Abstract
The potential use of sorbents to manage ultrafine ash aerosol emissions from residual oil combustion was investigated using a downfired 82 kW laboratory-scale refractory-lined combustor. The major constituents were vanadium (V), nickel (Ni), iron (Fe), and zinc (Zn). The overall ash content of residual oil is very low, resulting in total ash vaporization at 1725 K with appreciable vaporization occurring at temperatures as low as 1400 K. Therefore, the possibility of interactions between ash vapor and sorbent substrates exists. Kaolinite powder was injected at various locations in the combustor. Ash scavenging was determined from particle size distributions (PSDs) measured by a Scanning Mobility Particle Sizer. Impactor samples and X-ray fluorescence (XRF) analyses supported these data. Injection of kaolinite sorbent was able to capture up to 60% of all the ash in the residual fuel oil. However, captures of ∼ 30% were more common when sorbent injection occurred downstream of the combustion zone, rather than with the combustion air into the main flame. Without sorbent addition, baseline measurements of the fly ash PSD and chemical composition indicate that under the practical combustion conditions examined here, essentially all of the metals contained in the residual oil form ultrafine particles (∼0.1 μ m diameter). Theoretical calculations showed that coagulation between the oil ash nuclei and the kaolinite sorbent could account for, at most, 17% of the metal capture which was always less than that measured. The data suggest that kaolinite powders reactively capture a portion of the vapor phase metals. Mechanisms and rates still remain to be quantified.
INTRODUCTION
Airborne fine particulate matter (PM) is the subject of considerable environmental interest due to the results of a number of studies correlating short-term exposures to ambient levels of fine PM with acute adverse health effects (CitationWilson and Spengler 1996; CitationU.S. EPA 1996; CitationBachmann et al. 1996; CitationWolff 1996). Consequently, ambient concentrations and source emissions of PM smaller than 2.5 μm in aerodynamic diameter (PM2.5) face increased regulation (CitationFederal Register 1997). Numerous theories exist to explain the mechanisms causing physiological damage. Health effects studies have identified theories related to ambient particle composition that appear to exacerbate adverse health effects, including the presence of transition metals (e.g., vanadium (V), nickel (Ni), iron (Fe), zinc (Zn), and copper (Cu); CitationDreher et al. 1997) and aerosol acidity. In addition to particle composition, another apparent factor influencing health impacts is the presence of ultrafine particles (<0.1 μm diameter; CitationU.S. EPA 1996). All of these characteristics—transition metals, acidity, and ultrafine size—are exhibited by the PM generated from the combustion of residual fuel oils.
Whereas previous work by our group examined the physical and chemical characteristics of fine PM from residual fuel oil combustion (CitationMiller et al. 1998) and determined effects of combustor design on differences in emissions (CitationLinak et al. 2000), the research presented here focuses on the potential of an inorganic sorbent to reduce the emission of ultrafine metal ash nuclei from a residual oil-fired refractory-lined combustor. Particle size distributions (PSDs) and size-dependent chemical compositions were compared without and with sorbent injection. Experimental results were compared to predictions of an equilibrium chemistry model and also to an aerosol growth model. The primary metals contained in residual oil ash are V, Ni, Fe, and Zn. Previous research has indicated that injection of a sorbent can partially depress the mass of ultrafines emitted from residual oil combustion (CitationLinak et al. 2003), although the mechanisms governing the process are unclear.
The first objective of the current work was to investigate this process further by determining its response, in a practical combustion configuration, to various process parameters such as sorbent injection temperature and flow rate. With results from these parametric tests, it may be possible to determine whether the amount of ash captured could be increased from the low levels measured previously (CitationLinak et al. 2003), and consequently whether this approach has the potential to be a practical technology for controlling ultrafine residual fuel oil ash emissions from combustion sources. Another objective was to use parametric test results to shed new light on pertinent mechanisms.
While the occurrence of ash in residual fuel oils is quite different than that of coal, much can be learned from the results of the extensive studies on coal ash transformations. This research has been conducted by a large number of groups to predict the size-dependent composition of the ash aerosol evolving within pulverized coal combustion systems. The primary purpose of these efforts was to characterize and control the corrosion, fouling, slagging, and other detrimental effects ash species have on combustor materials and heat transfer surfaces. Data on the size-classified PM emission from full-scale coal combustion units were first published by CitationDavison et al. (1974), CitationKaakinen et al. (1975), CitationKlein et al. (1975), CitationGladney et al. (1976), CitationOndov et al. (1978, 1979), CitationNettleton (1979), and CitationSmith et al. (1979), to name but a few. These data indicated, in general, a bimodal particle size distribution and the conclusion that the submicron particles are enriched in a number of trace elements. Detailed mechanisms describing ash transformations and aerosol formation from pulverized coal combustion were first discussed by CitationSarofim et al. (1977), CitationFlagan and Friedlander (1978), and CitationFlagan (1979), and reviewed by CitationSmith (1980) and CitationDamle et al. (1982). These models propose that major ash inclusions inside the coal particle soften, melt, and coalesce during char combustion. As the carbon matrix recedes, these micron-scale structures are released to constitute the supermicron mode. A small fraction (∼1%) of the mineral matter, however, is vaporized. Most readily vaporized are ash species with higher vapor pressures than the common refractory oxides present in coal. The formation and subsequent vaporization of higher vapor pressure refractory metal suboxides due to the reducing environment around burning coal particles is used to account for their relatively high concentrations in the submicron aerosol.
In addition to full-scale field studies, numerous smaller scale combustion studies have made major advances in the understanding of inorganic ash transformations and aerosol formation from pulverized coal combustion. Early work by CitationSarofim et al. (1977), CitationMims et al. (1980), CitationNeville et al. (1981), CitationQuann et al. (1982), CitationHaynes et al. (1982), CitationQuann and Sarofim (1982), CitationNeville and Sarofim (1982), and CitationNeville et al. (1983) used well-defined drop-tube systems, while at the same time CitationFlagan and Taylor (1981) and CitationTaylor and Flagan (1982) used much larger laboratory-scale equipment. In general, these studies confirmed mechanisms of selective vaporization of volatile trace species and suboxides from the burning char particles, followed by nucleation or condensation of the less-volatile oxides and trace elements. Since then, a number of research groups have greatly expanded the scope of this research, so that today there exists a reasonable understanding of the mechanisms controlling the fate of ash species during pulverized coal combustion.
While the behavior of inorganic ash constituents within coal-fired power plants has been extensively studied, the study of residual oil combustion has focused primarily on the physical and chemical behavior of organic constituents, including the formation and combustion of large carbon cenosphere particles (CitationWilliams 1976). In fact, for small residual oil-fired process heaters and package boilers, particle emissions are often characterized by large (>10 μm diameter) cenospheric coke particles and high loss on ignition (LOI) values. This is in contrast to large utility-scale boilers where high thermal efficiencies and low values of LOI are more typical. For these systems, particle emissions are characterized by a large submicron ash mode and a much smaller unburned coke mode (CitationPiper and Nazimowitz 1985; CitationWalsh et al. 1991). CitationWalsh et al. (1991) show that, in contrast to pulverized coal, the majority of the sampled fly ash mass from residual fuel oil-fired utility boilers lies below 1 μm in diameter. This is consistent with the understanding that oil ash is primarily organically bound, while the major fraction of coal ash is contained within micron-scale mineral inclusions and excluded overburden (dirt) incorporated during mining (CitationGluskoter 1978). For coals, inherent (organically bound) ash typically constitutes only a small portion of the total. In general, the ash contents of residual oils are approximately 0.1%, while for coals ash contents are, in general, two orders of magnitude greater. For combustion typical of utility-scale residual oil boilers with low residual carbon in the fly ash, the majority of the emitted fly ash is found within particle sizes described by mechanisms of ash vaporization, nucleation, and growth (CitationLinak et al. 2000). At stack locations, these particles are present in a mode centered at ∼0.1 μm diameter. In contrast, typically less than 1% of coal fly ash is found in this ultrafine mode, with the vast majority being found in fragmentation modes, centered at ∼2 μm and ∼20 μm diameter (CitationLinak et al. 2002). The formation of primary ultrafine particles from U.S. utility coal and residual oil usage can be estimated using the information presented in (CitationU.S. DOE 1998; CitationKilgroe et al. 2002). These estimates indicate that for the U.S., coal combustion generates larger quantities of ultrafine PM than oil, but oil combustion generates larger quantities of ultrafine PM per equivalent unit of heat produced. Note that discussion here is for ultrafine PM, not PM2.5, which, for coal, is dominated by a fine fragmentation mode (CitationLinak et al. 2002). Clearly, as far as inorganic primary ultrafine particles are concerned coal still dominates, with 95.7% of total potential ultrafine emissions nationally, although the fraction of ultrafine to total coal ash is low. The potential release of oil ultrafines, however, is not negligible at 4.3% and may be even more important in particular locations. Residual oil fly ash also represents a prototype case for combustion-generated PM in which all of the mineral matter has been vaporized, without complications of possible interactions with the usually omnipresent alumino-silicate particles characteristic of coal combustion. Hence, combustion of residual fuel oil, with the systematic addition of alumino-silicate material downstream, allows interactions between organometallics and dispersed crystalline alumino-silicates to be investigated in a controlled and parametric manner, which was also the purpose of this work.
TABLE 1 Comparison of utility residual oil and coal usage and contribution of ash to ultrafine particle sizes (uncontrolled)
EXPERIMENTAL APPROACH
A new, vertically fired, 82 kW semi-industrial scale combustor shown in was the central experimental device used for this study. This rebuilt unit was based on the design of a previous horizontal unit that had been used for many years in this laboratory (CitationLinak et al. 1994). This 5.36 m tall modular, steel-shell, refractory-lined research combustor was designed to simulate the time–temperature histories typical of utility boilers. The 0.52 m inside diameter by 1.07 m long upper burner section equipped with an International Flame Research Foundation (IFRF) moveable-block variable air swirl burner provide for realistic near-flame fuel-air mixing processes, so that experimental results from this laboratory-scale unit might pertain directly to expected results from practical utility boilers. Following the burner section, the 0.27 m inside diameter lower sections are equipped with numerous access ports, permitting temperature measurement, gas and particle sampling, and the injection of sorbents. Temperatures, determined by a shielded type R thermocouple, ranged from a peak of ∼1750 K to 1300 K, 5 m and 2.6 s downstream. shows the relationship between measured temperatures, residence times, and location. This combustor was not designed to be an ideal reactor to study fundamental mechanisms pertaining to single particles. However, because the movable-block burner simulates the segregated air and fuel streams and subsequent contacting in practical residual oil flames, one could, for example, explore the effects of injecting metered amounts of sorbent into the air stream upstream of the burner, and still have confidence that results would replicate what might be expected in large practical boiler flames. In addition, as the postflame gases in this combustor were sufficiently laminar (Re ∼800), one could determine mean reaction times and temperatures for sorbent injected downstream of the flame and interpret the ensuing data in terms of fundamental mechanisms. Thus, the combustor was designed to yield experimental results that were both of immediate practical significance, as far as utility boilers are concerned, and also of use for elucidation of mechanisms.
FIG. 1 EPA downfired combustor with port temperatures and calculated residence times indicated. No. 6 residual fuel oil fired at a rate of 7.1 l/h.
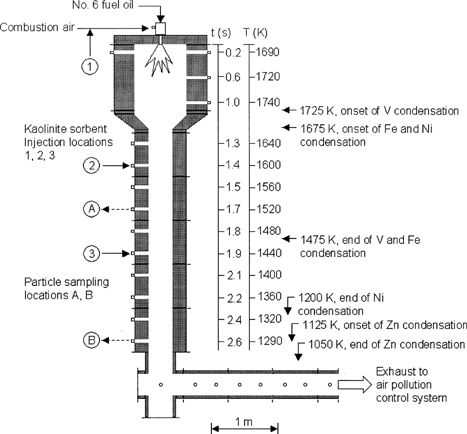
The IFRF burner was equipped with a Spraying Systems Co. (model Air Atom ¼-JSS) air-atomizing oil nozzle. Oil temperature and air pressure were maintained between 380–400 K and 200–240 kPa (gauge), respectively. Residual oil was fed at a rate of 7.1 l/h. These settings produced a relatively narrow residual oil droplet size distribution with a mean diameter between 30 and 40 μm. The burner was set to produce an IFRF Type 2 (Swirl Number = 1.48) turbulent diffusion flame. Powdered kaolinite sorbent (Burgess Pigment Co., Sandersville, GA, USA; No. 40, nominally 1.4 μm diameter) was introduced into the combustor using a K-Tron twin-screw loss-in-weight feeder and air eductor (model KCL 2420). Transport air introduced at the screw exit entrained the kaolinite and carried it into the combustor at one of three locations, namely into the air at the burner before combustion, where it would see the maximum temperature measured, or at two postflame locations corresponding to temperatures of 1600 and 1430 K (see ). Physical properties and chemical composition of the kaolinite used, as well as the No. 6 fuel oil, are shown in . Sorbent feed rates are reported based on sorbent equivalence ratio, Φ, defined as g-moles Al2O3-2SiO2 per g-moles of V + Ni + Fe + Zn. For this study, target values for Φ varied from 0 to 15. It should be noted, however, that measured sorbent concentrations at the exit of the furnace were always considerably lower that those directly based on feeder weight loss. The fine kaolinite powder used proved to be exceedingly difficult to handle, and was easily deposited within probes, transfer lines, and other surfaces. In fact, once sorbent had been fed into the system, a small residual amount seemed to be lodged in crevices and ledges, and continued to enter the flue gas for quite some time after a sorbent experiment had been completed. This turns out to be a problem with sorbent injection in larger and full-scale units as well (CitationBiermann et al. 2003).
TABLE 2 Ultimate analysis, heating value, and concentrations of four major metals in residual fuel oil examined
Scavenging of residual oil fly ash by dispersed sorbent was quantified using a modification of the aerosol fractionation method used by CitationGale and Wendt (2002). In this modification, the measured decrease in the ultrafine mode of the sampled aerosol yielded the fraction of the total ash that was scavenged. This method avoided uncertainties derived from ambiguities present in the absolute value of the sorbent mode in the PSD, as discussed below. Note that in this refractory-lined furnace, where carbon burnout approaches 100%, all of the ash aerosol is normally found in the ultrafine mode. This behavior has been shown previously (CitationLinak et al. 2000) for residual oil fly ash from a similar horizontal refractory-lined furnace but not from a fire-tube boiler, where steep temperature profiles and high quench rates produce significant amounts of carbon, in the form of unburned char, in the emitted particles.
PSDs were determined by electrical mobility, time-of-flight, and inertial impaction techniques for sampled aerosols. Extractive samples were taken using an isokinetic aerosol sampling system described elsewhere (CitationLinak et al. 1994). Diluted samples were directed to a TSI Inc. scanning mobility particle sizer (SMPS) configured to yield 54 channels, evenly spaced (logarithmically) over a 0.015 to 0.7 μm diameter range, and to a TSI Inc. aerodynamic particle sizer (APS), which uses time-of-flight principles to measure particles with diameters of 0.5–20 μm. Together, these two analyzers can measure PSDs over a range from approximately 0.01 to 20 μm diameter.
An MSP Inc. nine-stage, 30 l/min micro-orifice uniform deposit impactor (MOUDI) was used to size-classify and collect physical samples for chemical analysis. MOUDI stages (and afterfilters) were examined by X-ray fluorescence (XRF) spectroscopy to examine changes in metal PSDs as a result of sorbent injection. Raw XRF data is given as counts for a specific element, where the proportionality constant relating counts to concentration varies from element to element on a given stage, but not from stage to stage for a given element. Therefore, the metal mass fractions presented are equivalent to metal XRF count fractions and are determined by individual metal counts (V, Ni, Fe, or Zn) on a given stage divided by XRF counts for the same metal summed over all stages (and afterfilter). Absolute concentrations of μg metal per μg of particle are not available.
EQUILIBRIA
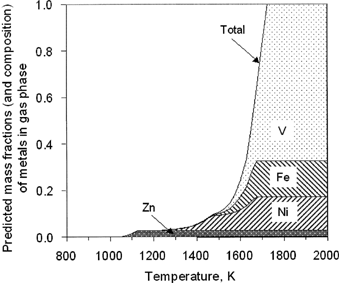
The Chemical Equilibrium Analysis (CEA) code (McCitationBride et al. 1993) was used to predict the element speciation and, more importantly, the fractions of vapor-phase species as a function of temperature for the four principal metals of interest (V, Ni, Fe, and Zn). References for the thermochemical properties and species data sets used are found in CitationLinak et al. (2003). These equilibrium predictions suggest vapor-phase V and Fe species above 1475 K and vapor-phase Ni species above 1200 K. Vapor-phase Zn species are predicted to be present above 1050 K. shows the temperatures and furnace locations at which each metal is expected to start being removed from the vapor (denoted as onset of condensation) and where it finishes being removed from the vapor (denoted as end of condensation).
It is useful to quantify these results further as shown in , in which the fraction of total ash predicted to be vapor at a point in the combustion zone is plotted against temperature, with that amount being divided into segments showing relative amounts of V, Ni, Fe, and Zn in the vapor phase. Thus, when 100% of all the ash is predicted to be vapor, the composition of the vapor is the composition of the original ash in the oil. shows that 100% of the metal is predicted to be vapor at temperatures of 1725 K and above. Thus, at 1740 K, the highest bulk temperature measured here, all of the oil ash metals are predicted to be vapor. However, at 1600 K, the highest postflame sorbent injection temperature, only 25% of the total metal is vapor, while at 1430 K the metal vapor fraction is less than 10%. If we neglect nuclei-sorbent coagulation and assume that only metal vapor can interact via chemical or physical processes with a solid substrate, these calculations suggest that, depending on sorbent injection location, anywhere from 10 to 100% of the metal might be captured in this particular system. However, this would only be the case if the interaction were sufficiently rapid and if the contact between metal and substrate were optimum. These equilibrium predictions suggest that only partial capture of residual fuel oil ash can be achieved if temperatures at which metal-sorbent interactions occur fail to exceed 1725 K. The equilibrium predictions are, however, sufficiently promising to suggest that appreciable residual oil ash scavenging is possible, leading to the experimental results described below.
FIG. 2 Equilibrium predictions of the mass fraction and composition of oil ash metals in the vapor phase.
RESULTS
Particle Size Distributions
presents PSDs measured for the baseline case (without sorbent) and also with sorbent addition. These PSDs are indicated by diamond and triangle symbols, respectively. , therefore, shows representative SMPS/APS data on the effects of sorbent injection. Experimental parameters, including sorbent equivalence ratio (Φ), sorbent injection temperature (T inj ), sampling temperature (T s ), and residence time (τ), are included. Sorbent was injected through the burner, and these conditions represent the largest capture observed in this work. These data are in agreement with previous literature (CitationLinak et al. 2000, 2002) and clearly suggest that essentially all metals in the residual oil did vaporize, and subsequently nucleated to form the ultrafine particles with a unimodal peak centered at 0.07 μm. The small hump in the large particle region for the base case (no sorbent) is an artifact caused by carryover of residual sorbent from crevices and ledges in the combustor from previous experiments. It was not observed in baseline experiments before sorbent injection experiments had been attempted. Total vaporization of all oil ash is consistent with the equilibrium predictions on , where 100% vaporization of all ash is predicted at temperatures greater than 1725 K. While the largest bulk temperature measured was 1740 K, higher temperatures may well be reached locally in regions surrounding burning oil droplets and within burning char particles. Similarly, local regions may also exhibit temperatures below the bulk temperature, resulting in less-effective sorbent capture potential. It should be noted that the ultrafine particle mode centered at 0.07 μm diameter cannot be the result of unvaporized residual ash from the vaporizing fuel droplets. Calculated residual ash particles determined from the fuel oil droplet size distribution (30–40 μm mean diameter), oil density (0.993 g/cm3), oil ash content (0.08%), and oil ash density (3.5 g/cm3) are predicted to have mean diameters between 2.0 and 2.3 μm.
FIG. 3 Measured volume PSDs using SMPS (0.015–0.7 μm) and APS (0.5–20 μm) instrumentation.
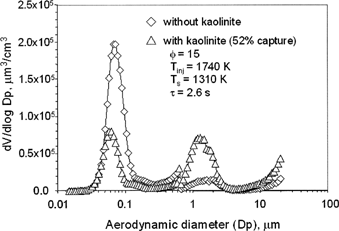
The triangle symbols on depict the bimodal PSDs measured by the SMPS and APS when kaolinite sorbent at an equivalence ratio (Φ) of 15 (Φ defined as g-moles Al2O3-2SiO2/g-moles V + Ni + Fe + Zn). These data represent conditions in which the sorbent was injected through the burner with the combustion air, making it likely to experience the peak temperatures of 1740 K or higher. The area under the ultrafine particle peak, as determined from all the SMPS channels, has decreased by 52%, which was among the largest decreases measured. In this work this difference is taken to reflect the amount of metal ash that was captured. Hence, in , approximately 50% of the oil ash was captured by the sorbent. Replicate experiments showed this to vary from 39 to 59% capture for a sorbent injection temperature equal to the maximum measured (i.e., 1740 K). It should be noted that these experiments are conducted in a semi-industrial–scale, swirling turbulent diffusion flame environment, and therefore some variability in contacting of sorbent and metals and local temperature is to be expected from experiment to experiment. This is especially true where the sorbent is introduced at the burner, where some degradation of burner performance with time was observed as sorbent deposited on and partially blocked the swirl vanes.
The area under the PSD for the supermicron particle size range cannot be used to calculate the fraction of ash scavenged since it changes very little due to metal sorption. Furthermore, it appears that the APS yields a PSD peak that is far too small to represent a sorbent loading of Φ = 15. A portion of this error is due to the considerable deposition of sorbent within the combustor and transfer lines referred to above. In addition, we do not believe the rising tail in the PSD above 10 μm reflects a real phenomenon, as the APS model used has manufacturer-identified problems with small-particle recirculation in the optics chamber, which results in false large-particle counts. This is further confirmed with additional data, as described below.
Composition PSDs were obtained using MOUDI samples, and XRF analysis. represent MOUDI-XRF data for sorbent injection at 1600 K (downstream of the flame). The mass fractions of V, Ni, Fe, and Zn measured on each impactor stage divided by the stage delta log Dp are presented. Similar mass fraction PSDs for Al and Si, found only in the kaolinite sorbent, are also shown on . Again, without sorbent () almost all of the V, Ni, Fe, and Zn formed (condensing) nuclei in the ultrafine particle size mode. This supports the data on and previous data (CitationLinak et al. 2000, 2002) showing (almost) complete vaporization of the oil ash, and, as noted above, is also consistent with equilibrium predictions provided the metals see temperatures higher than 1725 K.
FIG. 4 Measured elemental PSDs for four major residual oil metals and Al and Si present in sorbent: (a) baseline, without sorbent; (b) with sorbent.
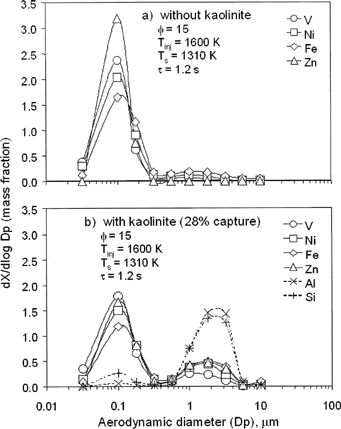
shows the bimodal metal fraction PSDs for V, Ni, Fe, and Z, indicating partial capture of each metal when sorbent is injected. Note that the Al and Si PSDs are (mostly) unimodal and can be used to track the sorbent. The small ultrafine peak for Si suggests the interesting observation that a small portion of silicon in the sorbent may have vaporized, which one would normally expect only under locally reducing conditions (CitationQuann and Sarofim 1982). Note that the MOUDI data show no “tail” reaching upwards at 10 μm, as does the APS. This suggests that this aspect of the APS data is indeed an artifact related to recirculation of small particles in the optics field and need not be considered further.
One can use the MOUDI-XRF data to calculate the percentage of metals in the oil that were captured by the sorbent and compare this to the SMPS/APS data. This can be done in two ways. Method A uses only a single PSD with sorbent injection and focuses attention on the weighted fractions of V, Ni, Fe, and Zn collected on the top six stages of the MOUDI (0.5 μm < D p < 20 μm) divided by the total amount of those four metals in the oil. Method B uses PSDs from two experiments (baseline data and sorbent injection data), and focuses attention on the weighted decrease of metals on the small particle stages of the MOUDI (comparing ). The two methods are only identical when the integral under the ultrafine particle mode of the baseline data is equal to one.
Specifically, the MOUDI-XRF data are analyzed as follows. For a specific experiment, let X A i be the number of XRF counts for metal A, (V, Ni, Fe, or Zn) on MOUDI stage i (or afterfilter, AF), W A the concentration (μg/g) of metal A in the oil, and M k l the mass fraction of all metals captured on all stages k through l of the MOUDI. Then, following Method A, the weighted fraction of all metals captured on the top six stages, M 1 6, is equal to the fraction captured (χ captured ) and is given by:
The data presented on also suggest that at this sorbent injection temperature (T inj =1600 K), greater fractions of Ni, Fe, and Zn were captured (about 25–35%), than V (15%). This is qualitatively consistent with the equilibrium predictions at 1600 K (see ). The MOUDI-XRF data suggest that at 1600 K the solid phase contains 23% of all metals (V +Ni +Fe + Zn) originally in the oil, of which 11% is V, 5% Ni, 6% Fe, and 1% Zn. The corresponding equilibrium values for metals in the vapor phase is 24% (total), of which 7% is V, 9% Ni, 5% Fe, and 3% Zn. According to both theory and experiment, these elemental percentages change with temperature. However, although there is some qualitative agreement between theory and experiment on trends with temperature, there is not quantitative agreement at the other temperatures, as shown in the subsection below.
Effects of Sorbent Equivalence Ratio, Injection Temperature (Location), and Available Residence Time on Residual Oil Ash Capture
The effects of sorbent equivalence ratio (Φ), sorbent injection temperature (T inj ) and available residence time (τ) are shown in . Data were selected so that results from changing one variable at a time (as closely as experimentally possible) are reported. does not report all the data collected, or necessarily all the data referred to in other sections of this article.
FIG. 5 Measured mass fraction of residual oil metal captured as a function of: (a) sorbent-to-metal equivalence ratio (Φ); (b) sorbent injection temperature (T inj ); and (c) sorbent residence time (τ).
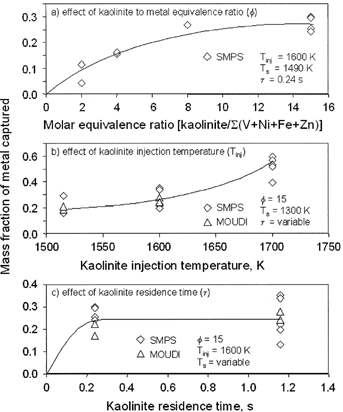
indicates the effect of increasing sorbent equivalence ratio (Φ), and is important since it shows that capture effects are seen even for small amounts of sorbent. However, the effects are not linear with sorbent addition. For T inj =1600 K and τ = 0.24 s, a sorbent equivalence ratio (Φ) of 8 was sufficient to reach the asymptotic yield of 24%, as measured by SMPS ultrafine mode decrease. This translates to 12 g sorbent per kg oil. It should be noted that values of Φ = 2, 4, 8, and 15 relate to target sorbent concentrations of approximately 200, 400, 800, and 1500 mg/m3. However, as noted above, much sorbent is lost in the combustion system. Replicate measurements at the bottom of the furnace indicate actual suspended sorbent concentrations between 450 and 500 mg/m3 at Φ = 15.
shows that an important variable is the injection temperature of the sorbent (T inj ), with Φ = 15 and T s = 1300 K. In general, the higher the sorbent injection temperature the greater the fraction captured. A maximum capture of 59% was achieved for one test with the sorbent injected with the burner air. The lowest capture at the same condition was 39%, and the average was 52%. No MOUDI samples were obtained for this condition. Injection of this amount of sorbent through the burner led to a significant fouling problem at the burner. This is not surprising, as the IFRF burner is not designed for solids, and its movable block design is known to have a high pressure drop and high impaction possibilities for particles introduced with the air. shows data from both the SMPS Method and from Method A for the MOUDI-XRF data (note diamond and triangle symbols). Agreement between both methods to determine capture is good for the two injection temperatures, for which both methods were performed. Not all data gathered are presented in , which focuses only in those data relating to changes of one variable at a time. However, some data not shown in are discussed in the text. At 1600 K the sorbent captures an amount of ash similar to amounts predicted by equilibrium to be vaporized (16–30% measured versus 24% predicted). At 1400 K the sorbent capture exceeds the amount predicted by equilibrium to be vapor (12–35% measured versus 10% predicted). For injection at the burner, with a maximum measured temperature of 1740 K, equilibrium predicts 100% of the ash to be vapor, while the data show 39–59% captured. Possibly the sorbent did not contact enough of the metal at the highest temperature, resulting in less-than-optimum capture. At 1600 K capture tracked the amount vaporized. However, if only vapor-phase metal reacted with sorbent, the equilibrium predictions at 1430 K appear to underestimate the amount vaporized. These results may also indicate contributions of additional mechanisms, such as sorbent deactivation at high temperatures, and perhaps additional physical mechanisms discussed below.
shows the effect of residence time (τ) for T inj = 1600 K and Φ = 15. In these experiments the sampling position was changed. Here, the SMPS data show almost no effect in the range of 0.3–1.2 s. The MOUDI data show somewhat scattered agreement with the SMPS data, but overall the conclusion to be drawn is that capture occurs within fractions of a second. Previous work (CitationDavis and Wendt 2000; CitationGale and Wendt 2002) shows rapid sorbent deactivation during sorption of sodium, lead, and cadmium, and scanning electron micrographs of samples here suggest that a similar process may also be occurring here.
DISCUSSION
It has heretofore been assumed that the primary capture mechanism is the reaction between sorbent substrate and metal vapor. However, metal capture can also occur through coagulation of nuclei with sorbent particles or through condensation of the metal vapor on the sorbent particles. Only when sampling occurs before the onset of metal condensation must the process be due to reaction (which may be slow or fast). In this work, sampling was never possible at temperatures above the onset of condensation of vanadium, the major metal in the ash.
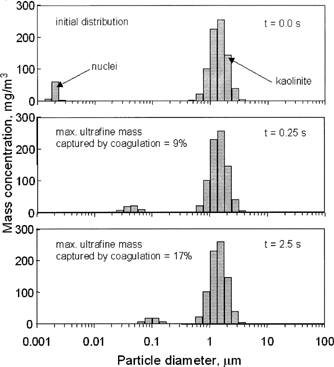
Coagulation was modeled using the multicomponent aerosol simulation (MAEROS) code (CitationGelbard and Seinfeld 1980). Input parameters include 71 mg/m3 of oil ash (measured), an initial nuclei diameter of 2 nm (geometric standard deviation = 1.1), a sorbent feed rate of 800 mg/m3, a sorbent particle mean diameter of 1.4 μm (geometric standard deviation = 1.4), and a temperature profile falling from 1600 to 1290 K in 2.5 s. Results of these calculations are shown in , in which the evolution of both modes with respect to time is depicted. The upper panel shows the initially assumed PSDs. After 0.25 s calculations show that approximately 9% of the oil ash nuclei had coagulated with the sorbent (second panel), while after 2.5 s approximately 17% of the nuclei had coagulated with the sorbent. Therefore, coagulation might explain a portion of the metal–sorbent interaction, but it cannot explain all the capture measured, especially at the more typical system residence times of less than 1 s. Coagulation is, surprisingly, not negligible, but neither does it seem to be the dominant mechanism to explain the partial captures observed. The coagulation calculations presented in assume that none of the vapor-phase oil ash interacts with the sorbent and that all this ash contributes to the nuclei mode. If one assumes that 35% of the vapor-phase oil ash interacts with the sorbent prior to nucleation, then model calculations predict 8 and 15% of the oil ash to coagulate with the sorbent at 0.25 and 2.5 s, respectively. This also results in a slight reduction in the nuclei mean diameter at 2.5 s from ∼0.09 to ∼0.08 μm, and this qualitative behavior is noted in the measured PSDs presented in .
FIG. 6 Predicted evolution of residual oil nuclei in the presence of sorbent. MAEROS2 was used to predict the amount of metal sorbent interaction as a result of coagulation. Input parameters include: oil ash (nuclei) concentration = 71 mg/m3, initial nuclei diameter = 2 nm (geometric standard deviation = 1.1), sorbent concentration = 800 mg/m3, initial sorbent diameter = 1.4 μm (geometric standard deviation = 1.4), and a temperature profile falling from 1600 to 1290 K in 2.5 s.
CitationFriedlander et al. (1991) examined conditions where coagulation between a fine aerosol and a coarse particle mode might be an important mechanism. They concluded that the half-life of the fine mode depends strongly on its initial nuclei mass concentration (ρ g ρ p v o ) and on the coarse-mode diffusion integral (M1) given by CitationFlagan and Friedlander (1978). of the CitationFriedlander et al. (1991) paper presents a stability diagram, determined for a system temperature of 1000 K and 1 nm nuclei. Using values for ρ g ρ p v o (71 mg/m3) and M1 (1.2× 106 m/kg) for our system, from the CitationFriedlander et al. (1991) paper indicates a fine mode half-life (due to coagulation) of ∼100 s. While this time scale is far larger than that available in the combustor, it is sufficiently small enough to indicate that measurable coagulation between the oil ash nuclei and the sorbent is possible. This is consistent with MAEROS calculations that were allowed to continue for 100 s, which predict oil ash nuclei–sorbent coagulation of 54%. As noted by CitationFriedlander et al. (1991), the major temperature dependence is incorporated into the gas density as well as a weak T 1/2 dependence for particle diffusion in the free molecular regime. MAEROS calculations performed at ambient temperatures predict much smaller coagulation rates of 2 and 4% at 0.25 and 2.5 s, respectively.
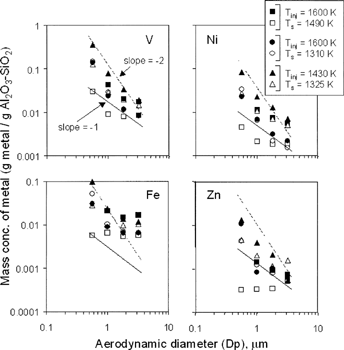
The bulk of the capture, therefore, occurs either by surface chemical reaction or surface physical condensation. If surface reaction (or pore diffusion) is controlling, the metal composition of supermicron particles should have 1/D p dependence. If gas-film diffusion is controlling (condensation or very fast, external surface, reaction) the metal composition should have 1/D p 2 dependence (CitationFriedlander 1977; CitationHaynes et al. 1982; CitationLinak and Wendt 1993). Coagulation might also lead to 1/D p 2 dependence. The MOUDI-XRF data show that particles on the four impactor stages to which the sorbent reports are in the applicable continuum regime. Results for all four metals for the four applicable particle sizes are plotted on . Results are inconclusive because the data are scattered, and so interpretation should be viewed as tentative at best. Logarithmic slopes vary from −2 (gas film diffusion controlled), through −1 (external surface reaction, or pore diffusion controlled), to zero (interior surface reaction controlled). In general, higher injection temperatures lead to less-sensitive particle diameter dependence, especially for Fe and Zn but also for V and Ni. This trend is consistent with mechanisms in which higher injection temperatures lead to an initial rapid formation of larger mesopores inside the kaolinite particles caused by greater separations between the crystalline alumino-silicate platelets in the freshly formed meta-kaolinite. This leads to greater penetration of metal vapors into the center of the particle and, consequently, to a possible mechanism transition to that of interior surface reaction control. However, melting can later close these mesopores and deactivate the process (CitationGale and Wendt 2003) as it proceeds, and this was observed (through scanning electron micrographs) in many but not all reacted particles.
FIG. 7 Mass concentration of residual oil metals versus particle diameter over the continuum regime where sorbent particles are present.
CONCLUSIONS
The potential for high temperature capture of metals in residual oil fuel ash by powdered kaolinite has been investigated. Partial capture of oil ash is possible, although capture efficiencies (determined three ways) greater than ∼30% could not be achieved when sorbent was injected downstream of the flame. This is roughly consistent with the partial ash vaporization predictions of equilibrium, although captures at lower temperatures were greater than those predicted by equilibrium. Injection through the burner caused fouling problems but led to significantly higher capture, in one case up to 59%. The through-the-burner injection process has yet to be optimized, and future work might address how flame aerodynamics may be exploited in order to enable oil ash to be captured by sorbent injected with the air. Since short residence times are effective, the key seems to be related to mixing between the sorbent and vaporized ash coming from the oil droplets. Sorbent deactivation by processes including sintering, melting, and pore closure of the kaolinite may be limiting metal captures at high temperatures, and these have been identified previously for high temperature kaolinite sorption of lead and cadmium (CitationDavis 2000). However, at low temperatures the effective metal sorption may be enhanced by nuclei-sorbent coagulation processes, causing metal capture greater than those predicted by metal volatility.
While coagulation between the oil ash nuclei and coarse sorbent particles is nonnegligible, it cannot account for the amounts of metal capture observed. Particle composition data do not support only film diffusion-controlled processes. Neither do they support only external surface-reaction–controlled processes, or only internal surface-reaction–controlled processes. Most likely some combination of all of these is appropriate, with an apparent transition to (interior) reaction control at the higher sorbent injection temperatures.
These data do not currently provide great encouragement for the practical utility of sorbent injection to remove more than 50% of the ultrafine oil ash particles. However, the fact that sorbent injection through the burner was able to vastly increase the fraction of ash captured, including vanadium, may provide some hope that future research in this direction may lead to substantially greater captures.
Acknowledgments
Portions of this work were conducted under EPA Purchase Order No. 1CR183NASA with J. O. L. Wendt and EPA Contract 68-C-99-201 with ARCADIS Geraghty & Miller, Inc. This work was also partially supported by the KEMCO academic research fund, No. 2002CCT03P01, in Korea. The authors would like to thank EPA's Shirley Wasson for her kind assistance with the XRF analysis. The research described in this article has been reviewed by the Air Pollution Prevention and Control Division, U.S. EPA, and approved for publication. The contents of this article should not be construed to represent agency policy, nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
Notes
a 1997 fuel oil sales (CitationDOE 1998).
b 1999 coal sales (Kilgroe et al. 2000).
REFERENCES
- Bachmann , J. D. , Damberg , R. J. , Caldwell , J. C. , Edwards , C. and Koman , P. D. 1996 . Review of the National Ambient Air Quality Standards for Particulate Matter: Policy Assessment of Scientific and Technical Information (OAQPS Staff Paper) Washington , DC EPA-452/R-96–013 (NTIS PB97–115406)
- Biermann , J. , Voogt , N. , Sorooshian , A. , Wendt , J. O. L. , Merritt , R. and Gale , T. K. 2003 . “ In-Flight Capture of Elemental and Oxidized Mercury in Coal Combustion Flue Gases by a Novel Mineral Sorbent Between 320 and 1540°F (160–840°C) ” . In DOE-EPRI-EPA-A&WMA Power Plant Air Pollutant Control Mega Symposium Washington , DC 19–22 May 2003
- Damle , A. S. , Ensor , D. S. and Ranade , M. B. 1982 . Coal Combustion Aerosol Formation Mechanisms: A Review . Aerosol Sci. Technol. , 1 : 119 – 133 .
- Davis , S. B. 2000 . Interactions Between Semi-Volatile Toxic Metals and Sorbents in Combustors , Tucson , AZ : University of Arizona . Ph.D. Dissertation
- Davis , S. B. and Wendt , J. O. L. 2000 . Mechanisms and Kinetics of Lead Capture by Kaolinite in a Downflow Combustor . Proceedings of the Combustion Institute , 28 : 2743 – 2749 .
- Davison , R. L. , Natusch , D. F. S. , Wallace , J. R. and Evans , C. A. Jr. 1974 . Trace Elements in Fly Ash Dependence of Concentration on Particle Size . Environ. Sci. Technol. , 8 ( 13 ) : 1107 – 1113 .
- Dreher , K. , Jaskot , R. , Lehmann , J. R. , Richards , J. H. , McGee , J. K. , Ghio , A. J. and Costa , D. L. 1997 . Soluble Transition Metals Mediate Residual Oil Fly Ash Induced Lung Injury . J. Toxicol. Environ. Health , 50 : 285 – 305 .
- r . 1997 . Federal Register , 18 July 62 FR 38652
- Flagan , R. C. 1979 . Submicron Particles from Coal Combustion . Proceedings of the Combustion Institute , 17 : 97 – 104 .
- Flagan , R. C. and Friedlander , S. K. 1978 . “ Particle Formation in Pulverized Coal Combustion—A Review ” . In Recent Developments in Aerosol Science , Edited by: Shaw , D. T. New York : John Wiley & Sons . In
- Flagan , R. C. and Taylor , D. D. 1981 . Laboratory Studies of Submicron Particles from Coal Combustion . Proceedings of the Combustion Institute , 18 : 1227 – 1237 .
- Friedlander , S. K. 1977 . Smoke Dust and Haze—Fundamentals of Aerosol Behavior , New York : Wiley .
- Friedlander , S. K. , Koch , W. and Main , H. H. 1991 . Scavenging of a Coagulating Fine Aerosol by a Coarse Particle Mode . J. Aerosol Sci. , 22 ( 1 ) : 1 – 8 .
- Gale , T. K. and Wendt , J. O. L. 2002 . High Temperature Interactions between Multiple Metals and Kaolinite . Combust. Flame , 131 : 299 – 307 .
- Gale , T. K. and Wendt , J. O. L. 2003 . Mechanisms and Models Describing Sodium and Lead Scavenging by a Kaolinite Aerosol at High Temperatures . Aerosol Sci. Technol. , 37 : 865 – 876 .
- Gelbard , F. and Seinfeld , J. H. 1980 . Simulation of Multicomponent Aerosol Dynamics . J. Colloid Interface Sci. , 78 : 485 – 501 .
- Gladney , E. S. , Small , J. A. , Gorden , G. E. and Zoller , W. H. 1976 . Composition and Size Distribution of In-Stack Particulate Material at a Coal-Fired Power Plant . Atmos. Environ. , 10 : 1071 – 1077 .
- Gluskoter , H. J. 1978 . “ An Introduction to the Occurrence of Mineral Matter in Coal ” . In International Conference on Ash Deposits and Corrosion from Impurities in Combustion Gases , Edited by: Bryers , R. W. Washington , DC : Hemisphere Publishing . In
- Haynes , B. S. , Neville , M. , Quann , R. J. and Sarofim , A. F. 1982 . Factors Governing the Surface Enrichment of Fly Ash in Volatile Trace Species . J. Colloid Interface Sci. , 87 ( 1 ) : 266 – 278 .
- Kaakinen , J. W. , Jorden , R. M. , Lawasani , M. H. and West , R. E. 1975 . Trace Element Behavior in Coal-Fired Power Plant . Environ. Sci. Technol. , 9 ( 9 ) : 862 – 869 .
- Kilgroe , J. D. , Sedman , C. B. , Srivastava , R. K. , Ryan , J. V. , Lee , C. W. and Thorneloe , S. A. 2002 . “ Contol of Mercury Emissions from Coal-fired Electric Utility Boilers: Interim Report ” . In EPA-600/R-01–109 (NTIS PB2002–105701) , Research Triangle Park , NC : U.S. Environmental Protection Agency . April, 2002
- Klein , D. H. , Andren , A. W. , Carter , J. A. , Emery , J. F. , Feldman , C. , Fulkerson , W. , Lyon , W. S. , Ogle , J. C. , Talmi , Y. , VanHook , R. I. and Bolton , N. 1975 . Pathways of Thirty-Seven Trace Elements Through Coal-Fired Power Plant . Environ. Sci. Technol. , 9 ( 10 ) : 973 – 979 .
- Linak , W. P. , Miller , C. A. , Santoianni , D. A. , King , C. J. , Shinagawa , T. , Wendt , J. O. L. , Yoo , J. and Seo , Y. 2003 . Formation of Fine Particles from Residual Oil Combustion: Reducing Nuclei through the Addition of Inorganic Sorbent . Korean J. Chem. Eng. , 20 ( 4 ) : 664 – 669 .
- Linak , W. P. , Miller , C. W. and Wendt , J. O. L. 2000 . Fine Particle Emissions from Residual Fuel Oil Combustion: Characterization and Mechanisms of Formation . Proceedings of the Combustion Institute , 28 : 2651 – 2658 .
- Linak , W. P. , Miller , C. W. , Seames , W. S. , Wendt , J. O. L. , Ishinomori , T. , Endo , Y. and Miyamae , S. 2002 . On Trimodal Particle Size Distributions in Fly Ash from Pulverized-Coal Combustion . Proceedings of the Combustion Institute , 29 : 441 – 447 .
- Linak , W. P. , Srivastava , R. K. and Wendt , J. O. L. 1994 . Metal Aerosol Formation in a Laboratory Swirl Flame Incinerator . Combust. Sci. Technol. , 101 : 7 – 27 .
- Linak , W. P. and Wendt , J. O. L. 1993 . Toxic Metal Emissions from Incineration: Mechanisms and Control . Prog. Energy Combust. Sci. , 19 : 145 – 185 .
- McBride , B. J. , Gordon , S. and Reno , M. A. 1993 . Coefficients for Calculating Thermodynamic and Transport Properties of Individual Species , NASA Technical Memorandum 4513
- Miller , C. A. , Linak , W. P. , King , C. and Wendt , J. O. L. 1998 . Fine Particle Emissions from Heavy Fuel Oil Combustion in a Firetube Package Boiler . Combust. Sci. Technol. , 134 : 477 – 502 .
- Mims , C. A. , Neville , M. , Quann , R. J. , House , K. and Sarofim , A. F. 1980 . Laboratory Studies of Mineral Matter Vaporization during Coal Combustion . AIChE Symp. Ser. , 76 ( 201 ) : 188 – 194 .
- Nettleton , M. A. 1979 . Particulate Formation in Power Stations Boiler Furnaces . Prog. Energy Combust. Sci. , 5 : 223 – 243 .
- Neville , M. , McCarthy , J. F. and Sarofim , A. F. 1983 . Size Fractionation of Submicrometer Coal Combustion Aerosol for Chemical Analysis . Atmos. Environ. , 17 ( 12 ) : 2599 – 2604 .
- Neville , M. , Quann , R. J. , Haynes , B. S. and Sarofim , A. F. 1981 . Vaporization and Condensation of Mineral Matter During Pulverized Coal Combustion . Proceedings of the Combustion Institute , 18 : 1267 – 1274 .
- Neville , M. and Sarofim , A. F. 1982 . The Stratified Composition of Inorganic Submicron Particles Produced during Coal Combustion . Proceedings of the Combustion Institute , 19 : 1441 – 1449 .
- Ondov , J. M. , Ragaini , R. C. and Biermann , A. H. 1978 . Elemental Particle-Size Emissions from Coal-Fired Power Plants: Use of an Inertial Cascade Impactor . Atmos. Environ. , 12 : 1175 – 1185 .
- Ondov , J. M. , Ragaini , R. C. and Biermann , A. H. 1979 . Elemental Emissions from a Coal-Fired Power Plant: Comparison of a Venturi Wet Scrubber System with a Cold-Side Electrostatic Precipitator . Environ. Sci. Technol. , 13 ( 5 ) : 598 – 607 .
- Piper , B. and Nazimowitz , W. 1985 . High Viscosity Oil Evaluation, 59th Street Station—Unit 110 , Vol. 1 March 1985
- Quann , R. J. , Neville , M. , Janghorbani , M. , Mims , C. A. and Sarofim , A. F. 1982 . Mineral Matter and Trace-Element Vaporization in a Laboratory Pulverized Coal Combustion System . Environ. Sci. Technol. , 16 ( 11 ) : 776 – 781 .
- Quann , R. J. and Sarofim , A. F. 1982 . Vaporization of Refractory Oxides During Pulverized Coal Combustion . Proceedings of the Combustion Institute , 19 : 1429 – 1440 .
- Sarofim , A. F. , Howard , J. B. and Padia , A. S. 1977 . The Physical Transformation of the Mineral Matter in Pulverized Coal Under Simulated Combustion Conditions . Combust. Sci. Technol. , 16 : 187 – 204 .
- Smith , R. D. 1980 . The Trace Element Chemistry of Coal During Combustion and the Emissions from Coal-Fired Plants . Prog. Energy Combust. Sci. , 6 : 53 – 119 .
- Smith , R. D. , Campbell , J. A. and Nielson , K. K. 1979 . Characterization and Formation of Submicron Particles in Coal-Fired Plants . Atmos. Environ. , 13 : 607 – 617 .
- Taylor , D. D. and Flagan , R. C. 1982 . The Influence of Combustor Operation on Fine Particles from Coal Combustion . Aerosol Sci. Technol. , 1 : 103 – 117 .
- U.S. Department of Energy, Fuel Oil and Kerosene Sales 1997 . 1998 . DOE/EIA-0535(97) , Washington , DC : Energy Information Administration .
- U.S. Environmental Protection Agency . 1996 . National Ambient Air Quality Standards for Particulate Matter: Proposed Decision 40 CFR, Part 50
- Walsh , P. M. , Wei , G. and Xie , J. 1991 . “ Metal Oxide and Coke Particles Formed During Combustion of Residual Fuel Oil ” . In 10th Annual AAAR Meeting Traverse City , MI 7–11 October
- Williams , A. 1976 . Fundamental of Oil Combustion . Prog. Energy Combust. Sci. , 2 : 167 – 179 .
- Wilson , R. and Spengler , J. D. , eds. 1996 . Particles in Our Air: Concentrations and Health Effects , Cambridge , MA : Harvard Univ. Press .
- Wolff , G. T. 1996 . Closure by the Clean Air Scientific Advisory Committee (CASAC) on the Staff Paper for Particulate Matter , Washington , DC : U.S. Environmental Protection Agency . 13 June, 1996