Abstract
A stable numerical solution to the unsolved problem of nonequilibrium growth/evaporation at long time step of multiple dissociating acids (e.g., nitric acid, hydrochloric acid, carbonic acid, sulfuric acid, etc.) and a base (ammonia) is discussed. The solution eliminates most oscillatory behavior observed in previous solutions at long time step. The solution is applicable across the entire relative humidity range, both in the presence and absence of solids and among multiple aerosol size bins and distributions. It involves solving growth/evaporation of semivolatile acids with a dissolutional growth scheme at high liquid water content (LWC), semivolatile acids with a condensational growth scheme at low LWC, involatile acids with a condensation scheme at all LWCs, then equilibrating ammonia and pH simultaneously between gas and solution phases at all LWCs based on updated acid contents, and finally solving internal-aerosol composition, final pH, and LWC with an operator-split equilibrium calculation. The new method at long time step (150–300 s) compares well with, and is 10–60 times faster than, a previous solution at short time step over the entire size distribution. Solutions at short and long time steps converge to equilibrium solutions when unique equilibrium solutions exist, even in the presence of calcium, magnesium, potassium, and carbonate. The new scheme is referred to as PNG-EQUISOLV II, where PNG is Predictor of Nonequilibrium Growth. Analysis with the scheme suggests that, under some conditions of high relative humidity and concentration, some coarse-mode particles (< 6 μ m diameter) may reach equilibrium on time scales < 1 h.
INTRODUCTION
The transfer of a gas to and from a population of particles is a time-dependent process. The numerical treatment of this process is complicated by the fact that several gases transfer simultaneously and may interact with each other chemically in particle solutions or on particle surfaces. In particular, the simultaneous transfer of acids such as nitric acid, hydrochloric acid, and sulfuric acid, and a base such as ammonia, results in the instantaneous adjustment to pH, particle composition, and vapor pressure that renders the resulting set of equations stiff and difficult to solve at long time step.
Several studies of the simultaneous nonequilibrium transfer of acids and bases have been carried out. CitationWexler and Seinfeld (1990) derived expressions and time scales to equilibrium for the transfer of NH3, HNO3, and HCl to a population of solid particles containing NH4Cl(s) and NH4NO3(s). CitationJacobson et al. (1996) derived expressions for the transfer of any soluble gas to a population of particles containing liquid water, where the solution accounted for the instantaneous change in vapor pressure over the surface due to dissolution, and for the operator-split hydration of liquid water and formation of solids and ions. The growth equations were solved with a sparse-matrix ordinary differential equation solver. CitationMeng and Seinfeld (1996) solved growth expressions of CitationWexler and Seinfeld (1990) at short time step and found that the fine mode of particles was closer to equilibrium than the coarse mode.
CitationJacobson (1997a, b, c) derived and applied to Los Angeles a noniterative, mass-conserving, and unconditionally stable solution to dissolutional growth of a gas to a population of particles containing any amount of liquid water on their surfaces. The growth solution allowed vapor pressures to adjust instantaneously as a gas dissolved in solution and was operator split from a chemical equilibrium solution that calculated liquid water content (LWC), pH, ion dissociation, activity coefficients, and solid composition within each particle size bin. After gases dissolved in even a tiny amount of water during the growth step, solids formed during the equilibrium step. It was found that, when the operator-split time interval between growth and equilibrium was < 15 s, the numerical solution was relatively smooth. At longer time step, though, the solution “produced oscillations” due to delays in feedback between the equilibrium and growth calculations. For example, at long time step the delay in the update to pH and sulfuric acid dissociation caused too much or too little growth during the step that was overcompensated for during the next time step. Another drawback was that, at low liquid water contents (e.g., < 0.01 μ g m− 3, summed over all particles), solids took too long to reach equilibrium because the LWC allowed only a small amount of gas to dissolve each growth time step, and a 15 s time step between growth and equilibrium delayed the gas-to-solid conversion. CitationJacobson (2002) tried to address this latter issue by solving growth of solid-forming gases as a condensation instead of dissolution process in size bins where solids already existed.
CitationMeng et al. (1998) solved nonequilibrium growth in a modeling study of Los Angeles. Vapor pressures, which were fixed during the growth calculation, were determined from an operator-split equilibrium calculation. The time step for growth was variable and dependent on the stiffness of the system, thereby limiting it.
CitationSun and Wexler (1998a, b) discuss the same time step limitation found in CitationJacobson (1997c), stating that a “very small time step” is necessary to integrate the stiff growth equations for HCl, HNO3, and NH3. They proposed calculating the growth rates of all three gases independently, then recalculating the growth rate of one of the gases assuming that pH stays constant during growth. CitationPilinis et al. (2000) also discussed the oscillatory behavior of acid/base growth at long time step. They postulated that holding pH constant to resolve this issue might cause errors in the growth solution and proposed calculating a “flux” of hydrogen ion to each particle size bin equal to the net change in ion concentration due to the fluxes of all acid and base gases, including HNO3, HCl, NH3, and H2SO4. To minimize oscillations, the hydrogen ion flux was limited, forcing a limit on the flux of one or more acid or base gases as well. They solved the growth equations with a stiff ODE solver, which still required a short time step. The time constraint was significant enough that CitationCapaldo et al. (2000) developed a hybrid scheme that solved fine-mode sizes with an equilibrium solution and coarse-mode sizes with a nonequilibrium solution. CitationKoo et al. (2003) compared these two approaches with a pure equilibrium approach.
Here, a scheme is developed that solves nonequilibrium growth operator split from equilibrium chemistry with remarkably stability and accuracy at long time step (e.g., 150–300+ s), at all relative humidities across the entire size distribution. The growth portion of the scheme is referred to as the predictor of nonequilibrium growth analytical predictor of dissolution (PNG) scheme. The equilibrium scheme is EQUISOLV II. The coupled scheme is PNG-EQUISOLV II.
NUMERICAL METHOD
The PNG-EQUISOLV II scheme involves the solution of five processes: (Equation1) dissolutional growth of semivolatile acid gases at high and moderate LWCs; (Equation2) condensational growth of semivolatile acid gases at low LWCs; (Equation3) condensational growth of involatile gases at all LWCs; (Equation4) equilibration of NH3/NH4 +and pH between the gas phase and all particle size bins while conserving charge and moles; and (Equation5) equilibration of internal-aerosol liquid, ion, and solid composition, pH, liquid water content, and activity coefficients.
The method described here differs from that of CitationSun and Wexler (1998a, b) in that this method allows vapor pressures to adjust instantaneously as a gas dissolves, and it equilibrates ammonia and pH with all particle sizes following acid growth rather than fixing pH and readjusting the growth rate of an acid or NH3 at the fixed pH, as was done in that study. The method also differs from that of CitationPilinis et al. (2000) in that this method equilibrates ammonia and pH after acid growth, whereas that method allowed acids and NH3 to grow independently and then adjusted the pH based on the change in charge-balance alone, while limiting the pH change and growth rates of acids and bases with a stability limiter. Here no stability limits are necessary, and the pH adjustment conserves charge among acid gases following their growth, and it also satisfies mole conservation and the NH3/NH4 + equilibrium constraint among all size bins. In addition, the ammonia equilibration technique here is applied during both liquid and solid formation. Below, the five steps in the procedure are described.
Dissolutional Growth of Semivolatile Gases at High LWC
When a liquid solution pre-exists on a particle surface, the growth of semivolatile gases onto the particle is controlled by dissolution. The growth equation for a semivolatile acid gas, such as hydrochloric acid (HCl), that dissolves then dissociates (in this case to the hydrogen ion, H+, and the chloride ion, Cl−) in solution, may be written as
The gas concentration of each growing species is linked to its aqueous concentration among all size bins through the mole-conservation relationship,
The mole concentration of any dissolved species q in size bin i is related to its molality (m q,i , mol kg− 1) by
In the case of HCl, its saturation mole concentration is calculated by considering the equilibrium equations, HCl ⇔ HCl(aq) and HCl(aq) ⇔ H+ + Cl−. The equilibrium coefficient relations for these equations are
Integrating EquationEquation (10) gives the growth solution for the chloride ion to a size bin as
An alternative to the exponential solution given in EquationEquations (11) and (12) is the semiimplicit solution, given here as
Growth equations for other semivolatile acid gases, such as nitric acid, carbon dioxide, formic acid, etc. are solved in the same manner as those for hydrochloric acid. In the case of carbon dioxide, the adjusted equilibrium coefficient (EquationEquation (9)) must include a second dissociation constant. Equations for all gases can be written to treat dissolution into multiple aerosol size distributions in a manner similar to that done in CitationJacobson (2002).
Condensational Growth of Semivolatile Gases at Low LWCs
EquationEquations (13) and (14) can be solved for any finite amount of liquid water on a particle surface. However, when the total (over all sizes) aerosol LWC decreases to below about 0.01 μ g m− 3, solid formation from dissolution proceeds slowly at long time step since solids form only during the operator-splitting equilibrium calculation. If the time-interval between growth and equilibrium is long, the little gas that dissolves in a small amount of water during each growth step cannot convert to a solid rapidly each equilibrium calculation. This problem is addressed here by treating HCl and HNO3 growth as condensation rather than dissolution processes when the LWC is below 0.01 μ g m− 3. At such low water contents, growth of CO2 and other acids are still modeled as dissolution processes since (Equation1) such acids are less important with respect to solid formation than are HCl or HNO3, and (Equation2) the dissolution scheme, which still allows solids to form through the equilibrium calculation, is unconditionally stable for any nonzero LWC.
When an acid gas grows onto particles to eventually form a solid, it is modeled as first condensing as a liquid, but with its saturation mole concentration determined from gas-solid equilibrium to ensure that the correct amount of acid gas transfers to particle surfaces for solid formation. Unlike with dissolution where the SMC of the gas is an instantaneous function of its molality in solution, the SMC for a condensing gas is held constant during a growth time step.
During condensation, liquids (or their dissociation products) enter into the small (nonzero) amount of liquid water on the particle surface. The amount of ammonia that condenses simultaneously is discussed in section “Equilibration of NH3 with Growing Acids at all LWCs” below. Because the particle solution is now supersaturated with respect to the solid phase, solids readily form from the solution during the subsequent internal-aerosol equilibrium calculation. During that calculation, the molalities of the solution components (e.g., liquids and ions) quickly drop (due to solid formation) to satisfy solution–solid equilibrium constraints so that they are well within the range of molalities for which solute activity coefficients are valid. This holds true even if the LWC drops to machine precision (10− 40μ g m− 3 on some computers and 10− 250μ g m− 3 on others) because the numerical equilibrium solver (EQUISOLV II) always converges, and the converged solution between ions and solids requires that the solubility product of a given solid equals the product of the activities of the ions making up the solid. At equilibrium, these activities (molalities multiplied by activity coefficients) are relatively small or modest, regardless of the LWC.
It should be noted here that, although the method of acid nonequilibrium growth is changed from a dissolution to a condensation process at a fixed LWC (0.01 μ g m− 3), the water content may decrease to machine precision during the subsequent equilibrium calculation. In such cases, all liquids or ions effectively convert to solids while solid-solution equilibrium is maintained. Because equilibrium is maintained, the solute concentration (e.g., moles cm− 3-air) in the trivial amount of liquid water is also trivial, whereas the solute molality (moles kg− 1-water) is nontrivial and satisfies equilibrium constraints. In sum, this numerical solution is physically realistic, since it allows the water content naturally to tend toward zero. All other numerical treatments of solid formation assume that water either exists or doesn't exist in the particle, which results in an error in composition when this assumption is incorrect.
The SMCs of HCl, and HNO3 are solved here simultaneously by considering the two gas–solid equilibrium reactions, NH4NO3(s) ⇔ NH3 + HNO3 and NH4Cl(s) ⇔ NH3 + HCl. Either solid may form within aerosol particles when either (1) the relative humidity (RH) is increasing and less than the deliquescence relative humidity (DRH) of the solid, or (2) the RH is decreasing and less than the crystallization relative humidity (CRH) of the solid. DRHs and CRHs and references are given in CitationJacobson (1999a, Table 18.4). Although a solid can form when the RH is below the solid's DRH or CRH, the solid may or may not form. When multiple ions are present in solution and multiple solids can form, some may form below their DRH/CRH and others may not. When solids do form, all equilibrium constraints are satisfied (solution charge balance, solution–solid equilibrium, solution–solution equilibrium, mass balance).
For determining whether acid gases will be allowed to condense in the model eventually to form solids, one other criterion must be satisfied. Namely, for ammonium nitrate and/or ammonium chloride to form, the equations,
If only ammonium nitrate can form, EquationEquation (20) is solved by removing K NH4Cl and setting C 0 = C NH3, t − h − C HNO3, t − h . If only ammonium chloride can form, the equation is solved by removing K NH4NO3 and setting C 0 = C NH3, t − h − C HCl,t − h .
Once the SMCs for HNO3 and HCl are calculated, they are substituted into condensational growth equations and solved with the noniterative, unconditionally stable condensation solver described by EquationEquations (13)–(17) of CitationJacobson (2002) with D Ni = 0.
Condensational Growth of Involatile Gases At All LWCs
Condensation/evaporation of relatively involatile (extremely low vapor pressure) gases, such as sulfuric acid and high molecular weight organics, is solved as a condensation process among all size bins at low and high LWC (EquationEquations (13)–(17)) of CitationJacobson 2002 with D Ni = 0). In this case, the SMCs are determined a priori from data or parameterizations that are a function of temperature.
Equilibration of NH 3 with Growing Acids At All LWCs
Following the growth calculation for all acid gases, ammonia is equilibrated with all size bins, conserving charge among all ions, including those entering solutions during the dissolution and condensation steps of the three preceding sections. The calculation conserves moles and satisfies the gas–aerosol equilibrium constraint for ammonia. With the present method, the quantity of ammonium added to each size bin each time step is the exact amount necessary to balance charge among all ions in solution as determined after the growth calculation for such ions. Because the equilibration of ammonia is calculated after the diffusion-limited growth of all acids, ammonia growth is effectively a nonequilibrium growth process. This technique allows smooth solutions and convergence at long time step among all growing species. The alternative to equilibrating ammonia is to solve the same nonequilibrium equations for HNO3 or HCl, but this leads to oscillations in the solution at long time step, as discussed in the introduction.
With the present method, the charge balance equation for ammonium within each size bin is
The mole balance equation between gas-phase ammonia and the ammonium ion within each aerosol size bin is
Finally, the gas-aerosol equilibrium relations for ammonia are NH3(g) ⇔ NH3(aq) and NH3(aq) + H+ ⇔ NH4 +. The equilibrium coefficient relations for these equations are
In mole concentration units, EquationEquations (25) and (26) simplify to
Combining EquationEquations (28) and (29) gives an expression for the hydrogen ion concentration as
Substituting EquationEquation (31) and c NH3 (aq),i,t = C NH3,t H′NH3,i,t − h into the mole balance equation, EquationEquation (24), gives
This procedure works for any LWC above zero. Thus, when HNO3 and/or HCl condense (section “Condensational Growth of Semivolatile Gases at Low LWCs” below) at negligible LWC, the procedure calculates the amount of ammonia transferred to each bin to ensure mole and charge balance and satisfaction of the ammonia/ammonium equilibrium constraint. During the subsequent equilibrium calculation supersaturated nitrate, chloride, and ammonium may crystallize to form solids through the ion–solid equilibrium equations, NH4NO3(s) ⇔ NH4 + + NO3 − and NH4Cl(s) ⇔ NH4 + + Cl−. Alternatively, the ions may hydrate liquid water if conditions are right. Thus, the resulting particle can consist of both aqueous and solid components.
Operator-Split Equilibrium Calculation
Following the ammonia calculation, an operator-split internal aerosol equilibrium calculation is performed to recalculate aerosol ion, liquid, and solid composition, activity coefficients, and LWC, accounting for all species in solution in each size bin. The equilibrium solution is obtained with EQUISOLV II (CitationJacobson 1999b), which is iterative, positive-definite, and mole and charge conserving, regardless of the number of iterations taken. Since all growth solutions discussed are positive-definite and mole conserving between the gas phase and all particle size bins, and since equilibrium is solved after growth, the entire PNG scheme is mole and charge conserving and positive-definite regardless of the time step size for growth or between growth and equilibrium.
RESULTS
The PNG-EQUISOLV II scheme is analyzed here by comparing time-dependent results from it at long time step with results from the scheme under the same conditions at short time step and with results from simulations in which only gas-aerosol equilibrium (except as described shortly) was considered. In all cases, including the equilibrium-only cases, 60 aerosol size bins were used. In the equilibrium-only cases, sulfuric acid was allowed to condense in a nonequilibrium manner onto all size bins, but all other gases were equilibrated with the size distribution during and after condensation. A unique solution exists to equilibrium among multiple size bins in all cases except when a solid forms from two gases (e.g., NH4NO3(s) and NH4Cl(s)). In such cases, there is no unique size bin to which the solids go (e.g., CitationWexler and Seinfeld 1990). This constraint does not apply to solids such as NaNO3(s) or (NH4)2SO4(s) that form from one gas and one involatile liquid/ion (CitationJacobson 1999b). When NH4NO3(s) or NH4Cl(s) form, the equilibrium solution is also not the correct solution with respect to total mass (although it is close) because the formation of these solids in random size bins affects the rate of sulfuric acid condensation, which depends on particle size, and the relative amount of sulfate in a bin affects the equilibrium partitioning of bases and other acids to that bin.
shows the initial number and mass aerosol distribution for all cases except one (). Aerosol particles were initialized with 10 μ g m− 3 NaCl(s) and 20.4 μ g m− 3 of inert material. The temperature in all cases was 298 K.
FIG. 1 (a) Initial model size distribution for all simulations, except those for . The distribution consists of 10 μ g m− 3 NaCl(s) and 20.4 μ g m− 3 of inert particulate matter, spread over 60 size bins.
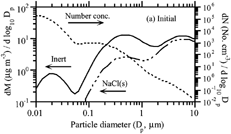
FIG. 7 (a) and (b) Same as , respectively, except when the initial particle size distribution contained 0.5, 0.5, and 0.5 μ g m− 3 K2CO3(s), CaCO3(s), and MgCO3(s), respectively, in the accumulation mode and 1.0, 3.0, and 3.0 μ g m− 3 K2CO3(s), CaCO3(s), and MgCO3(s), respectively, in the coarse mode.
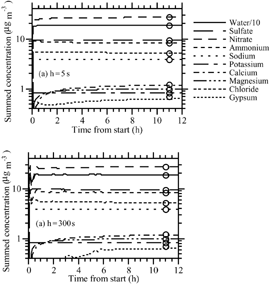
Several simulations were run for different initial gas concentrations and relative humidities. shows results for the first case, in which initial gas concentrations were 30 μ g m− 3 HNO3(g), 10 μ g m− 3 NH3(g), 0 μ g m− 3 HCl(g), and 10 μ g m− 3 H2SO4(g), and the RH was 90%. During the simulation period, H2SO4 condensed, HCl and HNO3 dissolved/dissociated, and NH3 equilibrated with dissolved and dissociated species, as described in section “Numerical Method” above. Water uptake and aerosol composition were calculated during each operator-split equilibrium calculation. show time-series of the aerosol concentrations, summed over all size bins, when the operator-split time interval between growth and equilibrium was 5 s and 300 s, respectively. The figures also show results from the equilibrium-only calculation. Results from both operator-split time steps matched each other and the equilibrium solution fairly accurately, although the longer time step resulted in a small error during the first 15 min. Equilibrium was reached within about 6 h in both simulations.
FIG. 2 (a) Time series of aerosol component concentrations, summed over all size bins, from a simulation of dissolutional growth coupled with chemical equilibrium (solved with PNG-EQUISOLV II) when the operator-split time interval between growth and equilibrium was h = 5 s. The circles are equilibrium solutions, found from EQUISOLV II alone (except that sulfuric acid condensed as a nonequilibrium process). Initial gas concentrations were 30 μ g m− 3 HNO3(g), 10 μ g m− 3 NH3(g), 0 μ g m− 3 HCl(g), and 10 μ g m− 3 H2SO4(g). In addition, RH = 90% and T = 298 K. (b) Same as , but with h = 300 s.
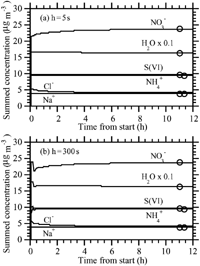
shows results under the same conditions as , except the initial nitric acid gas concentration was 0.1 μ g m− 3 instead of 30 μ g m− 3. Whereas, examined how the scheme performed under high nitrate conditions, this test examines how it performed under low nitrate conditions. In this case, the scheme demonstrated near-perfect accuracy at 300 s, even during the first 15 min. show the mass size distributions of individual components after 12 h when h = 5 s and h = 300 s, respectively. The distributions also matched accurately.
FIG. 3 (a) Same as (h = 5 s case), except HNO3(g) = 0.1 μ g m− 3 initially. (b) Same as (h = 300 s case), except HNO3(g) = 0.1 μ g m− 3 initially. (c) Size distribution after 12 h of the h = 5 s case shown in . (d) Size distribution after 12 of the h = 300 s case shown in .
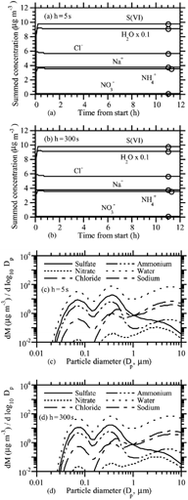
shows results under the same conditions as , except the initial ammonia gas concentration was 0.1 μ g m− 3 instead of 30 μ g m− 3. This test examines how the scheme performed under extremely low ammonia conditions. This test is of particular interest and possibly the toughest test of the code, since the PNG scheme is premised on the assumption that ammonia equilibrates with all sizes following acid growth. If ammonia is extremely low, one might think that the scheme would fail or result in severe oscillations. shows that, with a 5 s time step, the solution was perfectly smooth and matched the equilibrium solution within 6–8 h. shows that, with a 150 s time step, the solution was slightly oscillatory but not unstable. It was also slightly less accurate than the 5 s solution after 12 h. Despite the slight errors, this test demonstrates the ability of the code to perform well under an extreme condition. At a 300 s time step (not shown) the solution converged to values similar to those at 150 s but with greater oscillation for some species. show the size distribution from the equilibrium calculation and from the 5 s nonequilibrium calculation after 12 h. The figures are nearly identical, suggesting that the nonequilibrium solution converged to the equilibrium solution at all sizes.
FIG. 4 (a) Same as (h = 5 s case), except NH3(g) = 0.1 μ g m− 3 initially. (b) Same as , except NH3(g) = 0.1 μ g m− 3 initially and h = 150 s. (c) Size distribution from the equilibrium calculation corresponding to the circles in . (d) Size distribution after 12 h of the h = 5 s case shown in . (e) Size distribution of nitrate and chloride ions after 15 mins, 1 h, and 12 h of the h = 5 s case shown in . (Continued on next page)
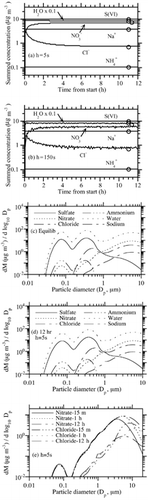
is also of interest physically for two reasons. First, it shows the time dependence of the well-known process of sea-spray acidification. Sea-spray acidification is the depletion of chloride from sea-spray drops when acid gases dissolve into them, reducing their pH and causing HCl to become supersaturated and evaporate (e.g., Ericksson 1960; CitationHitchcock et al. 1980). shows that the addition of these acids caused a 60% reduction in the chloride ion after 15 min, a 76% reduction after 1 h, and an 87% reduction after 12 h. Thus, chloride loss was relatively fast under the conditions tested (RH = 90%; high acid concentrations). shows that the greatest percentage loss in chloride occurred in submicron-sized particles. Several studies have shown that chloride depletion from acidification occurs more in small than large particles (e.g., CitationJunge 1957; CitationWilkniss and Bressan 1972; CitationMartens et al. 1973).
Second, shows that, under the conditions tested, particles smaller than 3 μ m diameter reached equilibrium within 15 min, particles smaller than 6 μ m diameter reached equilibrium within an hour, and the rest reached equilibrium within 12 h. Several studies have suggested or calculated that small particles are more likely to be in equilibrium than large particles (e.g., CitationMeng and Seinfeld 1996; CitationCapaldo et al. 2000; CitationPilinis et al. 2000; CitationFridlind and Jacobson 2000, CitationMoya et al. 2001). supports these results but also suggests that, in some situations (e.g., at high RH and at high gas concentration), large particles smaller than 6 μ m in diameter may reach equilibrium on the time scale of less than an hour. Thus, some exceptions may exist to the previously established result that coarse-mode particles are not in equilibrium. This conclusion is not unexpected because previous studies did not examine all possible cases.
shows results under the same initial concentration conditions as , but with RH = 50% and assuming solids formed only if the RH was less than the crystallization RH of the solid (e.g., the RH was decreasing). In this case, NaCl(s) was present initially and gradually disappeared, replaced in part by Na2SO4(s). The nonequilibrium solutions at both time steps (5 s and 150 s) approached the equilibrium solution nearly exactly, not only with respect to solid composition but also with respect to ion composition and LWC. The solution at a time step of 150 s showed small oscillations for Na2SO4(s) and a small error but remained stable. Oscillations grew at time steps > 150 s (not shown), but the solution converged to final values similar to those at 150 s.
FIG. 5 (a) Same as (h = 5 s case), except with RH = 50%. (b) Same as , except with RH = 50% and h = 150 s.
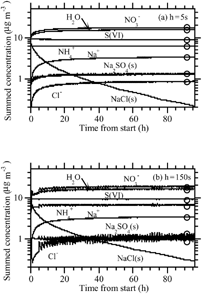
shows results under the same initial concentration conditions as , but at RH = 10%. At this RH, the LWC was always trivially small, and growth of ammonia and nitric acid was calculated as a condensation process (section “Condensational Growth of Semivolatile Gases at Low LWCs” below). Solids formed during the operator-split equilibrium calculation. The figure shows that the result with a 300 s growth time step and growth-equilibrium splitting interval was nearly the same as at 5 s. The equilibrium solution matched the nonequilibrium results relatively well. However, as discussed at the beginning of this section, the equilibrium solution is not the correct solution in this case, since NH4NO3(s) and NH4Cl(s) formed, and there is no unique equilibrium solution to the size bins in which they form.
FIG. 6 (a) and (b) Same as , respectively, except with RH = 10%.
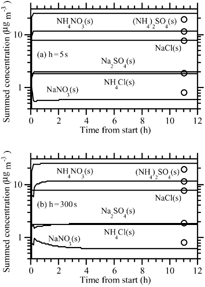
Finally, shows a case under the same conditions as , except the initial particle size distribution contains K2CO3(s), CaCO3(s), and MgCO3(s) in the accumulation and coarse modes in addition to NaCl(s) and inert material. As acids entered the particles, the carbonates dissolved, forcing carbon dioxide to the gas phase. Some of the calcium reassociated with sulfate to form gypsum (CaSO4). The rest remained unassociated. The numerical solution with a 300 s time step approached that at 5 s, and both nearly matched the equilibrium solution after about 8 h, although complete equilibrium did not occur until about 24 h in this case.
SUMMARY
A new method of solving nonequilibrium acid/base gasparticle transfer at long time step was derived. The method involves solving dissolutional or condensational growth equations for acids, then equilibrating ammonium and pH between the gas phase and all aerosol size bins at the end of each growth time step for acids, then updating aerosol composition, pH, and LWC with an operator-split internal-aerosol equilibrium calculation. The scheme was applied to cases at several RHs and concentration regimes, including in the presence of calcium, magnesium, potassium, and carbonate. Solutions at long time step (150 or 300 s) were compared with solutions at 5 s time step. Results in all cases matched remarkably well and converged to equilibrium solutions. Since nonoscillatory solutions to nonequilibrium growth across an entire size distribution for liquids and solids were limited previously to time steps of 5–15 s, the new scheme allows for a factor of 10–60 speedup in comparison with such solutions. A physical finding from this study was that under at least some conditions (e.g., high RH, high concentrations, and in the absence of solid formation) particles smaller than 6 μ m diameter reached equilibrium within less than 1 h and those smaller than 3 μ m in diameter reached equilibrium within less than 15 min. Equilibrium took longer for coarse particles in several other cases. Thus, in some cases, some coarse-mode particles may be in equilibrium, suggesting that some caveats may exist to the result found in previous studies that coarse-mode particles are not in equilibrium.
Acknowledgments
This work was supported, in part, by the NASA Earth Sciences Program, the Environmental Protection Agency Office of Air Quality Planning and Standards, the National Science Foundation Atmospheric Chemistry Division (ATM 0101596), and the Global Climate and Energy Project.
REFERENCES
- Capaldo , K. P. , Pilinis , C. and Pandis , S. N. 2000 . A Computationally Efficient Hybrid Approach for Dynamic Gas/Aerosol Transfer in Air Quality Models . Atmos. Environ. , 34 : 3617 – 3627 .
- Eriksson , E. 1960 . The Yearly Circulation of Chloride and Sulfur in Nature; Meteorological, Geochemical and Pedological Implications. Part II . Tellus , 12 : 63 – 109 .
- Fridlind , A. M. and Jacobson , M. Z. 2000 . A Study of Gas-Aerosol Equilibrium and Aerosol pH in the Remote Marine Boundary Layer During the First Aerosol Characterization Experiment (ACE 1) . J. Geophys. Res. , 105 : 17,325 – 17,340 .
- Hitchcock , D. R. , Spiller , L. L. and Wilson , W. E. 1980 . Sulfuric Acid Aerosols and HCl Release in Coastal Atmospheres: Evidence of Rapid Formation of Sulfuric Acid Particulates . Atmos. Environ. , 14 : 165 – 182 . http://dx.doi.org/10.1016%2F0004-6981%2880%2990275-9
- Jacobson , M. Z. 1997a . Development and Application of a New Air Pollution Modeling System. Part II: Aerosol Module Structure and Design . Atmos. Environ. , 31A : 131 – 144 . http://dx.doi.org/10.1016%2F1352-2310%2896%2900202-6
- Jacobson , M. Z. 1997b . Development and Application of a New Air Pollution Modeling System. Part III: Aerosol-Phase Simulations . Atmos. Environ. , 31A : 587 – 608 . http://dx.doi.org/10.1016%2FS1352-2310%2896%2900201-4
- Jacobson , M. Z. 1997c . Numerical Techniques to Solve Condensational and Dissolutional Growth Equations When Growth is Coupled to Reversible Reactions . Aerosol Sci. Technol. , 27 : 491 – 498 .
- Jacobson , M. Z. 1999a . Fundamentals of Atmospheric Modeling , New York : Cambridge University Press .
- Jacobson , M. Z. 1999b . Studying The Effects of Calcium and Magnesium on size-Distributed Nitrate and Ammonium with EQUISOLV II . Atmos. Environ. , 33 : 3635 – 3649 . http://dx.doi.org/10.1016%2FS1352-2310%2899%2900105-3
- Jacobson , M. Z. 2002 . Analysis of Aerosol Interactions with Numerical Techniques for Solving Coagulation, Condensation, Dissolution, and Reversible Chemistry among Multiple Size Distributions . J. Geophys. Res. , 107 ( D19 ) : 4366
- Jacobson , M. Z. , Tabazadeh , A. and Turco , R. P. 1996 . Simulating Equilibrium within Aerosols and Nonequilibrium between Gases and Aerosols . J. Geophys. Res. , 101 : 9079 – 9091 . http://dx.doi.org/10.1029%2F96JD00348 http://www.csa.com/htbin/linkabst.cgi?issn=0148-0227&vol=101&iss=&firstpage=9079
- Junge , C. E. 1957 . Chemical Analysis of Aerosol Particles and of Gas Traces on the Island of Hawaii . Tellus , 9 : 528 – 537 .
- Koo , B. , Gaydos , T. M. and Pandis , S. N. 2003 . Evaluation of the Equilibrium, Dynamic, and Hybrid Aerosol Modeling Approaches . Aerosol. Sci. Technol. , 37 : 53 – 64 . http://dx.doi.org/10.1080%2F02786820300893
- Martens , C. S. , Wesolowski , J. J. , Hariss , R. C. and Kaifer , R. 1973 . Chlorine Loss from Puerto Rican and San Francisco Bay Area marine aerosols . J. Geophys. Res. , 78 : 8778 – 8792 .
- Meng , Z. , Dabdub , D. and Seinfeld , J. H. 1998 . Size-Resolved and Chemically Resolved Model of Atmospheric Aerosol Dynamics . J. Geophys. Res. , 103 : 3419 – 3435 . http://dx.doi.org/10.1029%2F97JD02796 http://www.csa.com/htbin/linkabst.cgi?issn=0148-0227&vol=103&iss=&firstpage=3419
- Meng , Z. and Seinfeld , J. H. 1996 . Time Scales to Achieve Atmospheric Gas-Aerosol Equilibrium for Volatile Species . Atmos. Environ. , 30 : 2889 – 2900 . http://dx.doi.org/10.1016%2F1352-2310%2895%2900493-9
- Moya , M. , Pandis , S. N. and Jacobson , M. Z. 2001 . Is the Size Distribution of Urban Aerosols Determined by Thermodynamic Equilibrium? An Application to Southern California . Atmos. Environ. , 36 : 2349 – 2365 . http://dx.doi.org/10.1016%2FS1352-2310%2801%2900549-0
- Pilinis , C. , Capaldo , K. P. , Nenes , A. and Pandis , S. N. 2000 . MADM—A New Multicomponent Aerosol Dynamics Model . Aerosol Sci. Technol. , 32 : 482 – 502 . http://dx.doi.org/10.1080%2F027868200303597
- Sun , Q. and Wexler , A. S. 1998a . Modeling Urban and Regional Aerosols-Condensation and Evaporation Near Acid Neutrality . Atmos. Environ. , 32 : 3527 – 3531 . http://dx.doi.org/10.1016%2FS1352-2310%2898%2900059-4
- Sun , Q. and Wexler , A. S. 1998b . Modeling Urban and Regional Aerosols Near Acid Neutrality-Application to the 24–25 June SCAQS Episode . Atmos. Environ. , 32 : 3533 – 3545 . http://dx.doi.org/10.1016%2FS1352-2310%2898%2900060-0
- Wexler , A. S. and Seinfeld , J. H. 1990 . The Distribution of Ammonium Salts among a Size and Composition Dispersed Aerosol . Atmos. Environ. , 24A : 1231 – 1246 .
- Wilkniss , P. E. and Bressan , D. J. 1972 . Fractionation of the Elements F, Cl, Na, and K at the Air-Sea Interface . J. Geophys. Res. , 77 : 5307 – 5315 .