Abstract
We have designed a multiplex PCR, which allows for fast and high throughput demonstration of the BCL-1/IGH and BCL-2/IGH fusion DNA observed primarily in mantle cell- and follicular non-Hodgkin's lymphoma (NHL). Blood (PB) and/or bone marrow (BM) from 258 patients suspected of NHL have prospectively been evaluated. Eleven patients (4%) were found t(11;14)+ and 37 patients (14%) t(14;18)+. Comparing these results to standard diagnostic methods of PB and/or BM identified PCR+ samples that were normal by morphology (BCL-1/IGH: 1/11; BCL-2/IGH: 17/37). Equally important, patients who were not clonal in PB and/or BM by flow cytometry were identified as PCR+ (BCL-1/IGH: 3/11; BCL-2/IGH: 23/37). We conclude that this multiplex approach allows for easy and sensitive molecular determination of molecular lesions in NHL, which have diagnostic and prognostic importance.
Despite morphological and immunological similarities, more than 50 chromosomal translocations may be found in acute leukaemia (AL) and in non-Hodgkin's lymphoma (NHL) Citation[1], Citation[2]. Importantly, some of these aberrations have been identified as independent prognostic parameters, especially in acute myeloid leukaemia. These chromosomal aberrations can be identified by different methodologies e.g. by karyotyping, fluorescence in situ hybridization, or – increasingly – by PCR. An valuable extension of the latter approach is its capability to serve as a platform for the construction of minimal residual disease (MRD) assays Citation[3].
The balanced translocations reported Citation[4], Citation[5] in NHL's are shown to delineate distinct disease entities Citation[6], Citation[7]. Consequently, upfront identification of such lesions should facilitate a more accurate diagnosis in the single patient and allow for a more risk-based therapy.
The t(11;14)(q13;q32) found in the majority of mantle cell lymphomas (MCL) and the t(14;18)(q32;q21), identified primary in the follicular lymphomas (FL) are the most common translocations in NHL. The t(11;14) juxtaposes the BCL-1 gene on chromosome 11 to the immunoglobulin heavy chain gene (IGH) on chromosome 14 leading to overexpression of the cyclin D1 gene located approximately 120 kb upstream the BCL-1 locus Citation[4]. Likewise, the t(14;18) places the BCL-2 gene on chromosome 18 next to the IGH on chromosome 14 leading to deregulation of the BCL-2 gene thus inhibiting apoptosis Citation[8].
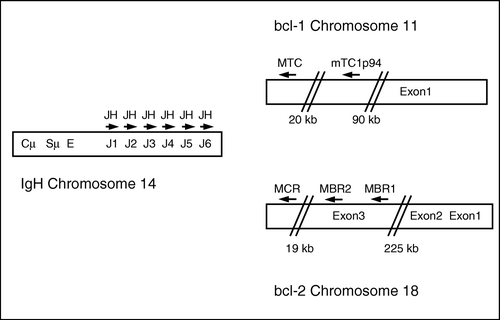
The breakpoints in these lesions are mainly clustered in certain regions, while a minor part is found scattered over a larger region Citation[9–11]. For BCL-1 a major translocation cluster (MTC) has been found to cover the majority of breakpoints Citation[12], while the mTCp94 region is found with a minor frequency Citation[13]. The breakpoints on BCL-2 are in approximately 70% of cases described to occur in the major breakpoint region (MBR) while about 10% are found in the minor cluster region (MCR) Citation[14].
The purpose of this study was two-fold: first, by employing positive continuous cell lines and fresh patient material to develop and validate pre-clinically a multiplex PCR assay to detect fusion DNA from patients with the t(11;14) or t(14;18) aberrations simultaneously. Second, to validate this reaction clinically on prospectively accrued patient samples suspected for NHL using blood (PB) or bone marrow (BM) as target material. The latter approach was chosen in preference to the analysis of lymph node material, since PB and BM are more readily available and since such sampling allows for evaluation of the sensitivity directly compared to flow cytometry (FCM). Moreover, by studying the primary diagnostic setting allows for evaluation of false-positive samples, since non-malignant samples are part of the data set. In this report we compare molecular PCR data obtained from 258 patients to FCM data determining clonal B-cells by CD19/Ig light chain assays and to the histological evaluation of the PB and BM samples.
Materials and methods
Patients
Two hundred and fifty eight patients were enrolled from May 1st 2000 to December 31st 2001 based on a suspicion of NHL and submission of samples (at presentation, follow-up, or at suspected relapse) to our laboratory for FCM analysis. If a patient was sampled more than once only the first sample was used for this analysis. All experimentation was performed according to protocols approved by the Ethical Committee for the County of Aarhus.
Mononuclear cells, cell lines, and normal controls
Mononuclear cells (MNC) were obtained from PB and BM by Lymphoprep™ density centrifugation (Axis-Shield PoC AS, Oslo, Norway). The t(11;14)+ human chronic B-cell leukaemia cell line JVM-2 and the t(14;18)+ human B-cell lymphoma cell lines DOHH-2, Karpas 422, and WSU-NHL were acquired from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). Cell lines were grown in medium RPMI1640 in 10% foetal calf serum without antibiotics, 5% CO2, 37°C, and harvested in exponential growth phase. Blood samples from 20 normal individuals were collected; MNC was obtained and used as normal controls.
PCR
DNA was prepared by Trizol purification (Gibco BRL, Grand Island, NY, USA) or by a MagNa-Pure LC robot for automated nucleic acid purification (Roche Diagnostics, Basel, CH).
All primers for PCR amplification and sequencing were designed using the Oligo software (Primer Analysis Software, version 6.83, Molecular Biology Insights, CO, USA) and purchased from DNA Technology, Aarhus, Denmark. The multiplex assay was designed as a single-step PCR in order to minimize contamination and avoid amplification of the background population of fusion DNA arising from t(14;18) positive cells described to occur in healthy individuals Citation[15]. PCR was performed with a consensus primer (5′-ACTTACCTGAGGAGACGGTGAC-3′) covering all six J genes of IGH on chromosome 14 together with specific primers covering the MTC (5′-GGCTCAACCCTTCACCTACTG-3′) and the mTC1p94 (5′-GCATCCAAGGCTGATCTGAGAA-3′) region of the BCL-1 gene on chromosome 11 and the MBR (MBR1: 5′-TCTGTTGTCCCTTTGACCTTGT-3′, MBR2: 5′-GGCAACAGAGAACCATCCCTAT-3′), and MCR (5′-ATAGAGCAAGCGCCCAATAA-3′) regions of the BCL-2 gene on chromosome 18. The mutual location of the primers is depicted in . In addition, a set of control primers amplifying part of the TCF20 gene (accession no. AL031346) that encodes a transcription factor (forward primer: 5′-AAGGACCTTCCAAGTCCAGATAG-3′ and reverse primer: 5′-AACCCTCATGGACCCAAAAT-3′) was added in the same tube as a measure of DNA quality and quantity. This control PCR product was designed to be considerably larger than the fusion PCR products to avoid false negative reactions, as large PCR products are more difficult to amplify than smaller ones. Subsequently, for identification of positive samples a split-out PCR was performed with only one set of translocation primers in each tube together with the control primers.
Figure 1. Location of the primers on BCL-1, BCL-2 and IGH included in the multiplex PCR.
PCR was performed in a thermocycler (GeneAmp PCR System 9600, Applied Biosystems, Foster City, CA, USA) in a volume of 25 µL on either 0.5 µg genomic DNA measured using an optical density spectrophotometer (GeneQuant, Pharmacia Biotech, Uppsala, Sweden) when samples were Trizol purified (Gibco BRL) or on genomic DNA prepared from 50 000 cells when the samples were purified on the MagNa-Pure LC robot (Roche Diagnostics ). The reaction mixture consisted of 1×HotStar PCR buffer including Tris-HCl, KCl, (NH4)2SO4, and 1.5 mM MgCl2, 0.2 mM of each dNTP, 0.75 units of HotStarTaq DNA polymerase (Qiagen AS, Oslo, Norway), 5 pmol of each of the translocation primers, and 10 pmol of each of the control primers. The PCR conditions were standardized for both the multiplex PCR and for the split-out PCR to: 35 cycles of 30 s at 95°C, 60 s at 55°C, 60 s at 72°C, initiated by 10 min of denaturation at 95°C, and completed by 5 min of extension at 72°C. The PCR products were analyzed by a 2% agarose gel electrophoresis in 1×TBE buffer followed by UV-visualization of ethidium bromide stained DNA.
DNA sequencing
PCR bands were sliced out from the agarose gel, transferred to spin-x tubes (Costar, Corning Inc., NY, USA), incubated at –20°C for 5 min followed by centrifugation to obtain the fusion DNA without surplus of nucleotides and primers. Sequencing was performed by the ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems) on an ABI prism 377 (Applied Biosystems) using internal sequencing primers (MBR1-seq: 5’-TCCGCATTTAATTCATGGTATTC-3’, MBR2-seq: 5′-GGGATTCACATCTGCATCTTAAC-3′, MCR-seq: 5′-TTACTCTTGCAGGGTCTTTAAGC-3′, MTC-seq: 5′-GTGGGTTGCTTCCAAGTTTT-3′, and mTC1p94-seq: CCAAGGCTGATCTGAGAACTACT-3′).
Flow Cytometry
FCM was performed on Coulter XL flow cytometers (Beckman-Coulter, Luton, England) using identical acquisition protocols and employing a standard panel of monoclonal antibodies for defining mature monoclonal B-cell disorders (CD19/CD5, CD19/CD23, CD19/FMC7, and CD20/CD11c). Monoclonal B-cells were demonstrated by multiparameter CD19/Ig light chain assays where monoclonality was defined as a κ/λ ratio <0.5 or >3.
Pathology
PB, BM aspirates, and BM biopsies were analyzed morphologically and supplemented with immunohistochemical staining on aspirate smears and clots or biopsies in doubtful cases. In most cases, the nodal or extra nodal malignant locations were also diagnosed using standard morphology and immunohistochemistry. The diagnosis followed the WHO-classification of malignant lymphomas (ML's) Citation[16].
Results
Pre-clinical validation of the multiplex PCR
We first determined the sensitivity and the potential for false positive results in the multiplex PCR, since it entails the use of different primer sets in the same reaction mixture. In six serial dilution experiments of DNA from patients (no. 27, 28, and 59) and cell lines (Karpas 422, WSU-NHL, and JVM-2) positive for t(11;14) (MTC and mTC1p94), and for t(14;18) (MBR1 and MBR2) diluted in DNA from MNC of a normal individual a sensitivity down to 0.1% was consistently observed (data not shown). With this experiment we also defined the basis for labelling bands as weak, medium, and strong. While this approach has no relevance in the diagnostic setting, it nevertheless allows for comparisons to FCM (see below).
With respect to false positive reactions, no amplification of PCR products of fusion DNA was observed in 20 healthy individuals (data not shown). These experimental conditions were deemed to be sufficiently sensitive for further experimentation without detecting fusions DNA in non-lymphomatous patients.
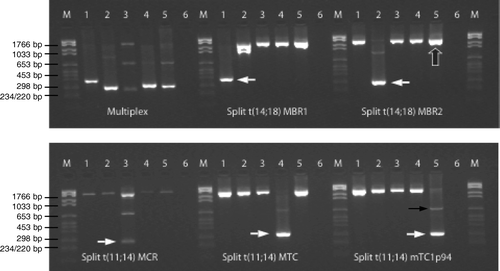
details an example of the t(11;14) and t(14;18) multiplex PCR including positive controls (cell lines) as well as patient samples. The control gene was co-amplified in all reactions apart from the ones where it was outcompeted by the PCR product of the fusion DNA. The appearance of the control gene ensured a sufficient DNA quantity and quality in each reaction. As expected, the cell lines DOHH-2, WSU-NHL, and JVM-2 were positive for t(14;18) MBR1 and MBR2, and for t(11;14) MTC, respectively and serve as positive controls for all subsequent experimentation. In addition, two patient samples (a lymph node biopsy from patient no. 261 and blood from patient no. 28) were employed as positive controls for t(14;18) MCR and t(11;14) mTC1p94 respectively since no cell lines with those breakpoints were accessible. In most of the positive reactions a weaker PCR band with a higher molecular size also appeared due to annealing of the IGH primer to the germline J gene most proximate to the J gene involved in the actual translocation. Probably due to a low content of malignant cells in the lymph node material or due to only a subclone carrying t(14;18) MCR the PCR band originating from the fusion DNA of t(14;18) MCR appeared weakly in . Breakpoints amplified with the MBR2 primer in the split reaction (e.g. WSU-NHL) were also detected by the MBR1 primer due the relatively short distance between these two primers.
Figure 2. Multiplex PCR for detection of t(11;14) and t(14;18). M: molecular marker. Lane 1: cell line DOHH-2, lane 2: cell line WSU-NHL, lane 3: lymph node biopsy from patient no. 261, lane 4: cell line JVM-2, lane 5: blood from patient no. 28, and lane 6: ddH2O. Multiplex PCR: primers amplifying MBR1, MBR2, and MCR in t(14;18) as well as MTC and mTC1p94 in t(11;14). Split reactions: 5 primer sets detecting t(14;18) MBR1, t(14;18) MBR2, t(14;18) MCR, t(11;14) MTC, and t(11;14) mTC1p94. A control gene (TCF20) is co-amplified in all lanes. White arrows show the translocation positive PCR products in the split reactions, large black arrow exemplifies a PCR product of the control gene, while the thin black arrow exemplifies a weak germline PCR band.
Clinical validation and value of the multiplex PCR
Over a period of 20 months, samples from 258 patients were received for FCM due to the suspicion of ML. DNA was extracted and subsequently analyzed by the multiplex PCR assay in PB (208 samples) and/or in BM (191 samples). In 141 patients both sources were analyzed. As shown in 11 patients were found positive for t(11;14). Eleven of the patients (4%) displayed a breakpoint in the BCL-1 gene; 10 were located in the MTC region, while only one patient had the breakpoint located in the region covered by the mTC1p94 primer. Thirty-seven patients (14%) exhibited an IGH/BCL-2 rearrangement (), 34 with breakpoints detected with the MBR1 primer and only three with the BCL-2 gene rearranged in the MCR region. When subtracting the number of patients subsequently diagnosed with other diseases than NHL (Hodgkin's disease, multiple myeloma, chronic lymphatic leukaemia, or not lymphoid malignancy (LM)) the frequencies of t(11;14)+ and t(14;18)+ patients were 6% and 22%, respectively.
Table I. PCR positive for t(11;14). Comparison between PCR, flow cytometry, and histology at the same time of sampling.
Table II. PCR positive for t(14;18). Comparison between PCR, flow cytometry, and histology at the same time of sampling.
Six of the t(11;14)+ patients examined both in PB and BM were PCR+ in both sources, while three were examined only in PB and two only in BM. Twenty-four of the t(14;18)+ patients were examined in both PB and BM. Twenty of these were positive in both sources, two were positive only in PB while two were positive only in BM.
We sequenced the fusion DNA found by the multiplex PCR in order to verify patient specific breakpoints and to exclude contamination from previously positive PCR products of fusion DNA. Sequences were obtained for all translocation positive patients with the exception of two t(14;18)+ patients (patient no. 64 and patient no. 101), where insufficient amounts of DNA was attained to allow sequencing. All sequences were unique showing a clone- and patient specific sequence of the breakpoint region. shows the location of the breakpoints scattered over the BCL-1 and BCL-2 genes. The breakpoints are, with a few exceptions, clustered in small regions.
Figure 3. Breakpoints from the fusion DNA found in patients included in this study. Arrows indicate breakpoints and numbers refer to patient IDs. A. Sequence of BCL-1 MTC region. Sequence start at position 431 in the Genebank sequence with accession no. s77049. B. Sequence of BCL-1 mTC1p94 region. Reverse complement of accession no. ap001824 in the Genebank database. Sequence start at position 41535. C. Sequence of BCL-2 MBR region. Sequence start at position 2981 in the Genebank sequence with accession no. m14745 Citation[8]. D. Sequence of BCL-2 MCR region. Sequence from Ngan, 1989 Citation[27]. Sequence starts at position 905 and ends at position 1432.
![Figure 3. Breakpoints from the fusion DNA found in patients included in this study. Arrows indicate breakpoints and numbers refer to patient IDs. A. Sequence of BCL-1 MTC region. Sequence start at position 431 in the Genebank sequence with accession no. s77049. B. Sequence of BCL-1 mTC1p94 region. Reverse complement of accession no. ap001824 in the Genebank database. Sequence start at position 41535. C. Sequence of BCL-2 MBR region. Sequence start at position 2981 in the Genebank sequence with accession no. m14745 Citation[8]. D. Sequence of BCL-2 MCR region. Sequence from Ngan, 1989 Citation[27]. Sequence starts at position 905 and ends at position 1432.](/cms/asset/65a67a2d-6b48-4b35-b41f-b7373c669088/ionc_a_168125_f0003_b.gif)
Comparison of molecular data to FCM and histology
We next compared the PCR results described above to standard methods for diagnosing NHL. The initial histological typing of the diagnostic lymph node material evaluated by experienced haematopathologists is shown in and for the patients found positive for fusion DNA from either t(11;14) or t(14;18). As shown in 10 of the t(11;14)+ patients were diagnosed as MCL (whereof one showed follicular growth at diagnosis – later classified as MCL; patient no. 30) while one was diagnosed as marginal zone lymphoma. Twenty-seven of the 37 t(14;18)+ patients were diagnosed as FL's, six as diffuse large cell B lymphomas (DLCL), one as a lymphoplasmacytic lymphoma (LL), two as NHL not otherwise specified (NHL NOS), while one patient did not have a diagnosis of LM but was diagnosed with a seminoma 17 years earlier (patient no. 102).
The microscopic examination of PB and BM showed that 5/11 of the t(11;14)+ patients in PB and 1/11 patients in BM were not diagnosed morphologically positive. For t(14;18)+ patients 25/37 and 15/37 were not morphologically positive in PB or BM, respectively. The patient with LL (no. 73) who by PCR was t(14;18)- in BM was found positive for lymphoma infiltration in BM by pathology.
With respect to FCM data, cases where the κ/λ ratio was not performed because of too few CD19+ cells were recorded as not done (ND), while cases that were not analyzed because of absent samples are denoted as not available (NA) in and . Seventy-three percent (8/11) of the t(11;14)+ patients and 38% (14/37) of the t(14;18)+ patients were found monoclonal by FCM in either PB or BM. Two of the t(11;14)+ patients were not found clonal in PB and three not clonal in BM by FCM. For t(14;18)+ samples 15 in PB and 15 in BM were not clonal according to FCM. One t(14;18)+ patient (patient no. 73, PB) diagnosed as LL was PCR negative in BM, but clonal in BM by FCM. One patient that was found positive by PCR for t(11;14) (patient no. 30) was not monoclonal by FCM or diagnosed positive by pathology, while 14 of the t(14;18)+ patients were only positive by PCR.
provides a summary of the patients, who were PCR-. When compared to microscopic examination, this group comprised 13 patients with MCL, 23 patients with FL, and 57 patients with DLCL. FCM detected monoclonality in nine of the PCR- patients with MCL, nine of the PCR- patients with FL, and 11 of the PCR- patients with DLCL. The morphological assessment showed malignant cells in nine of the MCL patients, in four of the FL patients, and in seven of the DLCL patients. Thus, if only considering the samples of the PCR- group where the histological type of the time of sampling were found to be either MCL or FL together with the PCR+ samples from and the frequencies of PCR+ patients were 53% (10/19) and 87% (27/31) of the MCL and FL subgroups, respectively.
Table III. PCR negative for t(11;14) and t(14;18). Patients and samples.
Discussion
In NHL, a large body of data has shown that balanced translocations are present in a large and heterogeneous number. The IGH/BCL-2 and the IGH/BCL-1 aberrations found by multiplex PCR constitute almost 30% of the NHL patients included in the present study. In addition, a number of other translocations have been demonstrated in smaller fractions of lymphoma patients (reviewed by Medeiros and Carr Citation[4]) including C-MYC/IGH (t(8;14)(q24;q32)), IGK/C-MYC (t(2;8)(p12;q24)), and c-MYC/IGL (t(8;22)(q24;q11)) in Burkitt lymphoma, BCL-6/VARIOUS (t(3;Var)(q27;Var)) and IGH/BCL-8 (t(14;15)(q32;q11-13)) in DLCL, API-2/MLT (t(11;18)(q21;q21)) and BCL-10/IGH (t(1;14)(p22;q32)) in MALT lymphoma, ALK/NPM-1 (t(2;5)(p23;q35)) in anaplastic large cell lymphoma, and PAX-5/IGH (t(9;14)(p13;q32)) in LL Citation[4].
Identifying a genetic marker, which can be used in tracing diminutive cell populations by molecular biology techniques such as real-time PCR, enables a quantitative assessment of MRD in haematological disorders Citation[17]. Several studies have shown that the amount of residual disease after induction therapy is directly correlated to the prognosis in childhood leukaemia Citation[18–20]. Moreover, the evaluation of PB and/or BM with e.g. PCR has shown to be valuable in predicting outcome in NHL after autologous bone marrow transplantation using fusion DNA from t(14;18) as genetic target Citation[21].
In designing multiplex PCR such as the one already published for acute leukaemias Citation[3], it is vital that the assay is condensed in as few tubes as possible to save material and to make the method applicable in a routine setting. In addition, patients with only a minor fraction of affected cells in PB or BM should be identified while, concomitantly, a background occurring in healthy individuals should not be picked up Citation[15]. Furthermore, it is important to design the reaction so that the fusion PCR products are in the same size range and thus amplified with similar efficiencies while the control PCR product should have a considerably larger size so that false negative reactions are avoided. Finally, unspecific PCR products as a result of the presence of multiple primers should be avoided.
Previous studies report on PCR detection of t(11;14) and/or t(14;18) in a multiplex setting. Simultaneous detection of t(11;14) and t(14;18) together with clonal IGH and TCR gene rearrangements using capillary electrophoresis have been reported by Meier et al., however, the mTCp94 region is not covered by this assay Citation[22]. A more recent report is the BIOMED-2 study Citation[23] where a multiplex PCR spanning three tubes have been developed for the detection of t(14;18) fusion DNA using nine different primers located in three different breakpoint regions of BCL-2 enabling a higher detection rate of t(14;18) aberrations than in our multiplex PCR assay, however, an internal control ensuring an acceptable DNA quality and quantity is not included and reactions are not limited to a single tube for greater convenience as in the present study.
We have developed a multiplex PCR assay that covers t(14;18) and t(11;14) within the most frequent breakpoint regions in one single PCR tube. The data presented here as well as our daily experience strongly suggest that it might be a useful adjunct to the diagnostic armamentarium in patients suspected of ML. We found the assay to be much more sensitive than FCM as monoclonality was detected in many more cases by using multiplex PCR than by FCM. Thus, more than half of the t(14;18)+ patients (62%) were not found to be monoclonal by FCM. These data show that a molecular diagnosis in NHL can be obtained employing PB or BM material. Comparing our data with the histological examination we identified t(14;18) in a patient (no. 102) that was evaluated as LM negative by histology but diagnosed with seminoma 17 years earlier. Patients with seminoma has previously been described to have an increased frequency of chromosomal translocations which might explain this findingCitation[24]. The PCR frequency of t(14;18)+ patients found in the present study (87%) also corresponds to what is found in the literatureCitation[14].
MCL constitutes a clinical entity most probably underlying a heterogeneous aetiology and with several markers that have been linked to it including immunohistochemical appearance, Cyclin D1 expression, (CD5, FMC7) double expression in addition to the translocation and its resulting BCL-1/IGH fusion DNA Citation[25]. Further studies should be designed to more closely define MCL with the aim of designing even more tailored treatment strategies, and a multiplex PCR assay should be helpful in this respect.
Collectively, the molecular data presented here add to the notion of extreme heterogeneity within the group of NHL patients. This is exemplified by the demonstration of PCR+ cases that were not found positive for lymphoma infiltration by histology, be it FL or MCL. In these cases, a molecular lesion is apparently not resulting in a universally recognizable disease phenotype, and such patients should be candidates for further molecular studies searching for other molecular lymphomatogenic events. The demonstration that t(14;18) seem to occur as a normal background in healthy individuals Citation[15] also point out that the translocation is not the only event leading to lymphomatogenesis. Irrespective of these considerations, upfront inclusion of molecular methods such as the one presented here should add diagnostic information enabling more uniform cytoreduction in both these important groups of NHL.
We chose validating the assay on PB and BM samples, as this material is readily available and often serve as the initial source of diagnosis. More importantly, demonstrating the value in this setting virtually ensures that it can also be applied to lymph node preparations, where the malignant clone is more prominent in the vast majority of cases. Indeed, preliminary testing in a retrospective fashion analyzing more than 100 lymph node samples confirms that this notion is correct.
The role of an assay like the one described here in the age of high-end techniques like comparative genomic hybridization and oligonucleotide expression arrays is clear. While the latter techniques are exploratory in nature identifying novel relations between potentially genetic aberrations and altered gene expressions, our assay focuses on known aberrations defining already known biological entities. Moreover, while expression signatures obtained from array data have been purported to be helpful in prognosticating patients, they are not a first step for minimal residual disease detection like the multiplex PCR assay. Thus, they cannot be used to evaluate treatment responses similar to what was accomplished for the BCL2/IGH aberration in a recent report using real-time quantitative PCR Citation[26]. Needless to state, the novel assays have unlimited potential for unravelling new and interesting genetic alterations in NHL.
We conclude that the NHL multiplex PCR described is an easy method to identify heterogeneous molecular disease markers for patients with FL and MCL and the condensation of the assay in a single PCR tube make the method convenient in a clinical setting. Application of this assay and identification of positive patients has the added advantage that they are candidates for quantitative real-time PCR to monitor the residual disease during treatment course. Further studies employing this assay are necessary to delineate the intricate interrelations between histology and molecular biology with the ultimate goal of better classifying patients with NHL.
We thank Hanne Tønder for excellent technical assistance and Niels Pallisgaard for helpful suggestions. This work was supported by grants from the Danish Cancer Society and the Danish Medical Research Council.
References
- Rabbitts TH. Chromosomal translocations in human cancer. Nature 1994; 372: 143–9
- Johansson B, Mertens F, Mitelman F. Clinical and biological importance of cytogenetic abnormalities in childhood and adult acute lymphoblastic leukemia. Ann Med 2004; 36: 492–503
- Pallisgaard N, Hokland P, Riishoj DC, Pedersen B, Jorgensen P. Multiplex reverse transcription-polymerase chain reaction for simultaneous screening of 29 translocations and chromosomal aberrations in acute leukemia. Blood 1998; 92: 574–88
- Medeiros LJ, Carr J. Overview of the role of molecular methods in the diagnosis of malignant lymphomas. Arch Pathol Lab Med 1999; 123: 1189–207
- Chaganti RS, Nanjangud G, Schmidt H, Teruya-Feldstein J. Recurring chromosomal abnormalities in non-Hodgkin's lymphoma: Biologic and clinical significance [In Process Citation]. Semin Hematol 2000; 37: 396–411
- Huang JZ, Sanger WG, Greiner TC, Staudt LM, Weisenburger DD, Pickering DL, et al. The t(14;18) defines a unique subset of diffuse large B-cell lymphoma with a germinal center B-cell gene expression profile. Blood 2002; 99: 2285–90
- Barrans SL, O'Connor SJ, Evans PA, Davies FE, Owen RG, Haynes AP, et al. Rearrangement of the BCL6 locus at 3q27 is an independent poor prognostic factor in nodal diffuse large B-cell lymphoma. Br J Haematol 2002; 117: 322–32
- Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for BCL-2 and a hybrid BCL-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell 1986; 47: 19–28
- Cleary ML, Galili N, Sklar J. Detection of a second t(14;18) breakpoint cluster region in human follicular lymphomas. J Exp Med 1986; 164: 315–20
- Cleary ML, Sklar J. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci USA 1985; 82: 7439–43
- Williams ME, Meeker TC, Swerdlow SH. Rearrangement of the chromosome 11 BCL-1 locus in centrocytic lymphoma: Analysis with multiple breakpoint probes. Blood 1991; 78: 493–8
- Rimokh R, Berger F, Delsol G, Digonnet I, Rouault JP, Tigaud JD, et al. Detection of the chromosomal translocation t(11;14) by polymerase chain reaction in mantle cell lymphomas. Blood 1994; 83: 1871–5
- Fan H, Gulley ML, Gascoyne RD, Horsman DE, Adomat SA, Cho CG. Molecular methods for detecting t(11;14) translocations in mantle-cell lymphomas. Diagn Mol Pathol 1998; 7: 209–14
- Buchonnet G, Jardin F, Jean N, Bertrand P, Parmentier F, Tison S, et al. Distribution of BCL2 breakpoints in follicular lymphoma and correlation with clinical features: Specific subtypes or same disease?. Leukemia 2002; 16: 1852–6
- Yasukawa M, Bando S, Dolken G, Sada E, Yakushijin Y, Fujita S, et al. Low frequency of BCL-2/J(H) translocation in peripheral blood lymphocytes of healthy Japanese individuals. Blood 2001; 98: 486–8
- Jaffe ES, Harris NL, Stein H, Vardiman JW. WHO Classification of tumours: Pathology and genetics of tumours of haematopoietic and lymphoid tissues. IARC Press, Lyon 2001
- Iqbal S, Jenner MJ, Summers KE, Davies AJ, Matthews J, Norton AJ, et al. Reliable detection of clonal IGH/BCL2 MBR rearrangement in follicular lymphoma: Methodology and clinical significance. Br J Haematol 2004; 124: 325–8
- Nyvold C, Madsen HO, Ryder LP, Seyfarth J, Svejgaard A, Clausen N, et al. Precise quantification of minimal residual disease at day 29 allows identification of children with acute lymphoblastic leukemia and an excellent outcome. Blood 2002; 99: 1253–8
- Brisco MJ, Condon J, Hughes E, Neoh SH, Sykes PJ, Seshadri R, et al. Outcome prediction in childhood acute lymphoblastic leukaemia by molecular quantification of residual disease at the end of induction. Lancet 1994; 343: 196–200
- Izraeli S, Waldman D. Minimal residual disease in childhood acute lymphoblastic leukemia: Current status and challenges. Acta Haematol 2004; 112: 34–9
- Darby AJ, Johnson PW. Molecular remission and non-Hodgkin's lymphoma. Best Pract Res Clin Haematol 2002; 15: 549–62
- Meier VS, Rufle A, Gudat F. Simultaneous evaluation of T- and B-cell clonality, t(11;14) and t(14;18), in a single reaction by a four-color multiplex polymerase chain reaction assay and automated high-resolution fragment analysis: A method for the rapid molecular diagnosis of lymphoproliferative disorders applicable to fresh frozen and formalin-fixed, paraffin-embedded tissues, blood, and bone marrow aspirates. Am J Pathol 2001; 159: 2031–43
- van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003; 17: 2257–317
- Schmidberger H, Virsik-Koepp P, Rave-Frank M, Reinosch KR, Pradier O, Munzel U, et al. Reciprocal translocations in patients with testicular seminoma before and after radiotherapy. Int J Radiat Oncol Biol Phys 2001; 50: 857–64
- Yatabe Y, Suzuki R, Tobinai K, Matsuno Y, Ichinohasama R, Okamoto M, et al. Significance of cyclin D1 overexpression for the diagnosis of mantle cell lymphoma: A clinicopathologic comparison of cyclin D1-positive MCL and cyclin D1-negative MCL-like B-cell lymphoma. Blood 2000; 95: 2253–61
- Rambaldi A, Carlotti E, Oldani E, Della Starza I, Baccarani M, Cortelazzo S, et al. Quantitative PCR of bone marrow BCL2/IGH+ cells at diagnosis predicts treatment response and long-term outcome in follicular non-Hodgkin lymphoma. Blood 2005; 105: 3428–33
- Ngan BY, Nourse J, Cleary ML. Detection of chromosomal translocation t(14;18) within the minor cluster region of BCL-2 by polymerase chain reaction and direct genomic sequencing of the enzymatically amplified DNA in follicular lymphomas. Blood 1989; 73: 1759–62