Abstract
A denuder-based integrated organic gas and particle sampler (IOGAPS), designed to minimize artifacts often encountered during conventional filter pack (FP) sampling, was used in this study to examine seasonal and diurnal variations of particle-bound organic carbon (OC) and elemental carbon (EC) in Toronto, Canada. Daytime and nighttime PM 2.5 samples were collected using both the IOGAPS and FP in Toronto from 16–21 July 2001 and 17–24 March 2003. The average OC determined by the IOGAPS (IOGAPS-OC) was 6.7 μ g C/mbm3 in July 2001 and 5.3 μ g C/mbm3 in March 2003. The average EC determined by the IOGAPS (IOGAPS-EC) was 0.9 μ g C/mbm3 and 0.4 μ g C/mbm3, respectively, during these periods. The IOGAPS-OC concentrations were often higher during the night. This could be explained by the partitioning of the gas phase semivolatile organic carbon (SVOC) onto the existing aerosols due to decreased ambient temperatures. The average of the SVOC measured by the IOGAPS was 0.5 μ g C/mbm3 and 4.5 μ g C/mbm3 during the day and night in July 2001, respectively. In March 2003 the daytime average SVOC was 1.9 μ g C/mbm3 and the nighttime average SVOC was 2.5 μ g C/mbm3. The formation of secondary organic aerosols (SOA) contributed significantly to the summertime OC in July 2001, which was confirmed by a poor correlation between the IOGAPS-OC and IOGAPS-EC (Rbm2 = 0.01). In contrast, a relatively strong correlation between IOGAPS-OC and IOGAPS-EC (Rbm2 = 0.87) during the winter in March 2003 suggests that the main sources of the carbonaceous aerosols were primary.
INTRODUCTION
Carbonaceous materials associated with atmospheric particles are generally classified into organic carbon (OC) and elemental carbon (EC). There is a continuing interest in understanding the concentrations and temporal variations of OC and EC due to their relatively large concentrations and potential impact on the environment and human health (CitationFinlayson-Pitts and Pitts 2000). EC is effective in reducing visibility and altering radiative forcing due to its strong light-absorbing properties (CitationLarson and Cass 1989). OC, which can also influence visibility and radiative transfer, is a mixture of hundreds, if not thousands, of organic compounds with various chemical and physical properties. Some of them (e.g., polycyclic aromatic hydrocarbons, polychlorinated biphenyls, polychlorinated dibenzofurans) are potentially mutagenic and carcinogenic (CitationSeinfeld and Pandis 1998).
Integrated measurement of particle-bound OC is hindered by artifacts encountered during conventional filter pack (FP) sampling using single or multiple filters (e.g., quartz filter). Positive artifacts result from the adsorption of gas-phase organics by the filter or accumulated particles on the filter. Negative artifacts arise from the evaporation of particulate organics on the filter. Both positive and negative artifacts occur concurrently during the sampling, thus it is difficult to design an experiment to estimate their relative contributions to the measured OC concentration or to obtain a true measure of the amount of particulate OC. In addition, some of the particulate chemicals collected on the filter may undergo reactions during sampling (e.g., reactions with ozone; CitationVolckens and Leith 2003). Measurement of OC is further complicated by the fact that some portion of particulate OC is semivolatile (SVOC). These SVOC species can readily change phase depending upon the quantity and nature of the atmospheric particles, their physical and chemical properties, and ambient conditions such as temperature, solar radiation, and relatively humidity (CitationFinlayson-Pitts and Pitts 2000; CitationVolckens and Leith 2003). Sampling itself also disturbs the gas–particle equilibrium, causing mass transfer between the phases. In fact, some SVOC species (e.g., PAHs and oxygenated PAHs) are typically not at gas–particle equilibrium in urban areas (CitationAllen et al. 1996, Citation1997). Due to the complex behavior of SVOC and sampling limitations highlighted above, current understanding of particle-bound OC is limited, particularly in urban atmospheres of eastern North America. The goal of the research described in this article is to improve understanding by examining the seasonal and day–night differences in OC and SVOC in downtown Toronto.
Double quartz filters (i.e., QQ combination) have been used to exclude adsorption-induced artifacts. The front filter collects essentially 100% of the particulate matter (and some gas-phase species as well), and the backup filter is only exposed to gas-phase material, some of which is adsorbed. If the amount of this backup adsorption is the same as the amount of the gas-phase adsorption on the front filter and there is no loss of particle-phase SVOC from the front filter, then the OC on the backup filter can be subtracted from the front filter OC to obtain a better measure of particulate OC. However, the amount of gas-phase OC adsorbed on the second filter may be different from that adsorbed on the front filter, and the magnitude of the evaporative artifacts remains unknown (CitationSalma et al. 2004; CitationViidanoja et al. 2002). Consequently, there is still debate as to whether the OC found in the second filter comes from the gas phase in the atmosphere, from the evaporation off particles collected on the front filter, or both (CitationTurpin et al. 2000). An alternative to the QQ combination is to run, in parallel, a separate FP with a Teflon filter followed by a quartz filter (i.e., Q and TQ combination). Since the Teflon filter does not adsorb gas-phase SVOC, the amount on the backup quartz filter is believed to be a better measure of the positive artifact. However, the amount of particulate SVOC evaporated off the quartz filter cannot be determined, and thus this approach is still not ideal for the measurement of particulate OC (CitationTurpin et al. 2000).
More recently, several approaches to scrub gas-phase SVOC using a diffusion-based denuder prior to the collection of the particles have been reported. These have included the Particle Concentrator–Brigham Young University Organic Sampling System (PC–BOSS; CitationEatough et al. 2001) and the integrated organic gas and particle sampler (IOGAPS; CitationGundel et al. 1995; CitationLewtas et al. 2001; CitationPang et al. 2002c). The IOGAPS, which was used in this study, collects the particles (i.e., PM2.5) by a quartz filter placed downstream of a XAD-resin-coated denuder that functions to remove gas-phase organics. This minimizes the possibility of artifacts arising from interaction of gas-phase organics with the particle-laden filter. Evaporation of semivolatile organic carbon (SVOC) from the filter, which may be enhanced by the upstream denuder, is quantified through the use of backup sorbent (i.e., XAD-4 resin)-impregnated filters (SIFs) designed to retain the off-gassing of organic compounds.
Previously published OC and EC concentrations in Toronto are limited. CitationBrook et al. (2002) measured OC and EC in PM2.5 in Toronto at a traffic-impacted site in spring and summer 1998 and found the average OC and EC concentrations were about 5 and 2 μ g C/m3, respectively. Recently, CitationLee et al. (2003) used one-year daily OC and EC from downtown Toronto for source apportionment. The median concentrations during this period (Feb. 2000–Feb. 2001) were 3.7 and 0.6 μ g C/m3 for OC and EC, respectively. The much lower EC values in this more recent work compared to CitationBrook et al. (2002) is expected to be due to sampling location and time period as well as analytical differences. The 1998 measurements were collected 75 m from a busy highway, and the method of thermal optical reflectance (TOR; CitationChow et al. 1993) used in that study tends to produce higher EC concentrations than the method of thermal optical transmittance (TOT) utilized by CitationLee et al. (2003).
These earlier Toronto data, like much of the available OC and EC data, are potentially biased due to the sampling artifacts mentioned earlier. In addition, the samples were collected on a daily basis, and thus the observation of diurnal variation of OC and EC are not available. Our study was undertaken to measure the concentrations of OC and EC associated with PM2.5 in downtown Toronto using the IOGAPS method so that sampling artifacts could be minimized. Two sampling campaigns were launched: one in the summer of 2001 (July) and the other in late winter of 2003 (March). Throughout the rest of the article we refer to the July 2001 period as “July” and the March 2003 period as “March.” During both periods, samples were collected separately during the day and night, allowing diurnal and seasonal variations of OC and EC to be examined.
EXPERIMENTAL
IOGAPS
The IOGAPS has been described in detail elsewhere (CitationGundel et al. 1995; CitationLane et al. 2000; CitationPeters et al. 2000) and is briefly summarized here. It consists of a cyclone inlet with 2.5 μ m size-cut followed by an XAD-4 resin-coated glass denuder (8-channel, 60 cm long; from URG, Chapel Hill, NC, USA). A prefired quartz filter is placed downstream of the denuder to collect the particles (i.e., PM2.5), followed by three stages of XAD-4 resin-impregnated quartz filters (SIFs) designed to capture the SVOC that evaporates from the front quartz filter. Coating procedures for the denuder and SIFs were based upon the patent of CitationGundel et al. (1998). Blank prefired quartz filters and SIFs, handled in the same manner as the sampled filters including placing them in the sampling box in their petri slides throughout the sample collection period, were collected with every sample. The denuder was cleaned by extraction after each sample collection period using a mixture of hexane, dichloromethane, and methanol (1:1:1, v/v). The IOGAPS was operated at the rate of 16.7 l/min. The performance of the IOGAPS was systematically evaluated previously in the field and laboratory, and it was found that the gas-phase SVOC can be completely removed by the denuder if sampling time is less than 16 h (CitationFan et al. 2003).
All filters used for the IOGAPS and filter pack sampler (discussed below) were 47 mm in diameter. Quartz filters (Type QM-A ultrahigh-purity quartz filters, from Whatman) were prefired at 700°C for 10 h before use. SIFs were prepared by coating fine XAD-4 particles onto prefired quartz filters based on the procedures described by CitationGundel et al. (1998). Briefly, each filter was dipped in a slurry of XAD in hexane, air dried, dipped again, air dried, rinsed twice with hexane, and air dried. The prepared SIFs were kept in an airtight jar and stored in a freezer (< −4°C).
Filter Pack Sampler
A filter pack (FP) sampler was collocated with IOGAPS for a comparison study. In July, the FP used one channel of the versatile air pollution sampler (VAPS; URG, Chapel Hill, NC, USA), which is described in detail elsewhere (CitationPinto et al. 1998) and briefly summarized here. The VAPS was a modified dichotomous sampler consisting of an inlet with a PM10 size-cut and a virtual impactor, which separates the air stream into two fine channels (PM2.5) and a coarse channel (PM2.5 − 10). One fine channel was used to collect PM2.5 (15.0 l/min) by a prefired quartz filter for OC and EC analysis (CitationLee et al. 2003). The coarse channel and the other fine channel were for other uses and will not be discussed here. The FP sampler employed in March consisted of a 2.5 μ m cyclone inlet (URG, Chapel Hill, NC, USA) followed by a single quartz filter and was operated at the rate of 16.7 l/min.
Field Sampling
Field sampling was conducted in downtown Toronto from 16–21 July 2001 (total: 12 samples) and from 17–24 March 2003 (total: 16 samples). Both the IOGAPS and FP samplers were installed on the roof of the Gage Occupational and Environmental Health Unit of the University of Toronto. The sampling inlet was about 15 m above the ground and 20 m south of College Street, which experiences weekday traffic volumes of ∼ 33,000 vehicles (CitationTan et al. 2002). All the flows were calibrated using DryCalreg DC-1 calibration meter (Bios International Co., Butler, NJ, USA). Both IOGAPS and FP samples were collected twice a day from ∼ 9:00 to ∼ 18:00 EDT during the day and from ∼ 18:30 to ∼ 8:30 EDT during the night, yielding volumes around 9 m3 and 14 m3, respectively. After sampling, each filter was stored in a petri dish (Analyslide Petri dish on a slide, 47 mm in diameter, from Pall Gelman) and kept refrigerated (< −4°C) until analysis.
Analysis of OC and EC on Quartz Filter
OC and EC concentrations were determined using a TOT aerosol carbon analyzer (Sunset Laboratory, Inc., Forest Grove, OR, USA). The temperature program utilized was a combination of the NIOSH 5040 (CitationBirch and Cary 1996) and TOR (DRI) methods (CitationChow et al. 1993), which had dwell times at each temperature long enough to separate CH4 peaks. This method is described in detail by CitationSharma et al. (2002) and briefly summarized here. A punch (1.45 cm2) of quartz filter sample is heated in a stepwise fashion to temperatures of 250°C for 150 s (OC1), 450°C for 150 s (OC2), 550°C for 180 s (OC3), and 900°C for 90 s (OC4) in a pure helium atmosphere for OC determination. The sample is then combusted at 550°C for 240 s, 700°C for 210 s, and 800°C for 150 s in 10% O2 in 90% helium. The carbon evolved in these final combustion steps (with O2 added into the helium stream) is separated into EC and pyrolyzed carbon (PC) formed from the charring of some OC. The PC is defined as the carbon evolved from the filter after the initial introduction of oxygen and before the laser transmittance signal returns to its initial value (CitationBirch and Cary 1996). The method has a precision of ± 10% and a quantitation limit of 0.3 μ g C/cm2 on the filter. Therefore, the quantitation limit of atmospheric concentration of OC or EC is 0.4 μ g C/m3 if air is sampled for 8 h at 16.7 l/min. For quality assurance, a five-point calibration was performed prior to the analysis of field samples. Daily calibration was performed before and after sample analysis using a standard glucose solution that was prepared by dissolving highly pure glucose (Aldrich Chemical Co.) in 18 MΩ water (from Millipore filtration apparatus).
Analysis of SVOC Collected on the Backup SIFs
Since volatile organic carbon (VOC) has a greater potential to break through the denuder, the OC captured on the backup SIFs could contain the VOC that breaks through the denuder in addition to the SVOC that evaporates off the particles (CitationLewtas et al. 2001; CitationMader et al. 2001). We developed an analytical method that helps differentiate SVOC from VOC captured by the SIFs using the same TOT instrument for OC and EC analysis (CitationFan et al. 2003). The TOT instrument temperature program was modified as follows: 200°C for 240 s and 350°C for 300 s in pure helium. All of the VOC is desorbed from the SIF at the first stage (200°C), while SVOC is subsequently desorbed during the second stage (350°C). The highest temperature and length of time were carefully selected so that all SVOC can be desorbed from the SIF while the decomposition of XAD is minimized. This method, previously validated in our lab, has a precision of ± 13% and a quantitation limit of 0.3 μ g C/cm2 on the filter. It should be pointed out that there is no specific definition for SVOC. In this study, we define SVOC as the total OC thermally released from a SIF between 200°C and 350°C, but this is an instrument-operational definition. Spike and recovery tests on the SIFs were performed using 14 organic compounds covering a wide range of subcooled vapor pressures, and we found that if the subcooled vapor pressure was less than 0.1 Pa (at 25°C), the average recovery of SVOC fraction of these compounds was more than 82% (CitationFan et al. 2003).
One major concern using the IOGAPS is whether the backup SIFs can completely collect all SVOC evaporated from the particles. Our field measurements show that even three SIFs were not enough to collect all SVOC desorbed from the particles during heavy pollution episodes in July. This suggests that some SVOC species may not have strong affinity with XAD-4 resin coating or that some SIFs are saturated. Using the same approach of linear extrapolation based on the SVOC concentrations on the second and third SIF as the one used by Letwas et al. (2001), we estimated the amount of the SVOC that broke through the three SIFs. We found that if the SVOC concentration was less than 3.8 μ g C/m3, the extrapolated SVOC concentration on the fourth SIF was below the detection limit (∼ 0.4 μ g C/m3) (CitationFan et al. 2003). Therefore, for March samples we simply analyzed total SVOC by combining three punches—one from each SIF—since the highest SVOC concentration during that period was 3.4 μ g C/m3.
RESULTS AND DISCUSSION
Comparison of EC Determined by the IOGAPS and FP
During IOGAPS sampling, small particles may be lost due to their diffusion onto the denuder walls. Such particle loss can be tested by comparison of EC determined by the IOGAPS (IOGAPS-EC) and FP (FP-EC), since EC is exclusively from incomplete combustion of organic matter and is nonvolatile. If particle loss is significant, then the IOGAPS-EC should be lower than FP-EC. There was no significant difference between the EC determined by the IOGAPS and FP in March (), but based on the paired ttest at 95% confidence level, the IOGAPS-EC was significantly higher than the FP-EC in July (). The daytime average IOGAPS-EC and FP-EC in July were 0.94 (0.33–1.59) and 0.75 (0.37–1.43) μ g C/m3, respectively. The nighttime average IOGAPS-EC and FC-EC in July were 0.79 (0.42–1.07) and 0.64 (0.39–0.92) μ g C/m3, respectively. A larger EC from the IOGAPS is consistent with results reported by CitationLewtas et al. (2001) and is contrary to the hypothesis that there is particle loss in the denuder. One possible explanation is that the gas-phase organics adsorption on the filter during FP sampling may enhance pyrolyzed carbon (PC). Our measurements show that the IOGAPS-PC was significantly lower than the FP-PC in July (), but there was no significant difference between the IOGAPS-PC and FP-PC in March (). This leads to the hypothesis that in July semivolatile secondary organic aerosols (SOA) in the gas phase contributed to the positive artifact and that these SOA compounds had a higher potential to char during thermal analysis. Most SOA materials are polar and water soluble, such as low molecular weight acids (CitationCocker et al. 2001; CitationRohrl and Lammel 2001; CitationSaxena et al. 1995), and CitationYu et al. (2002) showed that water-soluble OC (WSOC) associated with ambient aerosols accounted for a large fraction (13–66%) of charring (i.e., the formation of PC) during thermal analysis. Reducing this charring by the use of a denuder-based sampler such as the IOGPAS can be expected to lead to a more accurate EC measurement due to a smaller PC correction. However, this hypothesis needs further investigation.
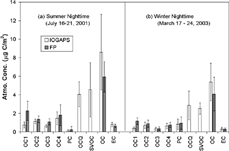
FIG. 1 Comparison of average daytime OC fractions determined by the IOGAPS and FP. IOGAPS-OCQ is the sum of OC1 to OC4 and PC determined from the IOGAPS quartz filter. IOGAPS-OC is the sum of IOGAPS-OCQ and IOGAPS-SVOC. (a) Summer daytime (16–21 July 2001), (b) Winter daytime (17–24 March 2003).

FIG. 2 Comparison of average nighttime OC fractions determined by the IOGAPS and FP. (a) Summer nighttime (16–21 July 2001), (b) Winter nighttime (17–24 March 2003).
Diurnal and Seasonal Variations of SVOC
The evaporation of particulate SVOC from the filter, which often causes negative artifacts, can be substantial under certain ambient conditions (CitationEatough et al. 2001; CitationKavouras et al. 1999; CitationLewtas et al. 2001; CitationPang et al. 2002a, b). In our experiment, we used three stages of SIFs placed downstream of the quartz filter to capture any SVOC off-gassing from the particle-laden filter. The removal of gas-phase organics by the denuder can enhance the evaporation of the SVOC from the filter. Therefore, the SVOC measured from the SIFs is expected to be an upper limit of the actual concentration of particulate SVOC initially captured on the front quartz filter. The IOGAPS results are thus not expected to provide a realistic measure of the actual partitioning of SVOC between the gas phase and the particle phase. However, the sum of the OC on the quartz filter (IOGAPS-OCQ) and IOGAPS-SVOC is expected to represent the true total particle-bound OC (IOGAPS-OC).
The diurnal variation in SVOC was significant during both sampling campaigns but was much more pronounced in July (). On average, in July the SVOC concentration was 4.53 (1.24–8.43)μ g C/m3 during the night, about 8 times higher than the daytime SVOC of 0.49 (0.12–1.4)μ g C/m3, while in March the average nighttime SVOC concentration was 2.52 (1.42–3.36) μ g C/m3 compared to 1.91 (1.29–2.74) μ g C/m3 during the day. Since the nighttime sampling stopped at 9:00 am, emissions from the morning rush-hour traffic may have contributed to the SVOC attributed to nighttime conditions. Another potential contributor to the diurnal variation in SVOC is the formation of SOA materials. These compounds would have more likely condensed onto pre-existing particle surfaces at night due to the decreased ambient temperatures, even though they were formed and remained in the gas phase during the day. A decrease in temperature, even of a few degrees, can exponentially lower the vapor pressure of a SVOC species and thus enhance its partitioning towards the particle phase (CitationLiang et al. 1997; CitationOdum et al. 1997; CitationPeters et al. 2000). The temperature ranged from 17 to 30°C in July (average 23°C) and from −5 to 15°C in March (average 7°C). Major photochemical reactions occur between OH radicals and reactive organic gases such as hydrocarbons. The main source of ground-level OH radicals is from the photo-dissociation of ozone:
FIG. 3 The OC on the IOGAPS quartz filter (IOGAPS-OCQ) and the SVOC captured by the backup SIFs (IOGAPS-SVOC) determined by the IOGAPS. Also shown is the OC determined by the FP sampler (FP-OC).
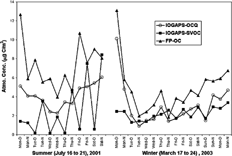
Therefore, higher SOA in July is not surprising given that photochemically related oxidants (e.g., mainly OH radicals) are more prevalent in the summer due to stronger solar radiation and higher temperatures and humidities, as well as elevated ozone concentrations. This typically leads to about a factor of two higher daytime OH concentrations in summer than in winter (CitationSeinfeld and Pandis 1998). Hourly ozone concentration peaked at over 60 ppb on 19, 20, and 21 July, with the maximum hourly level of 79 ppb in the mid-afternoon of 20 July. Upwind of Toronto in rural areas (e.g., Simcoe, ON) hourly ozone peaked at 86 ppb on 20 July. The elevated ozone concentration episodes on 19, 20, and 21 July coincided with higher nighttime SVOC (). In contrast, during the March period the hourly ozone was always below 45 ppb, except for 1 h of 48 ppb on the evening of 17 March. The correlation between OC and EC is often used as an indicator of SOA formation. A poorer correlation is expected when there is significant formation of SOA (CitationTurpin and Huntzicker 1991). During our sampling periods there was no correlation between IOGAPS-OC and IOGAPS-EC in July (R2 = 0.01), while the correlation was relatively strong in March (R2 = 0.78). Attributing the larger amount of July SVOC to SOA formation is also consistent with previous receptor model results for Toronto reported by CitationLee et al. (2003). They showed that the water-soluble, low molecular weight organic acids (LMW acids) and associated organic matter, which are known to be the photochemical products of hydrocarbons in the atmosphere (CitationCocker et al. 2001; CitationRohrl and Lammel 2001; CitationSaxena et al. 1995), contributed 23 and 6% to the total PM2.5 mass in the summer and winter of 2000, respectively.
Diurnal and Seasonal Variations of OC Determined by the IOGAPS and FP
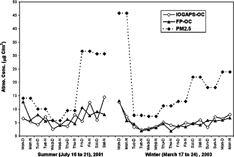
Due to accentuated evaporation of SVOC associated with the particles trapped by the filter, the OC on the IOGAPS quartz filter (IOGAPS-OCQ) is mostly nonvolatile, and thus its concentration is expected to be lower than the OC determined by the FP sampler (CitationLong et al. 2003). In addition, positive artifacts are expected to lead to higher FP-OC concentrations. shows that for each sampling event the IOGAPS-OCQ was lower than FP-OC. On average, the IOGAPS-OCQ was 70% lower during the day and 38% lower during the night in July, and 53% lower during the day and 35% lower during the night in March ().
The detailed TOT temperature profiles are consistent with the hypothesis that the gas-phase organics lead to positive artifacts during the FP sampling. The different OC fractions (i.e., OC1–OC4 and PC) were all higher, on average, during FP measurements compared to the IOGAPS. However, the biggest discrepancy was associated with OC1, which represents the OC with relatively high volatility (). For example, in July the daytime average FP-OC1 was 2.81 μ g C/m3, compared to an average IOGAPS-OC1 of 0.64 μ g C/m3; the average nighttime FP-OC1 was 2.26 μ g C/m3, compared to an average IOGAPS-OC1 of 0.73 μ g C/m3. In March, the daytime average FP-OC1 and IOGAPS-OC1 were 1.54 and 0.39 μ g C/m3, respectively. The average nighttime FP-OC1 was 1.15 μ g C/m3, compared to an average IOGAPS-OC1 of 0.37 μ g C/m3. Our findings are consistent with results reported by CitationPang et al. (2002c). They used two denuder-based samplers and a FP sampler to collect indoor PM2.5 samples in May 2001 in Seattle, WA. One was the IOGAPS equipped with an eight-channel, 27 cm long denuder; another was a personal particle organic and mass sampler (PPOMS), which used activated carbon-impregnated foam that functioned as both an inlet for PM2.5 and a denuder. The concentrations of different OC fractions (i.e., OC1–OC4) determined by these two samplers were close but were all lower than those determined by the FP sampler, with the OC1 concentrations significantly lower than the FP-OC1. However, it is important to note that since positive artifacts were also shown in OC2, OC3, and OC4 of FP samples (), excluding the low temperature peak(s) is not a viable approach for excluding the artifacts during FP measurements.
Although the daytime FP-OC was often higher than nighttime FP-OC, the IOGAPS-OC often showed the reverse pattern. shows that such diurnal variations in either FP-OC or IOGAPS-OC were more obvious in July. As mentioned earlier, higher nighttime IOGAPS-OC can be attributed to higher nighttime particulate SVOC, which was captured by the SIFs. Higher daytime FP-OC was caused by the adsorption of gas-phase SVOC onto the FP filters, which was removed by the denuder during IOGPAS sampling. It should be pointed out that if FP sampling is on a daily (24 h) basis or an even longer period, the effects of the positive and negative artifacts on OC measurement may cancel each other out or lead to significant error, depending on their relative magnitudes.
FIG. 4 Comparison of particulate OC determined by the IOGAPS and FP. Shown also is PM2.5 mass (μ g/m3) determined by a Partisol sampler on a daily basis.
OC Percentage in PMbm 2.5
PM2.5 mass was also measured on a daily basis during the two sampling campaigns by a Partisol sampler (i.e., USEPA FRM). The Partisol sampler used a single Teflon filter to collect ambient particles. The average PM2.5 mass concentrations, which are shown in , were 17.0 (5.8–31.8) and 18.6 (7.3–45.8) μ g/m3 in July and March, respectively. Statistical testing (t test) indicated that at 95% confidence level there was no significant difference in average PM2.5 mass between July and March. However, the average percentage of IOGAPS-OC mass in PM2.5 was significantly higher in July than that in March. Using a factor of 1.4 to convert IOGAPS-OC into organic matter (OM) (CitationLim and Turpin 2002), the average of OM percentage in PM2.5 was 65% (40–85%) in July and 45% (30–60%) in March. Use of a constant factor of 1.4 to adjust for the molecular form of the carbon species introduces some uncertainty in this percentage because its value varies in time and space. However, while a range of 1.2–2.6 has been used, 1.4 is a reasonable estimate for urban fine particles (CitationTurpin et al. 2000). Another uncertainty in the OM percentage in PM2.5 arises from a potential underestimation in PM2.5 measured by the Partisol sampler, which is caused by the loss of semivolatile materials (SVM) from the Teflon filter. Pang and coworkers operated their denuder-based PC-BOSS in Riverside and Bakersfield, CA (CitationPang et al. 2002a, b) and found the total PM2.5 mass determined by the USEPA FRM averaged ∼ 34% lower than that reconstructed from the PC-BOSS measurements (CitationPang et al. 2002b). This underestimation in PM2.5 was more significant during the summer due to a larger amount of SVM. These loses from the Partisol PM2.5 samples, if significant in Toronto, would lead to an over-estimate in our OM percentage values because the IOGAPS captures any lost SVOC but the Partisol would not.
CONCLUSION
Carbonaceous species (i.e., OC and EC) associated with PM2.5 were measured by both the integrated organic gas and particle sampler (IOGAPS) and filter pack (FP) sampler in July and March in downtown Toronto. Although it is difficult to estimate the relative contributions of positive or negative artifacts to particulate OC concentrations during FP sampling, the IOGAPS approach yields more-accurate measurements in OC due to its ability to remove gas-phase organics and its collection of particulate SVOC. The IOGAPS may also yield more-accurate measurements in EC due to the efficient removal of gas-phase SVOC. Such gas-phase SVOC is often adsorbed on FP filters and enhances charring during thermal analysis for OC and EC. The diurnal variation in the OC was largely affected by the SVOC, which is often not collected by a FP sampler or leads to artifact problems. Dramatic day–night differences in SVOC were observed during the summer campaign in July, evidence of the likelihood of significant SOA formation. In addition, a higher nighttime SVOC suggests that there is a “time lag” between the formation of SVOC in the gas phase and subsequent partitioning onto the existing aerosols. Consequently, subdaily temporal resolution in sampling is needed in order to study the SOA formation (CitationLim and Turpin 2002). Furthermore, sampling artifacts should be minimized so that accurate measurement of the amount of SVOC, and hence potential SOA materials, can be achieved. Although there was no significant difference in average daily PM2.5 mass during July and March, the average OC percentage in PM2.5 was significantly higher during July, which was largely caused by the higher SVOC concentrations.
Acknowledgments
The financial support for this study was from the Toxic Substances Research Initiative, Government of Canada. We thank Dr. Douglas A. Lane and Dr. Cristian Mihele of Meteorological Service of Canada (MSC) of Environment Canada for the training of denuder coating and SIF preparation, and Dr. Lara A. Gundel of Lawrence Berkeley National Laboratory for technical discussion. We also thank Melynda Bitzos of Ontario Ministry of Environment for providing us with ozone data collected in downtown Toronto.
Related Research Data
REFERENCES
- Allen , J. O. , Dookeran , K. M. , Smith , K. A. , Sarofim , A. F. , Taghizadeh , K. and Lafleur , A. L. 1996 . Measurement of Polycyclic Aromatic Hydrocarbons Associated with Size-Segregated Atmospheric Aerosols in Massachusetts . Environ. Sci. Technol. , 30 : 1023 – 1031 .
- Allen , J. O. , Dookeran , N. M. , Taghizadeh , K. , Lafleur , A. L. , Smith , K. A. and Sarofim , A. F. 1997 . Measurement of Oxygenated Polycyclic Aromatic Hydrocarbons Associated with a Size-Segregated Urban Aerosol . Environ. Sci. Technol. , 31 : 2064 – 2070 .
- Birch , M. E. and Cary , R. A. 1996 . Elemental Carbon-Based Method for Monitoring Occupational Exposures to Particulate Diesel Exhaust . Aerosol Sci. Technol. , 25 : 221 – 241 .
- Brook , J. R. , Lillyman , C. D. , Shepherd , M. F. and Mamedov , A. 2002 . Regional Transport and Urban Contributions to Fine Particle Concentrations in Southeastern Canada . J. Air and Waste Manage. Assoc. , 52 : 855 – 866 .
- Chow , J. C. , Watson , J. G. , Pritchett , L. C. , Pierson , W. R. , Frazier , C. A. and Purcell , R. G. 1993 . The DRI Thermal Optical Reflectance Carbon Analysis System Description, Evaluation and Applications in United States Air Quality Studies . Atmos. Environ. , 27 : 1185 – 1201 .
- Cocker , D. R. , Whitlock , N. E. , Flagan , R. C. and Seinfeld , J. H. 2001 . Hygroscopic Properties of Pasadena, California Aerosol . Aerosol Sci. Technol. , 35 : 637 – 647 .
- Eatough , D. J. , Eatough , N. L. , Obeidi , F. , Pang , Y. B. , Modey , W. and Long , R. 2001 . Continuous Determination of PM2.5 Mass, Including Semi-Volatile Species . Aerosol Sci. Technol. , 34 : 1 – 8 .
- Fan , X. , Brook , J. R. and Mabury , S. A. 2003 . Sampling Atmospheric Carbonaceous Aerosols Using an Integrated Organic Gas and Particle Sampler . Environ. Sci. Technol. , 37 : 3145 – 3151 .
- Finlayson-Pitts , B. J. and Pitts , J. N. 2000 . Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Application , San Diego : Academic Press .
- Gundel , L. A. , Daisey , J. M. and Stevens , R. K. 1998 . Quantitative Organic Vapor-Particle Sampler US Patent 5,763,360
- Gundel , L. A. , Lee , V. C. , Mahanama , K. R. R. , Stevens , R. K. and M. , D. J. 1995 . Direct Determination of the Phase Distributions of Semivolatile Polycyclic Aromatic Hydrocarbons Using Annular Denuders . Atmos. Environ. , 29 : 1719 – 1733 .
- Kavouras , I. G. , Lawrence , J. , Koutrakis , P. , Stephanou , E. G. and Oyola , P. 1999 . Measurement of Particulate Aliphatic and Polynuclear Aromatic Hydrocarbons in Santiago de Chile: Source Reconciliation and Evaluation of Sampling Artifacts . Atmos. Environ. , 33 : 4977 – 4986 .
- Lane , D. A. , Peters , A. J. , Gundel , L. A. , Jones , K. C. and Northcott , G. L. 2000 . Gas/Particle Partition Measurements of PAH at Hazelrigg, UK . Polycycl. Aromat. Comp. , 20 : 225 – 234 .
- Larson , S. M. and Cass , G. R. 1989 . Characteristics of Summer Mid-Day Low-Visibility Events in the Los Angeles Area . Environ. Sci. Technol. , 23 : 281 – 289 .
- Lee , P. K. H. , Brook , J. R. , Dabek-Zlotorzynska , E. and Mabury , S. A. 2003 . Identification of the Major Sources Contributing to PM2.5 Observed in Toronto . Environ. Sci. Technol. , 37 : 4831 – 4840 .
- Lewtas , J. , Pang , Y. B. , Booth , D. , Reimer , S. , Eatough , D. J. and Gundel , L. A. 2001 . Comparison of Sampling Methods for Semi-Volatile Organic Carbon Associated with PM2.5 . Aerosol Sci. Technol. , 34 : 9 – 22 .
- Liang , C. , Pankow , J. F. , Odum , J. R. and Seinfeld , J. H. 1997 . Gas/Particle Partitioning of Semivolatile Organic Compounds to Model Inorganic, Organic, and Ambient Smog Aerosols . Environ. Sci. Technol. , 31 : 3086 – 3092 .
- Lim , H. J. and Turpin , B. J. 2002 . Origins of Primary and Secondary Organic Aerosol in Atlanta: Results of Time-Resolved Measurements During the Atlanta Supersite Experiment . Environ. Sci. Technol. , 36 : 4489 – 4496 .
- Long , R. W. , Eatough , N. L. , Mangelson , N. F. , Thompson , W. , Fiet , K. , Smith , S. , Smith , R. , Eatough , D. J. , Pope , C. A. and Wilson , W. E. 2003 . The Measurement of PM2.5, Including Semi-Volatile Components, in the EMPACT Program: Results from the Salt Lake City Study . Atmos. Environ. , 37 : 4407 – 4417 .
- Mader , B. T. , Flagan , R. C. and Seinfeld , J. H. 2001 . Sampling Atmospheric Carbonaceous Aerosols Using a Particle Trap Impactor/Denuder Sampler . Environ. Sci. Technol. , 35 : 4857 – 4867 .
- Odum , J. R. , Jungkamp , T. W. P. , Griffin , R. J. , Forstner , H. J. L. , Flagan , R. C. and H. , S. J. 1997 . Aromatics, Reformulated Gasoline and Atmospheric Organic Aerosol Formation . Environ. Sci. Technol. , 31 : 1890 – 1897 .
- Pang , Y. B. , Eatough , D. J. and Eatough , N. L. 2002a . Effect of Semivolatile Material on PM2.5 Measurement by the PM2.5 Federal Reference Method Sampler at Bakersfield, California . Aerosol Sci. Technol. , 36 : 289 – 299 .
- Pang , Y. B. , Eatough , D. J. and Eatough , N. L. 2002b . PM2.5 Semivolatile Organic Material at Riverside, California: Implications for the PM2.5 Federal Reference Method Sampler . Aerosol Sci. Technol. , 36 : 277 – 288 .
- Pang , Y. B. , Gundel , L. A. , Larson , T. , Finn , D. , Liu , L. J. S. and Claiborn , C. S. 2002c . Development and Evaluation of a Personal Particulate Organic and Mass Sampler . Environ. Sci. Technol. , 36 : 5205 – 5210 .
- Peters , A. J. , Lane , D. A. , Gundel , L. A. , Northcott , G. L. and Jones , K. C. 2000 . A Comparison of High Volume and Diffusion Denuder Samplers for Measuring Semivolatile Organic Compounds in the Atmosphere . Environ. Sci. Technol. , 34 : 5001 – 5006 .
- Pinto , J. P. , Stevens , R. K. , Wills , R. D. , Kellogg , R. , Mamane , Y. , Novak , J. , Santroch , J. , Benes , I. , Lenicek , J. and Bures , V. 1998 . Czech Air Quality Monitoring and Receptor Modeling Study . Environ. Sci. Technol. , 32 : 843 – 854 .
- Rohrl , A. and Lammel , G. 2001 . Low Molecular Weight Dicarboxylic Acids and Glyoxylic Acid: Seasonal and Air Mass Characteristics . Environ. Sci. Technol. , 35 : 95 – 101 .
- Salma , I. , Chi , X. G. and Maenhaut , W. 2004 . Elemental and Organic Carbon in Urban Canyon and Background Environments in Budapest, Hungary . Atmos. Environ. , 38 : 27 – 36 .
- Saxena , P. , Hildemann , L. M. , Mcmurry , P. H. and Seinfeld , J. H. 1995 . Organics Alter Hygroscopic Behavior of Atmospheric Particles . J. Geophys. Res. Atmos. , 100 : 18755 – 18770 .
- Seinfeld , J. H. and Pandis , S. N. 1998 . Atmospheric Chemistry and Physics: From Air Pollution to Climate Change , New York : John Wiley and Sons, Inc. .
- Sharma , S. , Brook , J. R. , Cachier , H. , Chow , J. , Gaudenzi , A. and Lu , G. 2002 . Light Absorption and Thermal Measurements of Black Carbon in Different Regions of Canada . J. Gephys. Res. , 107 : AAC 11-1 – AAC 11-11 .
- Tan , P. V. , Evans , G. J. , Tsai , J. , Owega , S. , Fila , M. S. and Malpica , O. 2002 . On-Line Analysis of Urban Particulate Matter Focusing on Elevated Wintertime Aerosol Concentrations . Environ. Sci. Technol. , 36 : 3512 – 3518 .
- Turpin , B. J. and Huntzicker , J. J. 1991 . Secondary Formation of Organic Aerosol in the Los-Angeles Basin—A Descriptive Analysis of Organic and Elemental Carbon Concentrations . Atmos. Environ. , 25 : 207 – 215 .
- Turpin , B. J. , Saxena , P. and Andrews , E. 2000 . Measuring and Simulating Particulate Organics in the Atmosphere: Problems and Prospects . Atmos. Environ. , 34 : 2983 – 3013 .
- Yu , J. Z. , Xu , J. H. and Yang , H. 2002 . Charring Characteristics of Atmospheric Organic Particulate Matter in Thermal Analysis . Environ. Sci. Technol. , 36 : 754 – 761 .
- Viidanoja , J. , Sillanpaa , M. , Laakia , J. , Kerminen , V. M. , Hillamo , R. , Aarnio , P. and Koskentalo , T. 2002 . Organic and Black Carbon in PM2.5 and PM10: 1 Year of Data from an Urban Site in Helsinki, Finland . Atmos. Environ. , 36 : 3183 – 3193 .
- Volckens , J. and Leith , D. 2003 . Effects of Sampling Bias on Gas-Particle Partitioning of Semi-Volatile Compounds . Atmos. Environ. , 37 : 3385—3393