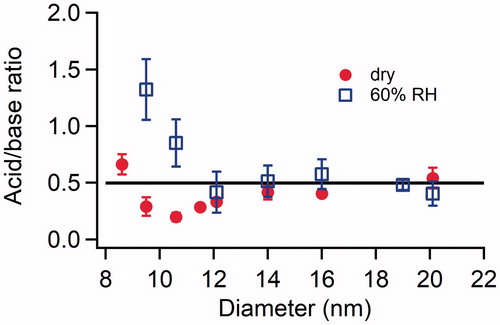
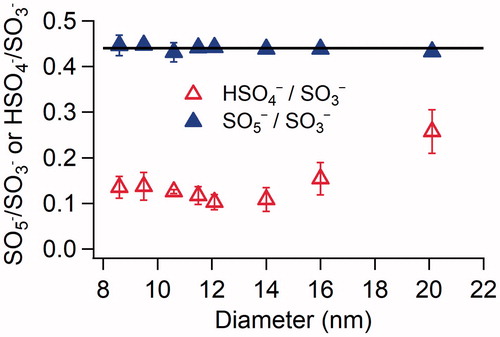
To further explore the size-dependent acid:base ratio of H2SO4-DMA particles, the ratios of different negative ions integrated over the desorption period were compared as shown in . Total ions of SO3− and SO5− are linearly correlated, showing a SO3−:SO5− ratio of ∼0.44 regardless of the particle size. The good linear correlation of SO3− and SO5− suggests their common sources, i.e., the thermal decomposition of sulfate salts as discussed earlier. The HSO4−:SO3− ratio, however, shows a strong dependence on size. The ratio decreases with size in the size range of 8.6–14 nm, and increases with size in the size range of 14–21 nm. While HSO4− was mainly from particle-phase H2SO4 and/or bisulfate salts, the higher HSO4−:SO3− ratio indicates that the smallest particles contain more H2SO4 and/or bisulfate salts, which decrease and then increase as particles grow. The trend is qualitatively similar to the size-dependent acid:base ratio shown in and provides a possible explanation for the observed variability.
Figure 5. Ratios of SO5−:SO3− and HSO4−:SO3− as a function of particle size for newly formed particles in the H2SO4-DMA system under dry conditions. The concentrations of H2SO4 and DMA introduced into the flow tube reactor were 2.5 × 1010 and 8.9 × 1010 cm−3, respectively. The error bars represent standard deviations of at least three repeated measurements. The horizontal black line represents a SO5−:SO3− ratio of 0.44.
The size-resolved acid:base ratio of H2SO4-NH3 particles generated under dry conditions and at 60% RH are shown in . For both the dry conditions and 60% RH, 7 nm diameter H2SO4-NH3 particles were highly acidic, showing an acid:base ratio that ranged from 3.1 ± 0.6 to 3.4 ± 0.3. The ratio gradually decreased as particles grew from 7 nm to 12 nm, reaching ∼2 and ∼1 for 12 nm particles generated under dry conditions and at 60% RH, respectively. Unlike H2SO4-DMA particles shown in , H2SO4-NH3 particles did not reach bulk sample thermodynamic equilibrium even for particles at the largest size collected in the experiment. Particles generated in the H2SO4-NH3 system were much smaller than those generated in the H2SO4-DMA system (), and may not have grown enough to reach bulk sample thermodynamic equilibrium for the experimental conditions that were employed in this study.
Figure 6. Size resolved acid:base ratio of newly formed particles formed in the H2SO4-NH3 system under dry conditions and at 60% RH. The concentrations of H2SO4 and NH3 introduced into the flow tube reactor were 2.5 × 1010 and 1.3 × 1012 cm−3, respectively, for both the dry conditions and 60% RH. The error bars represent standard deviations of at least three repeated measurements. The horizontal black line represents the acid:base ratio of fully neutralized H2SO4-DMA particles.
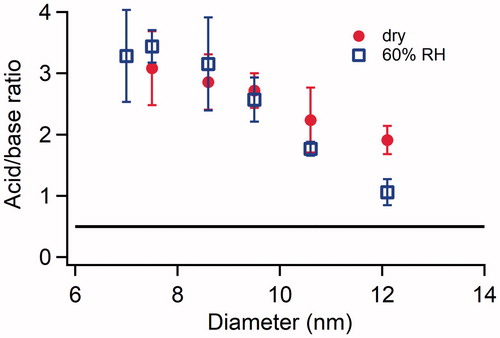
For both H2SO4-DMA and H2SO4-NH3 systems, the TDCIMS results indicate that smallest particles generated either under dry conditions or at 60% RH were acidic and did not reach a fully neutralized state even in excess of base. The base fraction in particles increased with size, but the incorporation of the base into particles shows different trends among individual experiments under different conditions.
It is important to consider the potential role that particulate water may play in these experiments. At 60% RH, H2SO4-NH3 nanoparticles always contain liquid water since their measured acid:base ratio is greater than that of ammonium bisulfate, the latter of which has a deliquescence RH of 40% (Tang and Munkelwitz Citation1977). As H2SO4-NH3 particles become more acidic, they increase in hygroscopicity and the deliquescence point eventually disappears completely. It is, therefore, possible that some H2SO4-NH3 particles may contain residual water even under “dry” conditions. In contrast, experiments with H2SO4-DMA particles show no clear deliquescence point over all observed acid:base ratios. Micron-sized particles with acid:base of 1:2 show a 50–75% increase in mass when exposed to 60% RH, and particles may contain some residual water at sub-3% RH (Chan and Chan Citation2013; Rovelli et al. Citation2017). Unlike the H2SO4-NH3 system, an aminium bisulfate particle is slightly less hygroscopic than its sulfate counterpart (Sauerwein et al. Citation2015). In summary, under “dry” conditions, particles from both systems may contain some residual water that may depend on acid:base ratio, whereas at 60% RH they likely contain significant water over all observed acid:base ratios.
During particle collection, particle-phase bases may be preferentially lost from the Pt filament compared to particle-phase H2SO4. This would lead to artificially high acid:base ratios. To investigate this possible artifact, a control experiment was carried out in which the collection time of particles was varied for the same size of H2SO4-DMA particles. This method was used previously to determine particulate compounds that were of intermediate volatility for nanoparticles generated by α-pinene ozonolysis (Winkler et al. Citation2012). The TDCIMS signals for both negative and positive ions showed a linear correlation with collection time (Figure S3), suggesting that losses of particle-phase H2SO4 and base from the Pt filament are minimal. Previous studies on ammonium or dimethylaminium sulfate aerosols also showed that the TDCIMS does not exhibit a strong tendency to lose bases prior to analysis (Lawler et al. Citation2016).
Another potential artifact that would affect observed acid:base ratios is that particles collected on the Pt filament were contaminated by H2SO4 that desorbed from the walls of the TDCIMS inlet system. However, the H2SO4 signal did not significantly change even after the TDCIMS inlet system was thoroughly cleaned. TDCIMS data for the calibration aerosol particles did not show any indication of H2SO4 contamination. We, therefore, conclude that contamination of H2SO4 was not an issue in the TDCIMS analysis.
The experimental evidence presented above supports the observation that the growth of nanoparticles above 3 nm differs from that of H2SO4-base clusters, which were proposed to grow through a stepwise addition of H2SO4 and base molecules (Kirkby et al. Citation2011; Almeida et al. Citation2013; Schobesberger et al. Citation2013; Bianchi et al. Citation2014; Kurten et al. Citation2014; Schobesberger et al. Citation2015). The size-dependent acid:base ratio of particles is consistent with previous observations in the CLOUD7 experiments (Kim et al. Citation2016; Lawler et al. Citation2016). The heterogeneous oxidation of SO2 (the H2SO4 precursor introduced into the CLOUD chamber) was offered as a possible explanation for the high H2SO4 in small particles in previous CLOUD7 experiments (Lawler et al. Citation2016). In this study, however, gas-phase H2SO4 was directly introduced into the flow tube, ruling out such a source of aerosol mass.
If oxidative kinetics is not the reason for the unexpected acid:base ratios, other factors must explain the deviations of the newly formed nanoparticles from bulk solution chemistry. One possibility is a kinetic limitation for the uptake of ammonia or amine. There is some evidence for the presence of an activation barrier to ammonia incorporation in charged clusters of ammonium and sulfate (Bzdek et al. Citation2013). While such a barrier for nanoparticles may explain their high acidity, it does not explain the size dependence of the acid:base ratio. In addition, thermodynamic modeling presented below suggests that a mere reduction in the particulate base cannot explain the size-dependent behavior of the HSO4−:SO3− ratio. Finally, the effect of an activation barrier to uptake would be most apparent when concentrations of base and sulfuric acid were of similar magnitude, whereas for our experiments with ammonia and sulfuric acid, the ammonia concentrations exceeded sulfuric acid levels by two orders of magnitude.
Aerosol phase state is known to play a major role in the equilibrium particle composition reached in systems of amines, ammonia, and sulfuric acid, due to the much more facile exchange for liquid particles compared with solid particles. In a study of micron-sized dimethylaminium sulfate particles, the experimental study by Chan and Chan (Citation2013) found that these particles were in a liquid phase, even at sub-3% RH. Cheng et al. (Citation2015) highlighted the challenge of predicting the phase of aerosol nanoparticles and concluded that below some threshold size (dependent on material, temperature, and water content) nanoparticles will be liquid. Specifically, their measurements of particle hygroscopicity suggested that ammonium sulfate nanoparticles are “molten” in the diameter range of 4–10 nm, depending on assumptions regarding the amount of residual particulate water. Lawler et al. (Citation2016) applied the assumption that nanoparticles formed in the ammonia-dimethylamine-sulfuric acid system were liquid and presented arguments for why a liquid phase may result in the measurement of more acidic nanoparticles. In the following section, we similarly assume that the present particles are “aqueous,” a term that we interpret as meaning a liquid state with varied amounts of residual water. With this assumption, we explore the possible roles of deviations of acid dissociation constants from bulk behavior using theoretical modeling.
Thermodynamic modeling of H2SO4-NH3 and H2SO4-DMA nanoparticles
The use of aqueous acid dissociation constants to describe thermodynamic equilibrium in the experimental systems may be insufficient, leading to the deviation between the observed size-dependent acid:base ratio and predictions based on bulk aqueous theory. It is known that individual pKa values vary with ionic strength and solvent dielectric constant (Reijenga et al. Citation2013; Farrokhpour and Manassir Citation2014; Hartono et al. Citation2014). Both ionic strength and solvent dielectric constant likely are size-dependent, as they depend on the amount of condensed-phase water and other components present in particles. Changes in electrostatic interactions, polarization, charge delocalization, and mixing state in nanoparticles may also lead to deviations in effective pKa values from bulk aqueous values.
Descriptions of the thermodynamic modeling cases and the results are shown in . In the Base Case and Sensitivity Case I, the acid:base ratio was assumed to be 0.5 to allow a fully neutralized solution and gas/particle partitioning was neglected. Relative to the Base Case, the pKa values were modified by 3.5 units in Sensitivity Case I, such that the H2SO4 becomes less acidic (pKa value increases by 3.5) and the bases become less basic (pKa value decreases by 3.5). Changing the pKa values accounts for the possibility of strongly inhibited proton transfer in nanoparticles due to steric or mixing effects. Compared to the Base Case, modifying the pKa values of the acid and the bases by 3.5 units results in an increase in the ratio of acidic to basic ions (([HSO4−] + [SO42−]): [BH+]), and a decrease in the ratio of SO42-:HSO4− (Sensitivity Case I, ). The deviation in the ratio of ([HSO4−] + [SO42-] to [BH+] is greater in H2SO4-NH3 particles than H2SO4-DMA particles. These modeling results reflect the observed trends in the laboratory measurements.
Table 1. Thermodynamic modeling of H2SO4-NH3 and H2SO4-DMA particles.
The ratio of protonated form of base to the total base ([BH+]:CB) in Sensitivity Case I () was less than 1, indicating the presence of neutral bases in the condensed phase. However, the relatively high Kelvin-corrected vapor pressures of NH3 and DMA preclude their presence as neutral compounds in nanoparticles. Thus, in Sensitivity Case II, the relative concentration of acid:base was assumed to be 2:1 to reflect the decreased availability of base that partitions from the particle phase to the gas phase. In Sensitivity Case II, the ratio of ([HSO4−] + [SO42−]):[B+] further increases, and the ratio of SO42−:HSO4− decreases, moving towards a better agreement with laboratory measurements.
In summary, the model best represents the laboratory measurements when: (1) the acid and the bases are assumed to be less efficient at acid-base chemistry (represented by a change in pKa values of 3.5 units); and (2) the fraction of the base in its neutral form is allowed to partition back to the gas phase and is not longer available to participate in acid-base chemistry. These could be the reasons for the strong size-dependent composition gradient of H2SO4-base nanoparticles observed in the experiments.
Conclusions
In summary, we measured the size-resolved chemical composition of newly formed particles in H2SO4-NH3 and H2SO4-DMA systems using TDCIMS. A strong size-dependent acid:base ratio in these particles was found, consistent with previous observations in the CLOUD7 experiments (Kim et al. Citation2016; Lawler et al. Citation2016). Smaller particles were generally more acidic and did not reach bulk sample thermodynamic equilibrium even in excess of gas-phase base. The acidity of particles decreased as particles grew to a larger size, but the incorporation of base into particles varied with base species, relative humidity, and particle size. Particles reached a fully neutralized state as they grew to 12 nm in the case of DMA. In contrast, 12 nm particles generated from H2SO4-NH3 were still highly acidic. Model results suggest that acid-base chemistry may be less efficient in nanoparticles than in bulk aqueous solutions. Modifying pKa values of H2SO4 and base by 3.5 units resulted in more acidic particles, leading to results consistent with observations. Due to the less efficient acid-base chemistry, a larger fraction of particle-phase base species present relatively volatile neutral molecules, allowing them to partition into the gas phase and resulting in acidic particles.
Our results suggest that small newly formed particles whose growth are driven by acid-base chemistry are acidic, which in turn affects physicochemical properties of particles like hygroscopicity and mixing states. These acidic particles can enhance acid-catalyzed uptake of organics (Jang et al. Citation2002), oligomerization of organics within particles (Holmes and Petrucci Citation2006), and other gas uptake pathways that contribute to particle growth. The size-dependent acidity together with the Kelvin effect and the potential phase transition of nanoparticles complicates the growth process of particles. While bulk phase solution thermodynamics provides insights into the underlying causes of the observed size-dependence of acid:base ratios of H2SO4-DMA and H2SO4-NH3 particles, it does not allow us to determine the underlying causes of the interesting local minimum in the size-dependent acid:base ratio of particles generated by the H2SO4-DMA system under dry conditions. This will be the subject of future experimental and modeling work.
Supplemental Material
Download Zip (75.3 KB)Acknowledgments
The authors sincerely thank Peter McMurry for his significant and pioneering contributions to understanding atmospheric nanoparticle formation and growth.
Additional information
Funding
Related Research Data
References
- Almeida, J., Schobesberger, S., Kürten, A., Ortega, I. K., Kupiainen-Määttä, O., Praplan, A. P., Adamov, A., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Donahue, N. M., Downard, A., Dunne, E., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Guida, R., Hakala, J., Hansel, A., Heinritzi, M., Henschel, H., Jokinen, T., Junninen, H., Kajos, M., Kangasluoma, J., Keskinen, H., Kupc, A., Kurtén, T., Kvashin, A. N., Laaksonen, A., Lehtipalo, K., Leiminger, M., Leppä, J., Loukonen, V., Makhmutov, V., Mathot, S., McGrath, M. J., Nieminen, T., Olenius, T., Onnela, A., Petäjä, T., Riccobono, F., Riipinen, I., Rissanen, M., Rondo, L., Ruuskanen, T., Santos, F. D., Sarnela, N., Schallhart, S., Schnitzhofer, R., Seinfeld, J. H., Simon, M., Sipilä, M., Stozhkov, Y., Stratmann, F., Tomé, A., Tröstl, J., Tsagkogeorgas, G., Vaattovaara, P., Viisanen, Y., Virtanen, A., Vrtala, A., Wagner, P. E., Weingartner, E., Wex, H., Williamson, C., Wimmer, D., Ye, P., Yli-Juuti, T., Carslaw, K. S., Kulmala, M., Curtius, J., Baltensperger, U., Worsnop, D. R., Vehkamäki, H., and Kirkby, J. (2013). Molecular Understanding of Sulphuric Acid-Amine Particle Nucleation in the Atmosphere. Nature, 502:359–363.
- Ball, S. M., Hanson, D. R., Eisele, F. L., and McMurry, P. H. (1999). Laboratory Studies of Particle Nucleation: Initial Results for H2SO4, H2O, and NH3 Vapors. J. Geophys. Res., 104:23709–23718.
- Barsanti, K., McMurry, P., and Smith, J. (2009). The Potential Contribution of Organic Salts to New Particle Growth. Atmos. Chem. Phys, 9:2949–2957.
- Benson, D. R., Erupe, M. E., and Lee, S.-H. (2009). Laboratory-Measured H2SO4-H2O-NH3 Ternary Homogeneous Nucleation Rates: Initial Observations. Geophys. Res. Lett., 36: L15818. doi:10.1029/2009GL038728
- Berndt, T., Stratmann, F., Sipilä, M., Vanhanen, J., Petäjä, T., Mikkilä, J., Grüner, A., Spindler, G., Lee Mauldin Iii, R., Curtius, J., Kulmala, M., and Heintzenberg, J. (2010). Laboratory Study on New Particle Formation from the Reaction OH + SO2: Influence of Experimental Conditions, H2O Vapour, NH3 and the Amine Tert-Butylamine on the Overall Process. Atmos. Chem. Phys., 10:7101–7116.
- Bianchi, F., Praplan, A. P., Sarnela, N., Dommen, J., Kürten, A., Ortega, I. K., Schobesberger, S., Junninen, H., Simon, M., Tröstl, J., Jokinen, T., Sipilä, M., Adamov, A., Amorim, A., Almeida, J., Breitenlechner, M., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Hakala, J., Hansel, A., Heinritzi, M., Kangasluoma, J., Keskinen, H., Kim, J., Kirkby, J., Laaksonen, A., Lawler, M. J., Lehtipalo, K., Leiminger, M., Makhmutov, V., Mathot, S., Onnela, A., Petäjä, T., Riccobono, F., Rissanen, M. P., Rondo, L., Tomé, A., Virtanen, A., Viisanen, Y., Williamson, C., Wimmer, D., Winkler, P. M., Ye, P., Curtius, J., Kulmala, M., Worsnop, D. R., Donahue, N. M., and Baltensperger, U. (2014). Insight into Acid-Base Nucleation Experiments by Comparison of the Chemical Composition of Positive, Negative, and Neutral Clusters. Environ. Sci. Technol., 48:13675–13684.
- Bianchi, F., Tröstl, J., Junninen, H., Frege, C., Henne, S., Hoyle, C. R., Molteni, U., Herrmann, E., Adamov, A., Bukowiecki, N., Chen, X., Duplissy, J., Gysel, M., Hutterli, M., Kangasluoma, J., Kontkanen, J., Kürten, A., Manninen, H. E., Münch, S., Peräkylä, O., Petäjä, T., Rondo, L., Williamson, C., Weingartner, E., Curtius, J., Worsnop, D. R., Kulmala, M., Dommen, J., and Baltensperger, U. (2016). New Particle Formation in the Free Troposphere: A Question of Chemistry and Timing. Science, 352(6289):1109–1112.
- Brus, D., Hyvärinen, A. P., Viisanen, Y., Kulmala, M., and Lihavainen, H. (2010). Homogeneous Nucleation of Sulfuric Acid and Water Mixture: experimental Setup and First Results. Atmos. Chem. Phys., 10:2631–2641.
- Bzdek, B. R., DePalma, J. W., Ridge, D. P., Laskin, J., and Johnston, M. V. (2013). Fragmentation Energetics of Clusters Relevant to Atmospheric New Particle Formation. J. Am. Chem. Soc., 135:3276–3285.
- Bzdek, B. R., Ridge, D. P., and Johnston, M. V. (2010). Amine Exchange into Ammonium Bisulfate and Ammonium Nitrate Nuclei. Atmos. Chem. Phys., 10:3495–3503.
- Chan, L. P., and Chan, C. K. (2013). Role of the Aerosol Phase State in Ammonia/Amines Exchange Reactions. Environ. Sci. Techno., 47(11):5755–5762.
- Chen, D.-R., and Pui, D. H., (1999). A High Efficiency, High Throughput Unipolar Aerosol Charger for Nanoparticles. J. Nanopart. Res., 1:115–126.
- Chen, H., Varner, M. E., Gerber, R. B., and Finlayson-Pitts, B. J., (2016). Reactions of Methanesulfonic Acid with Amines and Ammonia as a Source of New Particles in Air. J. Phys. Chem. B., 120:1526–1536.
- Chen, M., Titcombe, M., Jiang, J., Jen, C., Kuang, C., Fischer, M. L., Eisele, F. L., Siepmann, J. I., Hanson, D. R., Zhao, J., and McMurry, P. H., (2012). Acid–Base Chemical Reaction Model for Nucleation Rates in the Polluted Atmospheric Boundary Layer. Proc. Natl. Acad. Sci., 109:18713–18718.
- Cheng, Y., Su, H., Koop, T., Mikhailov, E., and Pöschl, U. (2015). Size Dependence of Phase Transitions in Aerosol Nanoparticles. Nat. Commun, 6:5923.
- Clegg, S. L., Brimblecombe, P., and Wexler, A. S., (1998). Thermodynamic Model of the System H+−NH4+−Na+−SO42−−NO3−−Cl−−H2O at 298.15 K. J. Phys. Chem. A, 102:2155–2171.
- Dawson, M. L., Varner, M. E., Perraud, V., Ezell, M. J., Gerber, R. B., and Finlayson-Pitts, B. J., (2012). Simplified Mechanism for New Particle Formation from Methanesulfonic Acid, Amines, and Water via Experiments and Ab Initio Calculations. Proc. Natl. Acad. Sci., 109:18719–18724.
- Donahue, N. M., Ortega, I. K., Chuang, W., Riipinen, I., Riccobono, F., Schobesberger, S., Dommen, J., Baltensperger, U., Kulmala, M., Worsnop, D. R., and Vehkamaki, H., (2013). How Do Organic Vapors Contribute to New-Particle Formation? Faraday Discuss., 165:91–104.
- E-AIM Model. Retrieved January 1, 2018, from http://www.aim.env.uea.ac.uk/aim/aim.php
- Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petaja, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F. (2014). A Large Source of Low-Volatility Secondary Organic Aerosol. Nature, 506:476–479.
- Erupe, M. E., Viggiano, A. A., and Lee, S. H. (2011). The Effect of Trimethylamine on Atmospheric Nucleation Involving H2SO4. Atmos. Chem. Phys., 11:4767–4775.
- Facchini, M. C., Decesari, S., Rinaldi, M., Carbone, C., Finessi, E., Mircea, M., Fuzzi, S., Moretti, F., Tagliavini, E., Ceburnis, D., and O’Dowd, C. D. (2008). Important Source of Marine Secondary Organic Aerosol from Biogenic Amines. Environ. Sci. Technol., 42:9116–9121.
- Farrokhpour, H., and Manassir, M. (2014). Approach for Predicting the Standard Free Energy Solvation of H+ and Acidity Constant in Nonaqueous Organic Solvents. J. Chem. Eng. Data, 59:3555–3564.
- Finlayson-Pitts, B. J., and Pitts, J. N. Jr. (2000). Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications. Academic press, San Diego.
- Freshour, N. A., Carlson, K. K., Melka, Y. A., Hinz, S., Panta, B., and Hanson, D. R. (2014). Amine Permeation Sources Characterized with Acid Neutralization and Sensitivities of an Amine Mass Spectrometer. Atmos. Meas. Tech., 7:3611–3621.
- Ge, X., Wexler, A. S., and Clegg, S. L. (2011). Atmospheric Amines – Part I. A Review. Atmos. Environ., 45:524–546.
- Glasoe, W. A., Volz, K., Panta, B., Freshour, N., Bachman, R., Hanson, D. R., McMurry, P. H., and Jen, C. (2015). Sulfuric Acid Nucleation: An Experimental Study of the Effect of Seven Bases. J. Geophys. Res. Atmos., 120:1933–1950.
- Hamed, A., Korhonen, H., Sihto, S.-L., Joutsensaari, J., Järvinen, H., Petäjä, T., Arnold, F., Nieminen, T., Kulmala, M., Smith, J. N., Lehtinen, K. E. J. and Laaksonen, A. (2011). The Role of Relative Humidity in Continental New Particle Formation. J. Geophys. Res. Atmos., 116:D03202.
- Hartono, A., Saeed, M., Kim, I., and Svendsen, H. F. (2014). Protonation Constant (pKa) of MDEA in Water as Function of Temperature and Ionic Strength. Energy Procedia, 63:1122–1128.
- Hemmilä, M., Hellén, H., Virkkula, A., Makkonen, U., Praplan, A. P., Kontkanen, J., Ahonen, L., Kulmala, M., and Hakola, H. (2018). Amines in Boreal Forest Air at SMEAR II Station in Finland. Atmos. Chem. Phys., 18:6367–6380.
- Henschel, H., Navarro, J. C. A., Yli-Juuti, T., Kupiainen-Määttä, O., Olenius, T., Ortega, I. K., Clegg, S. L., Kurtén, T., Riipinen, I., and Vehkamäki, H. (2014). Hydration of Atmospherically Relevant Molecular Clusters: Computational Chemistry and Classical Thermodynamics. J. Phys. Chem. A., 118:2599–2611.
- Herrmann, E., Brus, D., Hyva¨Rinen, A.-P., Stratmann, F., Wilck, M., Lihavainen, H., and Kulmala, M., (2010). A Computational Fluid Dynamics Approach to Nucleation in the Water − Sulfuric Acid System. J. Phys. Chem. A., 114:8033–8042.
- Holmes, B. J., and Petrucci, G. A. (2006). Water-Soluble Oligomer Formation from Acid-Catalyzed Reactions of Levoglucosan in Proxies of Atmospheric Aqueous Aerosols. Environ. Sci. Technol., 40:4983–4989.
- Jang, M., Czoschke, N. M., Lee, S., and Kamens, R. M. (2002). Heterogeneous Atmospheric Aerosol Production by Acid-Catalyzed Particle-Phase Reactions. Science, 298:814–817.
- Jen, C. N., McMurry, P. H., and Hanson, D. R. (2014). Stabilization of Sulfuric Acid Dimers by Ammonia, Methylamine, Dimethylamine, and Trimethylamine. J. Geophys. Res. Atmos., 119:7502–7514.
- Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V.-M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipilä, M. (2015). Production of Extremely Low Volatile Organic Compounds from Biogenic Emissions: Measured Yields and Atmospheric Implications. Proc. Natl. Acad. Sci. USA., 112:7123–7128.
- Kerminen, V.-M., Lihavainen, H., Komppula, M., Viisanen, Y., and Kulmala, M. (2005). Direct Observational Evidence Linking Atmospheric Aerosol Formation and Cloud Droplet Activation. Geophys. Res. Lett., 32. :L14803. doi:10.1029/2005GL023130.
- Keskinen, H., Virtanen, A., Joutsensaari, J., Tsagkogeorgas, G., Duplissy, J., Schobesberger, S., Gysel, M., Riccobono, F., Slowik, J. G., Bianchi, F., Yli-Juuti, T., Lehtipalo, K., Rondo, L., Breitenlechner, M., Kupc, A., Almeida, J., Amorim, A., Dunne, E. M., Downard, A. J., Ehrhart, S., Franchin, A., Kajos, M. K., Kirkby, J., Kurten, A., Nieminen, T., Makhmutov, V., Mathot, S., Miettinen, P., Onnela, A., Petaja, T., Praplan, A., Santos, F. D., Schallhart, S., Sipila, M., Stozhkov, Y., Tome, A., Vaattovaara, P., Wimmer, D., Prevot, A., Dommen, J., Donahue, N. M., Flagan, R. C., Weingartner, E., Viisanen, Y., Riipinen, I., Hansel, A., Curtius, J., Kulmala, M., Worsnop, D. R., Baltensperger, U., Wex, H., Stratmann, F., and Laaksonen, A. (2013). Evolution of Particle Composition in CLOUD Nucleation Experiments. Atmos. Chem. Phys., 13:5587–5600.
- Kim, J., Ahlm, L., Yli-Juuti, T., Lawler, M., Keskinen, H., Tröstl, J., Schobesberger, S., Duplissy, J., Amorim, A., Bianchi, F., Donahue, N. M., Flagan, R. C., Hakala, J., Heinritzi, M., Jokinen, T., Kürten, A., Laaksonen, A., Lehtipalo, K., Miettinen, P., Petäjä, T., Rissanen, M. P., Rondo, L., Sengupta, K., Simon, M., Tomé, A., Williamson, C., Wimmer, D., Winkler, P. M., Ehrhart, S., Ye, P., Kirkby, J., Curtius, J., Baltensperger, U., Kulmala, M., Lehtinen, K. E. J., Smith, J. N., Riipinen, I., and Virtanen, A. (2016). Hygroscopicity of Nanoparticles Produced from Homogeneous Nucleation in the CLOUD Experiments. Atmos. Chem. Phys., 16:293–304.
- Kirkby, J., Curtius, J., Almeida, J., Dunne, E., Duplissy, J., Ehrhart, S., Franchin, A., Gagné, S., Ickes, L., Kürten, A., Kupc, A., Metzger, A., Riccobono, F., Rondo, L., Schobesberger, S., Tsagkogeorgas, G., Wimmer, D., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Downard, A., Ehn, M., Flagan, R. C., Haider, S., Hansel, A., Hauser, D., Jud, W., Junninen, H., Kreissl, F., Kvashin, A., Laaksonen, A., Lehtipalo, K., Lima, J., Lovejoy, E. R., Makhmutov, V., Mathot, S., Mikkilä, J., Minginette, P., Mogo, S., Nieminen, T., Onnela, A., Pereira, P., Petäjä, T., Schnitzhofer, R., Seinfeld, J. H., Sipilä, M., Stozhkov, Y., Stratmann, F., Tomé, A., Vanhanen, J., Viisanen, Y., Vrtala, A., Wagner, P. E., Walther, H., Weingartner, E., Wex, H., Winkler, P. M., Carslaw, K. S., Worsnop, D. R., Baltensperger, U., and Kulmala, M. (2011). Role of Sulphuric Acid, Ammonia and Galactic Cosmic Rays in Atmospheric Aerosol Nucleation. Nature, 476:429–433.
- Kiyoura, R., and Urano, K. (1970). Mechanism, Kinetics, and Equilibrium of Thermal Decomposition of Ammonium Sulfate. Ind. Eng. Chem. Proc. Des. Dev., 9:489–494.
- Korhonen, P., Kulmala, M., Laaksonen, A., Viisanen, Y., McGraw, R., and Seinfeld, J. H. (1999). Ternary Nucleation of H2SO4, NH3, and H2O in the Atmosphere. J. Geophys. Res., 104:26349–26353.
- Kreidenweis, S. M., Flagan, R. C., Seinfeld, J. H., and Okuyama, K. (1989). Binary Nucleation of Methanesulfonic Acid and Water. J. Aerosol Sci., 20:585–607.
- Kuang, C., McMurry, P. H., and McCormick, A. V. (2009). Determination of Cloud Condensation Nuclei Production from Measured New Particle Formation Events. Geophys. Res. Lett., 36:L09822.
- Kulmala, M., Lehtinen, K. E. J., and Laaksonen, A. (2006). Cluster Activation Theory as an Explanation of the Linear Dependence between Formation Rate of 3nm Particles and Sulphuric Acid Concentration. Atmos. Chem. Phys., 6:787–793.
- Kulmala, M., Vehkamäki, H., Petäjä, T., Dal Maso, M., Lauri, A., Kerminen, V. M., Birmili, W., and McMurry, P. H. (2004). Formation and Growth Rates of Ultrafine Atmospheric Particles: A Review of Observations. J. Aerosol Sci., 35:143–176.
- Kurten, A., Jokinen, T., Simon, M., Sipilä, M., Sarnela, N., Junninen, H., Adamov, A., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., Dommen, J., Donahue, N. M., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Hakala, J., Hansel, A., Heinritzi, M., Hutterli, M., Kangasluoma, J., Kirkby, J., Laaksonen, A., Lehtipalo, K., Leiminger, M., Makhmutov, V., Mathot, S., Onnela, A., Petäjä, T., Praplan, A. P., Riccobono, F., Rissanen, M. P., Rondo, L., Schobesberger, S., Seinfeld, J. H., Steiner, G., Tomé, A., Tröstl, J., Winkler, P. M., Williamson, C., Wimmer, D., Ye, P., Baltensperger, U., Carslaw, K. S., Kulmala, M., Worsnop, D. R., and Curtius, J. (2014). Neutral Molecular Cluster Formation of Sulfuric Acid-Dimethylamine Observed in Real Time under Atmospheric Conditions. Proc. Natl. Acad. Sci. USA., 111:15019–15024.
- Kurtén, T., Loukonen, V., Vehkamäki, H., and Kulmala, M., (2008). Amines Are Likely to Enhance Neutral and Ion-Induced Sulfuric Acid-Water Nucleation in the Atmosphere More Effectively than Ammonia. Atmos. Chem. Phys., 8:4095–4103.
- Laaksonen, A., Kulmala, M., Berndt, T., Stratmann, F., Mikkonen, S., Ruuskanen, A., Lehtinen, K., E., J., D., Maso, M., Aalto, P., Petäjä, T., Riipinen, I., Sihto, S. L., Janson, R., Arnold, F., Hanke, M., Ücker, J., Umann, B., Sellegri, K., O'Dowd, C. D., and Viisanen, Y. (2008). SO2 Oxidation Products Other than H2SO4 as a Trigger of New Particle Formation. Part 2: Comparison of Ambient and Laboratory Measurements, and Atmospheric Implications. Atmos. Chem. Phys, 8:7255–7264.
- Lawler, M. J., Winkler, P. M., Kim, J., Ahlm, L., Tröstl, J., Praplan, A. P., Schobesberger, S., Kürten, A., Kirkby, J., Bianchi, F., Duplissy, J., Hansel, A., Jokinen, T., Keskinen, H., Lehtipalo, K., Leiminger, M., Petäjä, T., Rissanen, M., Rondo, L., Simon, M., Sipilä, M., Williamson, C., Wimmer, D., Riipinen, I., Virtanen, A., and Smith, J. N. (2016). Unexpectedly Acidic Nanoparticles Formed in Dimethylamine–Ammonia–Sulfuric-Acid Nucleation Experiments at CLOUD. Atmos. Chem. Phys., 16:13601–13618.
- Lawler, M., Whitehead, J., O'Dowd, C., Monahan, C., McFiggans, G., and Smith, J. (2014). Composition of 15-85 Nm Particles in Marine Air. Atmos. Chem. Phys., 14:11557–11569.
- Lehtipalo, K., Rondo, L., Kontkanen, J., Schobesberger, S., Jokinen, T., Sarnela, N., Kürten, A., Ehrhart, S., Franchin, A., Nieminen, T., Riccobono, F., Sipilä, M., Yli-Juuti, T., Duplissy, J., Adamov, A., Ahlm, L., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., Dommen, J., Downard, A. J., Dunne, E. M., Flagan, R. C., Guida, R., Hakala, J., Hansel, A., Jud, W., Kangasluoma, J., Kerminen, V.-M., Keskinen, H., Kim, J., Kirkby, J., Kupc, A., Kupiainen-Määttä, O., Laaksonen, A., Lawler, M. J., Leiminger, M., Mathot, S., Olenius, T., Ortega, I. K., Onnela, A., Petäjä, T., Praplan, A., Rissanen, M. P., Ruuskanen, T., Santos, F. D., Schallhart, S., Schnitzhofer, R., Simon, M., Smith, J. N., Tröstl, J., Tsagkogeorgas, G., Tomé, A., Vaattovaara, P., Vehkamäki, H., Vrtala, A. E., Wagner, P. E., Williamson, C., Wimmer, D., Winkler, P. M., Virtanen, A., Donahue, N. M., Carslaw, K. S., Baltensperger, U., Riipinen, I., Curtius, J., Worsnop, D. R., and Kulmala, M. (2016). The Effect of Acid–Base Clustering and Ions on the Growth of Atmospheric Nano-Particles. Nat. Commun., 7:11594.
- Liu, Y., Han, C., Liu, C., Ma, J., Ma, Q., and He, H. (2012). Differences in the Reactivity of Ammonium Salts with Methylamine. Atmos. Chem. Phys., 12:4855–4865.
- Loukonen, V., Kurtén, T., Ortega, I. K., Vehkamäki, H., Pádua, A. A. H., Sellegri, K., and Kulmala, M. (2010). Enhancing Effect of Dimethylamine in Sulfuric Acid Nucleation in the Presence of Water - a Computational Study. Atmos. Chem. Phys., 10:4961–4974.
- McMurry, P. H., Ghimire, A., Ahn, H.-K., Sakurai, H., Moore, K., Stolzenburg, M., and Smith, J. N. (2009). Sampling Nanoparticles for Chemical Analysis by Low Resolution Electrical Mobility Classification. Environ. Sci. Technol., 43:4653–4658.
- McMurry, P. H., Shan Woo, K., Weber, R., Chen, D.-R., and Pui, D. Y. H. (2000). Size Distributions of 3–10 Nm Atmospheric Particles: implications for Nucleation Mechanisms. Philos. Trans. Royal Soc. A Math. Phys. Eng. Sci., 358:2625–2642.
- Merikanto, J., Spracklen, D. V., Mann, G. W., Pickering, S. J., and Carslaw, K. S. (2009). Impact of Nucleation on Global CCN. Atmos. Chem. Phys., 9:8601–8616.
- Neitola, K., Brus, D., Makkonen, U., Sipila, M., Mauldin, R. L., Sarnela, N., Jokinen, T., Lihavainen, H., and Kulmala, M. (2015). Total Sulfate Vs. sulfuric Acid Monomer Concenterations in Nucleation Studies. Atmos. Chem. Phys., 15:3429–3443.
- Panta, B., Glasoe, W. A., Zollner, J. H., Carlson, K. K., and Hanson, D. R. (2012). Computational Fluid Dynamics of a Cylindrical Nucleation Flow Reactor with Detailed Cluster Thermodynamics. J. Phys. Chem. A., 116:10122–10134.
- Pratt, K. A., Hatch, L. E., and Prather, K. A. (2009). Seasonal Volatility Dependence of Ambient Particle Phase Amines. Environ. Sci. Technol., 43:5276–5281.
- Qiu, C., Wang, L., Lal, V., Khalizov, A. F., and Zhang, R., (2011). Heterogeneous Reactions of Alkylamines with Ammonium Sulfate and Ammonium Bisulfate. Environ. Sci. Technol., 45:4748–4755.
- Reijenga, J., van Hoof, A., van Loon, A., and Teunissen, B. (2013). Development of Methods for the Determination of pKa Values. Anal. Chem. Insights., 8:53–71.
- Riccobono, F., Schobesberger, S., Scott, C. E., Dommen, J., Ortega, I. K., Rondo, L., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Downard, A., Dunne, E. M., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Hansel, A., Junninen, H., Kajos, M., Keskinen, H., Kupc, A., Kürten, A., Kvashin, A. N., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Nieminen, T., Onnela, A., Petäjä, T., Praplan, A. P., Santos, F. D., Schallhart, S., Seinfeld, J. H., Sipilä, M., Spracklen, D. V., Stozhkov, Y., Stratmann, F., Tomé, A., Tsagkogeorgas, G., Vaattovaara, P., Viisanen, Y., Vrtala, A., Wagner, P. E., Weingartner, E., Wex, H., Wimmer, D., Carslaw, K. S., Curtius, J., Donahue, N. M., Kirkby, J., Kulmala, M., Worsnop, D. R., and Baltensperger, U. (2014). Oxidation Products of Biogenic Emissions Contribute to Nucleation of Atmospheric Particles. Science, 344:717–721.
- Rovelli, G., Miles, R. E. H., Reid, J. P., and Clegg, S. L. (2017). Hygroscopic Properties of Aminium Sulfate Aerosols. Atmos. Chem. Phys., 17:4369–4385.
- Sauerwein, M., Clegg, S. L., and Chan, C. K. (2015). Water Activities and Osmotic Coefficients of Aqueous Solutions of Five Alkylaminium Sulfates and Their Mixtures with H2SO4 at 25oC. Aerosol Sci. Technol., 49:566–579.
- Schobesberger, S., Franchin, A., Bianchi, F., Rondo, L., Duplissy, J., Kürten, A., Ortega, I. K., Metzger, A., Schnitzhofer, R., Almeida, J., Amorim, A., Dommen, J., Dunne, E. M., Ehn, M., Gagné, S., Ickes, L., Junninen, H., Hansel, A., Kerminen, V. M., Kirkby, J., Kupc, A., Laaksonen, A., Lehtipalo, K., Mathot, S., Onnela, A., Petäjä, T., Riccobono, F., Santos, F. D., Sipilä, M., Tomé, A., Tsagkogeorgas, G., Viisanen, Y., Wagner, P. E., Wimmer, D., Curtius, J., Donahue, N. M., Baltensperger, U., Kulmala, M., and Worsnop, D. R. (2015). On the Composition of Ammonia-Sulfuric-Acid Ion Clusters during Aerosol Particle Formation. Atmos. Chem. Phys., 15:55–78.
- Schobesberger, S., Junninen, H., Bianchi, F., Lonn, G., Ehn, M., Lehtipalo, K., Dommen, J., Ehrhart, S., Ortega, I. K., Franchin, A., Nieminen, T., Riccobono, F., Hutterli, M., Duplissy, J., Almeida, J., Amorim, A., Breitenlechner, M., Downard, A. J., Dunne, E. M., Flagan, R. C., Kajos, M., Keskinen, H., Kirkby, J., Kupc, A., Kurten, A., Kurten, T., Laaksonen, A., Mathot, S., Onnela, A., Praplan, A. P., Rondo, L., Santos, F. D., Schallhart, S., Schnitzhofer, R., Sipila, M., Tome, A., Tsagkogeorgas, G., Vehkamaki, H., Wimmer, D., Baltensperger, U., Carslaw, K. S., Curtius, J., Hansel, A., Petaja, T., Kulmala, M., Donahue, N. M., and Worsnop, D. R. (2013). Molecular Understanding of Atmospheric Particle Formation from Sulfuric Acid and Large Oxidized Organic Molecules. Proc. Natl. Acad. Sci., 110:17223–17228.
- Seinfeld, J. H., and Pandis, S. N. (2006). Atmospheric Chemistry and Physics: From Air Pollution to Climate Change. John Wiley & Sons, New York.
- Sihto, S. L., Kulmala, M., Kerminen, V. M., Dal Maso, M., Petäjä, T., Riipinen, I., Korhonen, H., Arnold, F., Janson, R., Boy, M., Laaksonen, A., and Lehtinen, K. E. J. (2006). Atmospheric Sulphuric Acid and Aerosol Formation: implications from Atmospheric Measurements for Nucleation and Early Growth Mechanisms. Atmos. Chem. Phys., 6:4079–4091.
- Sipilä, M., Berndt, T., Petäjä, T., Brus, D., Vanhanen, J., Stratmann, F., Patokoski, J., Mauldin, R. L., Hyvärinen, A.-P., Lihavainen, H., and Kulmala, M. (2010). The Role of Sulfuric Acid in Atmospheric Nucleation. Science, 327:1243–1246.
- Smith, J. N., Barsanti, K. C., Friedli, H. R., Ehn, M., Kulmala, M., Collins, D. R., Scheckman, J. H., Williams, B. J., and McMurry, P. H. (2010). Observations of Aminium Salts in Atmospheric Nanoparticles and Possible Climatic Implications. Proc. Natl. Acad. Sci., 107:6634–6639.
- Smith, J. N., Moore, K. F., McMurry, P. H., and Eisele, F. L. (2004). Atmospheric Measurements of Sub-20 Nm Diameter Particle Chemical Composition by Thermal Desorption Chemical Ionization Mass Spectrometry. Aerosol Sci. Technol., 38:100–110.
- Sorooshian, A., Padró, L. T., Nenes, A., Feingold, G., McComiskey, A., Hersey, S. P., Gates, H., Jonsson, H. H., Miller, S. D., Stephens, G. L., Flagan, R. C., and Seinfeld, J. H. (2009). On the Link between Ocean Biota Emissions, Aerosol, and Maritime Clouds: Airborne, Ground, and Satellite Measurements off the Coast of California. Global Biogeochem. Cycles, 23: GB4007. doi:10.1029/2009GB003464
- Spracklen, D. V., Carslaw, K. S., Kulmala, M., Kerminen, V.-M., Sihto, S.-L., Riipinen, I., Merikanto, J., Mann, G. W., Chipperfield, M. P., Wiedensohler, A., Birmili, W., and Lihavainen, H. (2008). Contribution of Particle Formation to Global Cloud Condensation Nuclei Concentrations. Geophys. Res. Lett., 35:L06808.
- Tang, I. N., and Munkelwitz, H. R. (1977). Aerosol Growth Studies—III Ammonium Bisulfate Aerosols in a Moist Atmosphere. J. Aerosol Sci., 8:321–330.
- Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U. (2016). The Role of Low-Volatility Organic Compounds in Initial Particle Growth in the Atmosphere. Nature, 533:527.
- Voisin, D., Smith, J. N., Sakurai, H., McMurry, P. H., and Eisele, F. L. (2003). Thermal Desorption Chemical Ionization Mass Spectrometer for Ultrafine Particle Chemical Composition. Aerosol Sci. Technol., 37:471–475.
- Weber, R. J., Chen, G., Davis, D. D., Mauldin, I. I. I. R. L., Tanner, D. J., Eisele, F. L., Clarke, A. D., Thornton, D. C., and Bandy, A. R. (2001). Measurements of Enhanced H2SO4 and 3-4 Nm Particles near a Frontal Cloud during the First Aerosol Characterization Experiment (ACE 1). J. Geophys. Res., 106:24107–24117.
- Weber, R. J., Marti, J. J., McMurry, P. H., Eisele, F. L., Tanner, D. J., and Jefferson, A. (1996). Measured Atmospheric New Particle Formation Rates: Implications for Nulceation Mechanisms. Chem. Eng. Commun., 151:53–64.
- Weber, R. J., Marti, J. J., McMurry, P. H., Eisele, F. L., Tanner, D. J., and Jefferson, A. (1997). Measurements of New Particle Formation and Ultrafine Particle Growth Rates at a Clean Continental Site. J. Geophys. Res., 102:4375–4385.
- Wexler, A. S., and Clegg, S. L. (2002). Atmospheric Aerosol Models for Systems Including the Ions H+, NH4+, Na+, SO42−, NO3−, Cl−, Br−, and H2O. J. Geophys. Res., 107:4207.
- Winkler, P. M., Ortega, J., Karl, T., Cappellin, L., Friedli, H. R., Barsanti, K., McMurry, P. H., and Smith, J. N. (2012). Identification of the Biogenic Compounds Responsible for Size-Dependent Nanoparticle Growth. Geophys. Res. Lett., 39:L20815.
- Wyslouzil, B. E., Seinfeld, J. H., Flagan, R. C., and Okuyama, K. (1991). Binary Nucleation in Acid-Water Systems. I. Methanesulfonic Acid–Water. J. Chem. Phys., 94:6827–6841.
- Yu, F. (2006). Effect of Ammonia on New Particle Formation: A Kinetic H2SO4-H2O-NH3 Nucleation Model Constrained by Laboratory Measurements. J. Geophys. Res., 111:D01204.
- Yu, H., Dai, L., Zhao, Y., Kanawade, V. P., Tripathi, S. N., Ge, X., Chen, M., and Lee, S.-H. (2017). Laboratory Observations of Temperature and Humidity Dependencies of Nucleation and Growth Rates of Sub-3 Nm Particles. J. Geophys. Res. Atmos., 122:1919–1929.
- Yu, H., McGraw, R., and Lee, S.-H. (2012). Effects of Amines on Formation of Sub-3 Nm Particles and Their Subsequent Growth. Geophys. Res. Lett., 39:L02807. doi:10.1029/2011GL050099
- Zhang, R., Khalizov, A., Wang, L., Hu, M., and Xu, W. (2012). Nucleation and Growth of Nanoparticles in the Atmosphere. Chem. Rev., 112:1957–2011.
- Zollner, J. H., Glasoe, W. A., Panta, B., Carlson, K. K., McMurry, P. H., and Hanson, D. R. (2012). Sulfuric Acid Nucleation: Power Dependencies, Variation with Relative Humidity, and Effect of Bases. Atmos. Chem. Phys., 12:4399–4411.