Abstract
The potential and the limitations of protein analysis of tissue samples are surveyed. The complexity, concentration range and dynamics of the human proteome are reviewed, as is the effect of handling and cryopreservation. Protein extraction, solubilization, resolution and detection are discussed, in relation to the properties of the human proteome.
Public opinion favors that surgically removed human tissue is put to the best possible use, not only for diagnosis, but also for research into the causes of diseases. Recent progress in genome sequencing and analysis, together with progress in protein analysis technology has opened new opportunities to derive important new information about disease mechanisms directly from tissue samples. Solid tissue biobanks have therefore been established Citation[1]. Protein analysis is demanding in that efficient extraction requires fresh, or fresh-frozen, tissue without fixation, and that no amplification method (such as PCR) exists, so that sufficient material for analysis has to be extracted from the tissue itself. To be relevant for proteomic analysis, the tissue needs to be handled in ways that reproducibly preserve important aspects of its proteome intact. Rationally based standard operating protocols are required for this purpose. These goals, and several others, are the mission of the Swedish National Biobank Program (www.biobanks.se). While recent progress in proteomic technology has increased its power, the entire proteome of a tissue cannot be analyzed with a single approach. Informed decisions therefore need to be made to focus the analysis onto the most informative tissue fractions and using the most appropriate techniques, for each purpose.
Why to analyze the proteome of fresh frozen tissue and blood?
Fresh freezing is currently the preservation method that is most compatible with subsequent proteomic analyses. Fixation by formaldehyde crosslinking, as is traditionally done in histopathology, is incompatible with some proteomic analyses, necessitating a change in the routines of histopathological sample handling, such that the tissue must be received unfixed.
Disease, such as cancer, may result from a relatively small number of underlying genetic or epi-genetic defects. These defects have phenotypic effect by resulting in proteins with altered function or regulation. The primary defect may have secondary functional consequences through its effect on signaling networks either inside cells or between cells. Some of these secondary effects may be possible to detect only at the protein level, and so can only be studied by proteomic techniques. Parameters that need to be detected include alterations in protein activity, of protein amounts, of posttranslational processing and of protein interaction partners.
There may exist several subgroups of a disease that clinically and histopathologically are difficult to distinguish but that can be distinguished by genomic and/or proteomic techniques Citation[2]. Knowledge of the nature of the subgroups is useful for understanding the mechanisms of disease, for diagnosis and for prognosis as well as to suggest new therapy targets Citation[3]. It is important to investigate a large number of independent cases both at the genetic and at the protein level to identify these subgroups. Thus tissue biobanks are of use as repositories of tissue so that a sufficiently large number of reproducibly handled samples of even rare diseases can be analyzed for deviation of both nucleic acid and protein constitution.
Blood samples are more easily obtainable than tissue biopsies. Tissue turnover leads to leakage of tissue degradation products, including proteins or peptides, into the bloodstream. If the type or amount of leakage is altered in disease, as may be a common occurrence, the altered protein pattern may be detectable in plasma Citation[4]. Likewise there may be a tissue reaction to disease, part of which may result in the presence of reactive proteins in the bloodstream. The proteome of blood samples can therefore be analyzed in the hope of discovering marker proteins that can be used for screening, staging or monitoring. Plasma markers may be sought either based on knowledge of the protein constitution of the diseased tissue or may be sought without prejudice. Early diagnosis, e.g. of cancer, through proteomic analysis of blood samples would be an important adjunct to tissue biopsy and histopathological analysis.
Advantages and disadvantages of analyzing solid tissue and blood
Human tissue is the real object of inquiry into the causes of health and disease and needs to be studied for the reasons given above, but it has several shortcomings from an analytical point of view. The tissue may not be homogenous, and so it is important to at least have a microscopic image to document the complexity present in a sample. The complexity of the disease can include presence of a mixture of cell types, including parenchyma cells, stroma cells, healthy and diseased cells, normal and reactive cells, inflammatory cells, extracellular matrix, necrosis, blood vessels and blood. In some cases it may be desirable or acceptable to obtain an integrated analysis of all those components. In other cases it may be important to only analyze a subset, by isolation using, for example, laser capture microdissection Citation[5], cell fractionation or cell culture. Fine needle aspiration of fresh resected epithelial tumors combined with enrichment by Percoll® centrifugation has proven to be useful since a majority of serum proteins, blood cells and connective tissue are removed Citation[6]. The often small amount of tissue available puts emphasis on efficiency of use and on sensitive analytical tools. Many types of experiments can only be performed in model organisms or in in vitro experiments. Even in cases where it is not possible, or desirable, to use human tissue directly for research purposes, the relevance of results from model organisms or in vitro systems will have to be tested by comparison with actual human tissue.
Blood contains tissue leakage products from many, if not all, tissues. These leakage products may serve as markers for disease screening, staging or for monitoring therapy response. Some such markers seem to be detectable by massively parallel techniques, such as 2D-PAGE or LC-MS/MS, while others may need to be detected by more sensitive, targeted, lower throughput techniques such as Western blot, ELISA or immuno-PCR. There is also a potential for a biological variability of the protein constitution within populations and within individuals that may confound plasma analysis. These, and other, parameters have started to be studied within the HUPO PPP Citation[7], Citation[8]. In addition, it is clear that preanalytical variables, such as sample handling, influences the protein composition of the blood sample Citation[9]. Initial studies of the nature of the altered composition due to preanalytical variables, as well as ways to limit the alteration or standardize it, have been performed Citation[9–12]. The ultimate goal would be to provide a basis for rational standard operating procedures for blood handling that would reliably preserve sufficient traces of the original protein constitution to be diagnostic, and avoid variable artefactual modifications, while being routinely applicable in a hospital setting.
The complexity, concentration range and dynamics of the human proteome
In order to determine which proteomic investigations are relevant it is essential to compare the protein complexity and concentration range of a human tissue to the resolution and sensitivity of currently available technology. The human genome gene number predictions fall in the range of 22 218 in Ensembl 35 and ∼28 913 genes Citation[13]. The DNA sequences of these genes provide a resource that has enabled and accelerated large-scale identification of isolated proteins by allowing identification of proteins based on short partial sequences or sets of specific protein fragments, primarily obtained by mass spectrometry. The analyzed genome sequences also provide a list of proteins, setting the outside limit of the proteome. Currently 39% of the human proteome can be assigned to known protein structures, while 77% has some functional annotation. As an example, the number of membrane spanning proteins is estimated to be ∼13% of the proteome. Alternative splicing of RNA, RNA editing, differential proteolysis and posttranslational modifications of amino acid side chains result in a larger diversity of physically different proteins than suggested by counting the number of estimated genes alone. An average of at least six protein variants per gene may exist Citation[14], and potentially many more.
Different cells and tissues express overlapping subsets of the total proteome. The current estimates are that individual cells express some 12 000 different open reading frames Citation[15], Citation[16]. The range of protein concentrations in solid tissue is considered to be about 1×106. This concentration range is outside the scope of current proteomic instrumentation, which has a concentration range of about 1×102 to 1×104 Citation[17]. Even for the yeast, Saccharomyces cerevisiae that expresses an estimated 6 139 genes, and has a range of protein concentrations of at least 105, only 282 proteins were identified by 2-D PAGE Citation[18] and 1 484 Citation[19] and 1 504 Citation[20], in two different studies, by LC-MS/MS. 4 251 TAP-tagged ORFs were detected by Western blot Citation[21], making Western blot the most sensitive current proteomic technology. In an investigation of human mammary epithelial cells by LC-MS/MS the estimated coverage of the human genome was 4% Citation[22]. It should be clear that current methods and instrumentation therefore unaided could not cover the full variety of proteins and range of concentrations present in human tissue in a single experiment. Diseased tissue, by definition, expresses a different proteome from healthy tissue. In terms of absolute protein amounts the differences may be small. Detection of the differences depends on the sensitivity and dynamic range of the methods and instruments used. Some important differences may be detected among the most abundant 4% of the proteins with current global methods. For complete coverage, however, experiments will need to be designed to home in on specific sub-proteomes that are within the analytical range of existing technology. Some ways to improve coverage of the total proteome are to subfractionate the tissue proteins, to deplete abundant proteins or to immunoaffinity purify proteins or protein complexes of interest. These approaches all bring their own challenges in increased workload, time expenditure, and the potential for protein losses. It is still not clear that any fractionation scheme, combined with protein identification analysis, has achieved a comprehensive identification of all proteins in a given human cell or tissue. It has been suggested Citation[23] that application of genome-wide affinity-ligands may be used to enhance the detection of low-abundance proteins. A genome wide project to generate antibodies Citation[24], Citation[25] may fulfill some of the necessary requirements.
The proteins as well as their posttranslational modifications turn over with varying half-lives, creating a proteome that is not only diverse, but also changing over time. Some of the dynamic modifications have important regulatory functions. Of particular interest are dynamic phosphorylation modifications that sometimes regulate protein function. Thirty percent of the proteins are estimated to be phosphorylated Citation[26]. About 0.1% of these are tyrosine phosphorylated Citation[27]. The phosphor groups are added with the help of protein kinases. Currently a total of 518 predicted kinases of various groups exist in the human genome, including an estimated 90 tyrosine kinases Citation[28]. The human genome also encodes approximately 100 phosphatases that belong to the protein tyrosine phosphatase (PTP) superfamily Citation[29], in addition to about 23 to 32 serine-threonine protein phosphatases Citation[30], Citation[31]. Direct or indirect alteration of the activity of a kinase or phosphatase will influence the steady state phosphorylation of its substrates and so potentially the substrate's activity. Alterations in protein phosphorylation are common in cancer and phosphorylation can be detected by proteomic techniques Citation[27], Citation[32–35]. It is a challenge to study a relevant snap-shot of this varied and dynamic proteome with a deep concentration span, by applying the appropriate technology to well preserved specimens.
The plasma and serum proteome
Blood can be considered the body's most complex proteome, since it contains not only proteins that function in the blood, ranging in concentration from albumin (10s of mg/mL) to cytokines (pg/mL), but also contains tissue leakage products from many, if not all, tissues. Blood is dominated by erythrocytes (about 45% of the volume). For the purpose of analyzing the presence of disease markers among tissue leakage proteins in plasma, the erythrocytes, and other blood cells, are uninformative and saturate the protein separation media. It is therefore logical that they be removed, by centrifugation. In this context it would be preferable if any leakage from not only the erythrocytes, but also white cells and platelets could be eliminated, or at least standardized, so that variable amount of such leakage does not confound the analysis of the plasma Citation[8], Citation[11], Citation[12]. Ninety nine percent of the protein complement in plasma is made up of the 22 most common proteins. The abundant plasma proteins are informative about those disease conditions that specifically involve those proteins, but also saturate existing analytical instrumentation. Therefore one should consider depleting them when rarer plasma proteins, and specifically tissue leakage proteins, are the objects of study. The possibility that relevant analytes may be inadvertently removed by being associated with abundant plasma proteins should be considered, however Citation[36], and so it may be relevant to study the composition also of the proteins associated with the abundant plasma proteins.
The observed concentration range of proteins in plasma exceeds that estimated for solid tissue and exceeds 1×1010, placing even greater demands on the sensitivity of the analytical technology. The numbers of identified proteins for the human serum proteome, prior to the PPP were 325 distinct proteins by 2-D PAGE Citation[14], and 490 proteins by LC-MS/MS Citation[37] to which should be added proteins identified by other means for a grand total of 1 175 distinct proteins. The PPP now catalogues a total of 3 020 proteins Citation[8]. Specific peptides, proteins or protein complexes can be concentrated by fractionation Citation[38], Citation[39], immuno-affinity chromatography or immuno-precipitation, yielding an enrichment of about 120-fold in the case of peptides Citation[40]. It is one of the ultimate goals of the HUPO PPP to comprehensively catalogue the proteins of plasma Citation[8]. As for solid tissue analysis, application of genome-wide affinity-ligands may be used to enhance the detection of low-abundance proteins Citation[23–25].
Even without prefractionation, crude serum samples can be applied to a protein-binding chip that is coated with a chromatographic matrix. The subset of the serum proteins that bind can then be analyzed by MS (SELDI Protein Chip technology, Ciphergen, Inc.). By trying various chromatographic matrices and conditions a protocol can sometimes be devised that detects a set of serum markers specific for a condition. In this way markers for ovarian cancer, prostate cancer and breast cancer were discovered Citation[41–45]. It should be noted that SELDI is still a relatively new technique, and that there are outstanding issues with respect to the influence of sample selection, sample handling, reproducibility, sensitivity and analytical paradigms on the results Citation[46]. The PPP recommends stringent standardization and pre-fractionation to increase the usefulness of this method Citation[8].
Protein degradation
Protein degradation is a normal part of digestion in the intestinal tract, and of tissue turnover in vivo, but may be a problem in the analysis of proteins. Ingested proteins are hydrolyzed in the gut by secreted proteases. Intracellular proteins are degraded by the ubiquitin-mediated proteolytic system, while membrane proteins and extracellular proteins that are engulfed by cells, and autophagosomes, are mainly digested by acid hydrolases in the lysosomes. Regulated cytoplasmic proteases are part of the apoptotic pathway. Proteolysis is a thermodynamically favored, potentially spontaneous, process. The predicted number of proteases in the human genome is estimated at about 553 Citation[47]. The proteases can be categorized mechanistically into aspartic, cysteine, metallo, serine and threonine proteases. Much remains to be known of their physiological functions.
For analytical investigation of a tissue, protein degradation, or rather proteolysis in excess of what occurs in vivo, i.e. artefactual proteolysis is one of the major concerns, since a degraded sample looses much its analytical utility. It is especially troublesome that proteolysis can be selective and subtle. In order to prevent proteolysis it is important to know about its mechanisms so that rational steps can be taken to avoid it. Secreted proteases and peptidases in the gut normally digest ingested proteins into peptides and amino acids for absorption. In vivo these proteases are either kept separate from cellular proteins or associate with specific inhibitors and so do not digest the host tissues. Isolated tissues that contain secreted proteases are perhaps at particular risk for artefactual proteolysis.
One of the main causes of artefactual proteolysis is admixture of the proteases with cellular compartments that are normally kept separate in vivo, thus allowing proteases access to substrates that they otherwise would not encounter. Causes of this kind of proteolysis are spontaneous Citation[48] or artefactual breakdown of cellular diffusion barriers, by, for example, treatment with detergent or by tissue homogenization (perhaps as initial steps in a fractionation procedure) Citation[49]. Tissue homogenization is often a necessary first step in a proteomic analysis, followed by extraction, solubilization and fractionation. The altered solution conditions that are part of a tissue extraction or protein solubilization may also artefactually dissociate proteases from their cognate inhibitors, thereby activating them. In other words, the experimental manipulations that are necessary for analysis may introduce proteolytic artifacts that were not present in the intact tissue. Thus there is an obvious conflict between the need to homogenize tissue and fractionate protein extracts and the need to preserve proteins intact.
Proteolysis, like many chemical reactions, is slowed by low temperature. Therefore short times and low temperature will minimize artefactual proteolysis during fractionation. Furthermore, as soon as proteases and protein substrates of interest have been resolved into separate fractions the proteins will be stable. Proteolysis during extraction, solubilization and fractionation is therefore often a problem during early and intermediate stages of the process, but not in late stages.
For those cases where fractionation is essential and where rapid processing and cold temperatures are not sufficient, protease inhibitors can be used Citation[50], see for a list of such inhibitors. Ideally the type of protease activity should be determined, and specific inhibitors directed against this activity used. In practice it is many times expedient to simply add a cocktail of protease inhibitors. A rational cocktail of protease inhibitors would be composed of irreversible inhibitors of each mechanistic class of protease. It should be kept in mind that some protease inhibitors, such as PMSF, have a short half-life in solution, and that other protease inhibitors, such as leupeptin, pepstatin or aprotinin, are peptide analogues and may confound analysis, especially when peptides are analyzed Citation[12]. Irreversible protease inhibitors that work by covalent modification, such as the serine protease inhibitor AEBSF, cause significant distortion of a 2-DE gel profile, by affecting the pI Citation[11]. Addition of protease inhibitors to samples that will be digested with trypsin as part of the analysis (see below under MALDI MS and ESI MS) would inhibit the enzyme and so has a negative impact on the analysis. For these reasons addition of protease inhibitors cannot be recommended as a routine addition to biobank samples, even if they can be shown to have a positive effect on the abundance of a specific analyte.
Table I. Common protease inhibitors and their specificities.
Intracellular proteins are degraded by the ubiquitin- and energy-dependent proteolytic system, as part of a quality control system or as part of a regulatory system. About 2 000 genes in the human genome encode proteins in the ubiquitin proteolytic pathway, about 1 500 of them encoding the specificity-determining E3 proteins. Energy is expanded in the ubiquitin proteolytic pathway to regulate the degradation of proteins, although proteolysis itself potentially is a spontaneous process. This creates the superficially counterintuitive situation that energy is expanded to carry out a degradation process. The energy expenditure may even be greater for protein degradation than that required for protein synthesis. Given the energy requirement for both protein synthesis and intracellular degradation, it is obvious that both protein synthesis and degradation will slow and eventually stop when metabolite levels fall after devascularization. While the ubiquitin- and energy-dependent proteolytic system is important in vivo, it is therefore not clear what its impact is, if any, on artefactual proteolysis.
Protein phosphorylation is frequently an important regulatory event. The phosphate groups of phosphoproteins turn over dynamically in vivo. Their very dynamic nature is an important part of their regulatory function. The turnover of the phosphorylation moiety demonstrates that kinases and phosphatases exist in the same subcellular compartment. There exists an estimated 518 kinases of varying specificity in the human genome, including about 90 tyrosine kinases Citation[28], about 13 phosphoprotein phosphatases of the PPP family and 10 of the PPM family Citation[30], and about 100 tyrosine phosphatases Citation[29]. PP1, PP2A, PP2B and PP2C are considered to be the major serine/threonine phosphatases Citation[30]. Devascularization with a concomitant lowering of ATP levels would tend to shift the equilibrium away from phosphorylation and towards artefactual dephosphorylation. The considerations to minimize dephosphorylation parallel those to minimize proteolysis, discussed above, i.e. short handling and cold temperatures should be beneficial. Phosphatase inhibitors (see ) should be considered during fractionations where dephosphorylation is observed or suspected. Once phosphatases and their substrates present in the sample have been resolved, the phosphate groups should be stable.
Table II. Common phosphatase inhibitors and their specificities.
Currently, it does not seem clear to what extent in vitro proteolysis has elements of admixture with secreted proteases, ubiquitin-mediated proteolysis, autophagy, necrosis and apoptosis, or indeed some other pathway. Current strategies to minimize degradation therefore includes general purpose precautions such as minimizing warm ischemia, quick handling, handling at temperatures just above freezing and, for specific purposes, the addition of protease or phosphatase inhibitors. Monitoring the integrity of the analyte of interest and developing validation protocols for that analyte would be necessary to determine if handling is adequate to preserve the analyte of interest.
Sample handling and cryopreservation
That tissue can be maintained intact ex vivo, in the absence of blood circulation, is obvious from the possibility of organ transplantation. A human heart can be maintained on ice for up to six hours and still be viable Citation[51]. It is reasonable to expect the proteome of a tissue that can be successfully transplanted to be functionally intact. Differences do exist in how long individual tissues can be maintained ex vivo and still remain intact, by this criterion. It is equally clear that there exist upper limits to how long the tissue proteome will remain intact. The onset and resolution of rigor mortis is an example: the onset of the rigidity following death, rigor mortis, is caused by an influx of calcium ions into muscle cells, down their electrochemical gradient 12–14 hours after death. The increased intracellular calcium causes the muscle fibers to contract, resulting in the characteristic symptom of rigor mortis. This contraction is only resolved after a further 15–25 hours, or more, at room temperature and is considered to be caused by lysosome-mediated proteolysis of the muscle fibers. Thus, the proteome in a muscle undergoing resolution of rigor mortis would be expected to be substantially degraded and so unsuited for certain kinds of molecular analyses.
Solid tissue handling
The exact time a tissue can be maintained after removal from the body without significant alterations in the proteome is still an outstanding issue. As soon as the tissue is cut from circulation it is potentially subject to artefactual degradation of both metabolites and macromolecules. Metabolite levels change within seconds or minutes. Preservation of metabolite levels would require “instant” enzyme inhibition, by microwaving live tissue Citation[52] or similarly quick methods, and cannot currently be incorporated into a standard human biobank scheme. Changes in macromolecules seem to take minutes, hours or days.
In a surgical setting, the tissue will need to be transported to a pathology department where a pathologist will inspect the tissue, cut out suitable samples for histopathological diagnosis and then freeze remaining tissue for future use. It is therefore inevitable that some time will pass between surgery and sample freezing. It is important to know if any degradation of the proteins occurs in the interim, and if so what type of degradation, and how fast. The outside limits of handling that are compatible with intact proteins need to be known so that the sample handling routines can accommodate these requirements. If degradation is unavoidable, then it is essential that the type and degree to which degradation has set in be known so that only unaffected parameters are studied. Careful investigations of the tissue handling parameters compatible with protein integrity are therefore necessary for relevant collection and handling protocols and for the evaluation of analytical results.
A need therefore exists to test the parameters that are actually measured in proteomics: specific protein and tissue integrity, protein quantity and state of posttranslational modification. The proteolytic changes may have a different kinetic in different kinds of tissue. The maximum allowable time before freezing would logically follow from the maximum time that results in intact proteins by the criteria listed in . Indications from human autopsy material show that human brain myelin basic protein is only subjected to minimal loss after 48 hours Citation[53], Citation[54]. The level of carboxymethylation of proteins from human post-mortem brain obtained within 24 hours of death is not significantly different to the level present at the time of death Citation[55]. Further studies of the temperature and time-dependence of the parameters listed in appear warranted.
Table III. Practical assays to analyze the integrity of tissue samples proteomes.
A study is currently under way within the Human Brain Proteome Project (within the HUPO) to, among other things; perform quantitative analysis of human brain samples to assess the protein stability over time in post mortem samples. In the interim it would seem advisable to hold tissue cold, but not frozen and for as short a time as possible prior to inspection by a pathologist, and to document the handling the tissue has been subjected to. Tissue for proteomic analysis should be snap frozen in liquid nitrogen at −196°C or in isopentane cooled with dry ice at −78°C as quickly as possible, in the absence of evidence that more relaxed freezing routines are adequate.
Blood handling
Blood, and blood derivatives, can be maintained in a functional state ex vivo for weeks, at least, as evidenced by the possibility of blood transfusions. This observation would suggest that at least the functionally important blood proteins could be maintained in a functional state for at least that amount of time. Total protein levels are stable in serum or plasma for up to four weeks at 4°C, although fresh specimens are recommended Citation[56]. Blood samples are also currently used to monitor certain cancer biomarker proteins, such as alpha-fetoprotein, chorionic gonadotropin-beta, CA19-9, CA125, CEA, thyroglobulin, Prostate Specific Antigen (PSA), CA15-3, CA27-29 or HER2/NEU Citation[57], demonstrating that at least some analytically essential features are in fact preserved. It is therefore clear that reliable analysis of at least some predefined analytes is adequate with existing blood handling paradigms. However, it is also clear that there exist intra-individual and inter-individual variations in the protein constitution of blood that would result in false positive correlations by a de novo massively parallel proteomic analysis, unless they are identified and compensated for Citation[8]. The HUPO PPP has set out to ultimately analyze the complete protein constituents in normal humans and determine the extent of variation in plasma proteins Citation[7].
It is equally clear that there exists significant pre-analytical variability that results from variation in handling that could also result in false positive correlations. Particularly spontaneous or accelerated clotting that is used to produce serum is difficult to standardize Citation[8] and the PPP for this, and other reasons, recommends plasma, particularly EDTA plasma.
There are currently four blood collection options: Blood samples could be allowed to clot spontaneously through the physiological clotting process that includes proteolytic modification and ultimately produces a clot through the conversion of fibrinogen to fibrin. Serum would then be produced as the liquid phase. A priori, a proteolytic event and a clotting event would not seem desirable in a standard protocol for collecting samples for proteomic analysis. In practice it turns out that up to 40% of the peptides in serum are specific to serum Citation[12] compared to plasma and so would seem to be generated through the clotting proteolytic process, or released from platelets or white blood cells. Alternatively three variants of plasma could be prepared by addition of potassium-EDTA, lithium heparin or sodium citrate, at the time of sampling. The PPP now recommends EDTA (or citrate) for anticoagulation, since the highly charged heparin interferes with some downstream applications Citation[8].
For the analysis of amounts of specific analytes, blood can be handled at 4°C, to improve protein integrity. However, because of the cold-activation of platelets Citation[58] and subsequent release of platelet content this may not be optimal for discovery proteomics, since variable release may cause false positive markers. An estimated 14% of peptides are platelet-derived in cold treated plasma Citation[12]. Reduction of platelet content can be achieved by re-centrifugation or filtration of the plasma, or by the addition of platelet stabilizer cocktail, at room temperature. It remains to be seen if the advantages of platelet reduction outweigh the potential of increased protein-degradation at room temperature, compared to handling at 4°C, or if addition of platelet stabilizer cocktail will be preferred. As discussed above, addition of protease inhibitors should only be done for specific purposes, where they do not interfere with the desired analysis.
Stability of frozen tissue
Frozen human cells can be stored in liquid nitrogen (−196°C) for years and still be revived (if provided with an appropriate cryoprotectant). Storage at −70°C shortens the shelf life. These common observations indicate that the proteome of frozen tissue may remain intact for years if stored at, or below, −70°C. Indeed, stability of blood enzyme marker proteins for 59 months at −70°C has been observed Citation[59]. A study by SELDI-TOF that compared blood stored at −20°C, −80°C and liquid in nitrogen over a two month period showed no major differences Citation[11]. While there currently does not exist evidence for proteolytic degradation of tissue during storage at −70°C, there exists a need for more investigations into the long-term preservation of frozen tissue samples, whether frozen intact or as extracts.
Risk of infection and contamination
To be useful for proteomic analysis, tissue needs to be fresh and unaltered. The handling of fresh human tissue (as opposed to traditional formaldehyde fixed tissue) requires that steps be taken to minimize exposure of personnel to known and unknown contagions. Conversely, contamination may make a sample less useful. To address these concerns, the NIH universal precautions (http://www.niehs.nih.gov/odhsb/biosafe/univers.htm) consisting of barrier protection should be applied to the design of rooms, ventilation, access, personal protection and handling routines.
Tissue extraction
Apart from immunohistochemistry or scanning mass spectrometry, no proteomic analysis can be performed on intact solid tissue. In order to perform a biochemical analysis of tissue proteins, they need to be effectively released from the tissue. The tissue needs to be disintegrated, proteins extracted and solubilized (if they are not already water soluble), resolved and detected.
Scanning mass spectrometry
A profile of the most abundant protein-masses in a thin tissue section can be obtained by spotting MALDI matrix on the tissue, followed by directing a laser beam onto successive matrix spots. The laser beam, together with the matrix, will desorb and charge proteins that will then be allowed to fly in the mass spectrometer to estimate their size, resulting in a profile of some of the most abundant protein species. By scanning from matrix spot to matrix spot a synthetic image of protein size distributions as a function of tissue location can be constructed Citation[60–62]. Such images may be diagnostic for a particular disease. In addition, the proteins being some of the most abundant proteins, give clues to what pathological proteins may be detectable in blood samples.
Protein folding, denaturation and protein-protein interactions
Protein extraction is in part based on disrupting protein folding and protein-protein interactions, and so extracting by solubilizing. Proteins fold onto themselves as they are translated, based on interactions between water molecules, amino acid side chains, and amino acid backbone and interactions with other protein subunits, including chaperones. The quantitatively most important atomic forces to stabilize protein folding are hydrophobic interactions Citation[63], Citation[64]. Hydrophobic interactions are due to the high energy required to move non-polar amino acids into a water solution. This causes hydrophobic amino acid residues to be most stable when buried in the protein interior. Proteins may have one or more stable states, only one of which may be associated with protein function, such as enzymatic activity or specific binding. This state is sometimes metastable in vivo if alternative states can be adopted. Proteins that adopt an alternative structural state than the functional state are non-functional and termed “denatured”. The quantitative dominance of hydrophobic interactions means that agents that break them, such as detergents, e.g. SDS, or chaotropes, e.g. urea, act as general protein denaturants and solubilizers.
Blood plasma, or serum, protein extraction
The liquid, water-soluble, component of blood, plasma or serum, can be obtained by centrifuging a blood sample at low speed. The sedimented solid component of EDTA-blood contains red blood cells, with a top layer of white blood cells, the so-called “buffy coat”. White blood cells can be collected separately from the plasma and DNA or RNA can be extracted from the cells by standard procedures, if desired. The plasma or serum liquid supernatant contains water-soluble proteins that can be subjected to fractionation or direct analysis (see below).
Solid tissue disintegration and protein extraction
Protein extraction involves mincing tissue so that the surface exposed to solvent is enlarged and cells are opened, to maximize extraction. A variety of homogenization methods exist, including handheld or motor driven homogenizers, sonication, French pressure cell, grinding with alumina or sand, glass bead vortexing, detergent lysis, or frozen tissue crushing Citation[65–67]. As cells are broken any proteases and other degrading enzymes gain access to cellular compartments they do not normally see, resulting in the potential for protein degradation Citation[49]. This risk is minimized by an as low a temperature above freezing as possible and by working quickly. The use of protease and other enzymatic inhibitors should also be considered Citation[49] as discussed above.
Water-soluble protein extraction
Extraction of water-soluble proteins without denaturation, allows some forms of functional assays. The proteins of the plasma component of blood exist as water-soluble proteins, which simplifies their study. In contrast, only the water-soluble fraction of solid tissue proteins will be extracted by exposure of disintegrated tissue to buffers. The buffer-insoluble residue can be sedimented by centrifugation and the soluble proteins can be fractionated or analyzed. A cocktail of detergents, for example the RIPA buffer that increases protein extraction while leaving some protein complexes apparently intact, can extract a larger fraction of solid tissue proteins. Care needs to be exercised to preserve protein stability during these procedures Citation[68]. These extracts, although incomplete, are suitable for some functional protein studies such as of enzymatic activity or of binding partners. Analysis of the subunits of protein complexes is of great interest.
Water-insoluble protein extraction
A large part of the total tissue protein is not soluble as individual polypeptide chains, directly after homogenization and extraction with hydrophilic buffer. They may be part of large or insoluble protein complexes, including lipid membranes and cellular and extracellular matrix and its associated proteins. In order to solubilize these proteins the complexes either need to be broken down into their constituent protein subunits, or digested by a sequence specific protease, such as trypsin, into water-soluble peptides. Two of the most efficient ways to extract water-insoluble protein subunits is solubilization in either the ionic detergent SDS or in the chaotrope urea. Both work by denaturing the proteins (although by different mechanisms) so that they cannot bind to other molecules and are set free in solution. Since these forces are of the same kind as those forces that cause proteins to fold into a functional structure, the individual subunit proteins are denatured and become non-functional upon solubilization. Thus functional studies are not normally possible on this material. Instead identification and quantitation benefits from the more complete solubilization than can commonly be achieved by hydrophilic extraction alone. SDS or urea will also effectively extract water-soluble proteins.
The very efficiency of SDS (or urea) solubilization suggests that it may be used as a standardized, “universal”, protein solubilization for identification and quantitation (but not for functional studies). Many analytical techniques do not tolerate the 2% SDS present in SDS-PAGE sample buffer, for example. However, low amounts of SDS are tolerated by several of the major proteomic techniques of 2-D PAGE, LC-MS and antibody array Citation[20], Citation[69], Citation[70]. Procedures to dilute the SDS or buffer change procedures may be used to make SDS extracts compatible with sensitive downstream analytical methods. An added benefit to SDS extraction is that SDS denatures and inactivates many, but perhaps not all, degrading enzymes, thus helping to preserve an intact proteome.
The analytical modalities that are compatible with each solubilization scheme are given in .
Table IVa. Compilation of tissue solubilization methods and compatible protein resolution methods.
Table IVb. Compilation of protein resolution methods and compatible protein analyses
Total protein assays
Several different quantitative protein assays exist. An important parameter in choosing a protein assay is sometimes its ability to tolerate the detergent and reducing agent that are part of an SDS extraction, so that the protein amount actually solubilized can be determined. The following common assays do not tolerate the amounts of SDS and reducing agent that typically are part of an SDS extraction buffer and therefore need to be used prior to their addition: absorbance at 280 nm, Lowry assay, Bradford assay, Bicinchoninic acid (BCA) assay and the Pierce assay. The Biorad RC DC assay is based on the Fohlin reagent of the Lowry assay, but includes a precipitation step that removes part of the detergent and reducing agent, thus allowing protein concentration determination of an SDS extract.
Protein resolution and detection
Protein fractionation
There are two basic reasons to fractionate a cell or tissue: 1. to reduce the complexity of an extract so as to enrich for low abundance proteins so that they can be detected Citation[71], , and 2. to isolate a particular organelle or complex to determine its composition. The disadvantages of fractionations are the risk of proteolysis, the risk of artefactual losses or gains and the risk of irreproducible fractionation outcomes, making comparisons difficult or impossible. Appropriate controls need to be made so that authentic subunits can be distinguished from contaminants. Numerous fractionation methods exist. Only some of the major methods are listed in . Please consult specialized manuals for details.
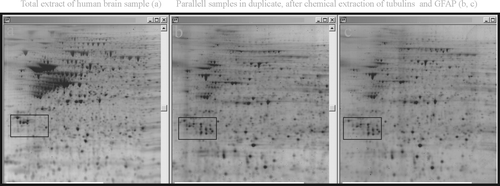
Figure 1. Demonstration of the effect of depleting highly abundant proteins, in this case by high salt extraction of tubulins and GFAP, on enhanced detection of less abundant proteins (compare, for example, contents of squares before and after extraction).
Protein resolution and detection methods
SDS-PAGE
SDS-PAGE Citation[72], Citation[73] is a one-dimensional electrophoresis system that separates proteins according to size. Because of the complexity in most cell extracts, SDS PAGE will not normally separate whole cell extracts into individual pure proteins. It has an advantage compared to 2-D PAGE in that there are no selective losses of charged or hydrophobic proteins. The load is practically limited to about 50–100 µg. The sensitivity is limited by the sensitivity of the detection techniques and by abundant proteins that saturate the loading capacity.
SDS is an ionic detergent that binds to proteins approximately evenly along their length. It solubilizes and denatures most proteins and dissociates most protein complexes. Since it is charged it imparts a net negative charge to proteins that is approximately proportional to their length. Electrophoretic sieving through a polyacrylamide matrix therefore separates the proteins approximately according to size. Proteins can be stained by general proteins stains such as Coomassie Brilliant blue, silver-staining, fluorescent ruthenium compounds such as Sypro® Orange (Molecular Probes) or Sypro® Ruby (Molecular Probes) Citation[74], Citation[75], or a natural fungal fluorophore, Deep Purple (Amersham Biosciences, Inc.) Citation[76]. Posttranslational modifications such as sugars can be stained by a version of the periodic acid Schiff reaction, such as Pro-Q™ 300 (Molecular Probes) Citation[77] or by labeled lectins. Phosphorylation can be detected by the proprietary Pro Q Diamond fluorescent stain (Molecular Probes) Citation[33]. Gel slices can be cut out, the proteins subjected to proteolytic digestion and the diversity of peptides analyzed by mass spectrometry (MALDI MS or ESI MS, see below) and identified by MS/MS.
Individual proteins can be identified and semi quantitatively quantified by Western blot Citation[78]. In addition to loading the same amount of protein on the different lanes, there needs to be an internal loading control detected in parallel. Typically GAPDH, actin or tubulin is used.
Differential protein expression can be studied even with one-dimensional SDS PAGE if there are very major differences between extracts. In most cases the increased resolution of a 2-D PAGE separation is needed when less abundant proteins are compared.
2-D PAGE
Two-dimensional electrophoresis separates according to the independent parameters isoelectric point and size Citation[79–81] and therefore has higher resolution than only SDS PAGE. Whole cell extracts can frequently be resolved into individual, relatively pure, proteins by this method. The capacity of the 2-D PAGE gel is limited primarily by the capacity of the first dimension immobilized isoelectric focusing strip to about 150 µg, but can be increased to milligram amounts Citation[82]. The sensitivity is governed by the same limitations as SDS-PAGE. Wide range isoelectric focusing can be used to obtain an overview over most detectable proteins. Use of a narrow pH range, that resolves only in a limited pI range, will increase the potential load of protein in this interval and will lead to increased sensitivity for low abundance proteins in this interval.
The proteins are dissolved in the chaotrope urea. Urea does not have a charge and does not interact with the proteins. It will therefore not interfere with the isoelectric focusing first dimension (as excess SDS does). Proteins with very high or low pI, high and low molecular weight proteins and integral membrane proteins are under-represented in 2-D PAGE. Very acidic and very basic proteins are underrepresented since the fixed pH gradient is technically limited to pH 3 to 10 (or 11). Membrane proteins may precipitate in the first dimension gel, at their isoelectric point. It may therefore be difficult or impossible to analyze some of them with this technique.
The proteins can be detected and identified with the same methods as proteins resolved by SDS-PAGE. Documentation, quantitation and gel comparisons are made with image analysis software. In addition, since 2-D PAGE can resolve many proteins into relatively pure individual proteins, mass spectrometry (typically MALDI mass spectrometry) can be used to peptide fingerprint and/or sequence the protein in question.
Matrix Assisted Laser Desorption Ionization (MALDI) mass spectrometry (MS)
Mass spectrometry can be used to determine the precise mass of individual proteins or peptides, whether pure or mixed. Normally the mass of a protein is not enough for positive identification, since there are many proteins with similar molecular weight. Digestion of a purified protein with a sequence specific protease, such as trypsin, generates a diagnostic set of peptides whose precise masses can be measured by a mass spectrometer. Typically isolated protein spots from a 2-D PAGE experiment are digested with trypsin and the digest embedded in an ionization matrix on a MALDI chip. The spectrum of peptide sizes is obtained by MS and DNA and protein sequence databases are searched to identify the protein or gene that would encode a protein whose calculated tryptic digest would generate the observed spectrum of peptides. With this approach, a more or less pure protein is needed as a starting point for digestion.
Mass spectrometry can also be used to determine directly the amino acid sequence of individual tryptic peptides. The masses of the peptides of interest are first determined. Then individual tryptic peptides are isolated and subjected to fragmentation, by collision-induced dissociation, into a series of peptides, each peptide differing from its parent by missing one amino acid residue. These fragments are fed into a second mass spectrometer (MS/MS) and their masses determined. The difference in mass between a fragment and its parent can be used to deduce the identity of the missing amino acid. By repeated application of this procedure an amino acid sequence is generated. The SEQUEST or other algorithm can be used to identify the original protein, by using the amino acid sequence to search sequence databases. This approach can be applied to complex protein mixtures.
Electro Spray Ionization (ESI) MS
ElectroSpray ionization ionizes soluble peptides that emerge from a capillary as a spray. ESI differs from MALDI in the practical respect that the peptides can be fed to the ESI MS directly in liquid form, without drying or matrix application. The ESI MS can therefore be coupled in real time to an HPLC and the eluate monitored by MS. Typically a sample is digested into specific peptides by trypsin, the tryptic mixture is chromatographically resolved on an HPLC and the resolved peptides eluted into an ESI MS. Specific peptides can be selected for sequencing by MS/MS. A powerful version of this idea is to use an ion exchange resin followed by a reverse phase resin (2D-LC or LC/LC) to absorb a trypsin-digested sample and then to gradually elute the sample. The sample is then analyzed by MS and MS/MS. The sequenced peptides are identified by a database search to generate a catalogue of identified proteins. The technique is variously referred to as 2D-LC MS/MS, LC/LC MS/MS or Multi Dimensional Protein Identification Technology (MuDPIT) Citation[19], Citation[20]. When applied to serum samples or yeast whole cell extracts, the technique identifies more proteins than does 2-D PAGE.
Surface Enhanced Laser Desorption/Ionization (SELDI) MS (Ciphergen, Inc.)
The proprietary SELDI MS (Ciphergen, Inc.) is a combination of a one-step chromatographic purification of whole proteins with MALDI MS. The sample chip is covered with a selection of one of various chromatographic matrices that selectively absorbs a subset of the proteins in solution. Loosely bound proteins are washed off. The bound proteins are then ionized, desorbed and analyzed for size distribution by MS. Characteristic markers for a specific sample is sought. In practice, mostly relatively low molecular weight proteins are discovered as biomarkers. Since intact proteins are analyzed, no peptide fingerprint or amino acids sequence is obtained initially. Typically the characteristic size of a marker is used as an assay to purify sufficiently large amounts of marker for generating a tryptic peptide fingerprint, or MS/MS amino acid sequence, for identification. Serum biomarkers for uterine cancer, ovarian cancer, prostate cancer and breast cancer were discovered by SELDI Citation[42–45], Citation[83]. Some outstanding issues and the limitations of SELDI have been discussed Citation[46].
Enzyme Linked Immuno Sorbent Assay (ELISA) and antibody microarray
The ELISA assay is a robust, sensitive and specific technique for identification and quantitation of specific protein analytes. In one variant of the method a specific antibody is bound to a solid matrix in a microtiter plate. The analyte is added in either pure form in solution or as part of a solubilized mixture. Unbound excess is washed off. Bound analyte is detected by enzyme-labeled antibodies, which are then detected by an enzymatic reaction. In another variant of the method the analyte mixture is chemically labeled with a fluorescent dye, such as CyDye, before being exposed to the bound antibodies. Excess labeled analyte mixture is washed off and bound analyte is detected and quantified, obviating the need for a second antibody reaction.
With increasing availability of antibodies and with an increasing interest in the parallel detection and quantitation of many proteins, it is relevant to attempt to increase the number of antibodies in an ELISA, to place the antibodies in an array, to use fluorescent labeling for detection and to miniaturize the array. This extended ELISA is called an antibody array or antibody microarray Citation[69]. With the sequencing of the human genome, and the concomitant availability of the sequence information of the predicted genes, it became possible to realistically conceive of making antibodies against all human proteins Citation[24], Citation[25]. It is clear that a genome-wide, or proteome-wide, antibody array would be a very powerful technique to simultaneously identify and quantify most, or even all, human proteins in any sample. It has however become obvious that scaling up the familiar ELISA in this way brings some major challenges:
Linearity of response is dependent on matching the affinity of the antibody to the concentration of the analyte. The currently observed concentration range is about 100 to 1000-fold Citation[69]. With a high dynamic range of analytes a single antibody is likely to be unable to accommodate the full dynamic range. Serial dilution of the analyte may solve this issue.
Specificity of an antibody may be insufficient Citation[84], especially if it is directed to a rare analyte. There will be present in the protein mixture proteins of similar or higher concentration than the specific analyte. Some of these proteins may show a smaller or larger degree of cross reactivity on a molar basis, which may obscure the specific analyte.
There exists a requirement for the antibodies to be used to be physically stable during the conditions encountered during fabrication, storage and use of the microarray.
Detection would either involve labeling every individual extract that is to be measured, or else involve creating a second set of detection antibodies directed against a second epitope of each analyte. Both approaches bring their own problems: Individually labeled extracts would be somewhat cumbersome. Creation of a second set of antibodies for each protein would double the effort that would have to go into the project and would result in additional crossreactivity and sensitivity problems.
Detection of phosphorylation
Protein phosphorylation can be an important regulatory event, whether it involves phospho-tyrosine, phospho-serine or phospho-threonine or another type of phosphorylation. Anti-phospho-tyrosine antibodies can be used both to detect tyrosine phosphorylation in Western blots and to affinity purify tyrosine phosphorylated proteins or peptides Citation[27]. Antibodies against serine- or threonine phosporylation have so far shown some peptide sequence specificity in addition to the phosphate specificity and have therefore been of limited utility as general reagents. Their use is therefore restricted to detect phosphorylation at specific sites on specific proteins.
Immobilized metal affinity chromatography (IMAC) can be used for a variety of chromatography purposes depending on the specific metal ion bound to the matrix. Iron ions can be used to relatively specifically purify phosphopeptides Citation[26]. Proteins need to be digested to their corresponding peptides for this purpose. Acidic amino acid residues are sufficiently similar to phosphate groups to copurify with the phosphopeptides. It is therefore important to methylate the glutamic acid, aspartic acid and the carboxy terminus of any peptides to neutralize their charge and so to increase the phosphopeptide specificity of an iron IMAC matrix.
While metabolic labeling with radioactive phosphor has been used for electrophoretic analysis of cultured cells Citation[34], no corresponding general reagent to detect phosphorylation has until recently existed for tissue samples. Molecular Probes now markets the proprietary Pro-Q™ Diamond Phosphoprotein gel stain () that can detect 1–16 ng of a phosporylated protein, depending on its phosphorylation state. It will detect serine-, threonine- and tyrosine-phosphorylated proteins resolved by SDS PAGE or 2D PAGE Citation[33].
Figure 2. Demonstration of the specific detection of phosphoproteins (by Pro-Q® Diamond staining) and total protein detection (by SYPRO Ruby staining) of a 2-D PAGE gel of cultured fibroblasts (B. Franzén, unpublished).

The increase in molecular weight that results when an amino acid sidechain is phosphorylated (+80 Da), can be detected by tandem mass spectrometry (MS/MS) Citation[85]. Sensitivity of detection is dependent on the fraction of the total specific protein that is phosphorylated at any given time.
By taking advantage of the chemical nature of the phosphorylated amino acid sidechain, a chemical derivative specific for phosphor groups can be attached Citation[86], Citation[87]. The modified phosphopeptides can be purified by chromatography. They can then be analyzed and sequenced by LC-MS/MS. The method holds the potential for an ICAT approach (see below) to study differential phosphorylation, if the method has the required sensitivity.
Detection of glycosylation
Glycosylated proteins can be detected in gels, either directly, or after blotting to a membrane, either by the periodic acid Schiff reaction (or a fluorescent derivative of the same, such as Pro-Q™ Emerald 300 glycoprotein gel stain (Molecular Probes)) or by a variety of fluorescently labeled lectins with different sugar recognition specificities.
Analysis of differential protein expression
Parallel assays can be used to compare specific analytes in normalized samples. To minimize inter-experimental error it is sometimes desirable to be able to mix two or more protein samples and simultaneously measure the relative concentration of the specific analyte. Proteomic tools have recently been established to compare the relative amounts of a specific protein analyte in two, or more, different samples. These techniques require differential labeling prior to analysis, either by chemical modification or by incorporating stable isotopes.
Relative quantitation
Difference Gel Electrophoresis (DIGE)
To compare two, or more, 2-D PAGE gels requires them to be virtually warped, using landmark spots, to match up the corresponding protein spots on the two different gels. This warping, and other potential inter-experimental variations in the execution of a 2-D PAGE experiment, opens for a variability that is sometimes undesirable. Instead, two or three extracts can be covalently labeled with different fluors, which allows a direct comparison of spots and quantities to be made in one and the same gel (2D Fluorescence Difference Gel Electrophoresis (2D Fluorescence DIGE)) Citation[88]. This eliminates the need to match different gels to each other and eliminates any differences in sample handling from the sample application and on. In addition, covalant prelabeling of protein extracts with a CyDye, or other fluor, may increase the sensitivity of detection.
Stable Isotope Labeling by Amino acids in Cellculture (SILAC) and Isotope Coded Affinity Tag (ICAT)
Differential labeling of protein extracts for mass spectrometry involves introducing a “light” isotopic tag into one sample and a “heavy” isotopic tag into a second sample. If the experiment is performed in cell culture, the light and heavy isotopes can be introduced as differentially labeled essential amino acids (SILAC) Citation[89], Citation[90]. Differential labeling of tissue extracts for mass spectrometry involves covalently modifying sulfhydryl groups (or other reactive groups) of proteins with chemical reagents that include stable isotopes, specifically an eightfold deuterated heavy version or an isotopically light version of a linker. In addition to the linker, a biotin affinity tag is added so that the modified peptides can be conveniently purified from non-modified peptides (those not containing any sulfhydryl groups), ICAT Citation[91], Citation[92]. The two differentially tagged extracts are pooled and digested to generate peptides. Irrespective of whether the peptides are labeled metabolically or by chemical modification, the labeled peptides (light and heavy, mixed) are applied to an LC-MS/MS system. The heights of paired peaks, corresponding to the peptides derived from the two extracts, can be compared for quantitation, and the amino-acid sequence of the corresponding peptide can be determined by MS/MS. In this way quantitative differences between extracts can be detected and the identity of the differentially expressed analyte determined.
These methods can be used both for broad comparisons of the composition of protein extracts Citation[91], Citation[92], and in analyzing the components of purified protein complexes, including signaling complexes in different functional states Citation[89], Citation[90], Citation[93]. The ratio between proteins from the two extracts in a multiprotein complex can be used to suggest which proteins are authentic members of the complex rather than non-specifically associated with it Citation[89], Citation[90], Citation[93].
Enzyme Linked Immuno Sorbent Assay (ELISA) or Antibody Array
Specific analytes in different extracts can be quantified by an ELISA-like antibody array assay Citation[69], Citation[94]. The extracts to be compared are differentially labeled, like an extract for 2-D PAGE, with different fluors, such as CyDye, and are then mixed. The mixed prelabeled extracts are applied to the antibody array, excess washed off and the bound material determined at two different wavelengths corresponding to the two different fluors. In this way both the identity and the relative quantity of an analyte in each of the samples can be determined simultaneously. The assay is similar to a Western blot assay in that it requires a specific antibody, that approximately equal amounts of the extracts to be compared need to be applied, and that there needs to be an internal control for actual loading. In comparison with Western blot the assay is quantitative, but lacks the built-in confirmation of specificity that a determination of the molecular weight of the analyte provides.
Absolute quantitation
Absolute quantitation, in this context, refers to the quantitation of a given analyte relative an exogenously added internal control that is mixed into the sample and can be distinguished from the endogenous analyte. Examples are differentially labeled, purified, proteins or peptides added to samples to be resolved by 2-D PAGE or an antibody array, or a stable isotopically labeled specific peptide that corresponds to a tryptic peptide to be quantified by mass spectrometry.
Absolute quantification (AQUA)
Internal standard peptides are synthesized that correspond to the expected native tryptic peptides of a protein of interest. The standards are made to incorporate stable isotope labeled amino acids, so that they are chemically identical to the native peptides, but can be distinguished from the native peptides by MS. Such synthetic peptides can also be prepared with postranslational modifications (e.g. phosphorylation, methylation or acetylation). Such internal standard peptides are then used to measure the absolute levels of proteins and posttranslationally modified proteins after proteolysis Citation[95].
Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA)
In a variant of the internal standard approach for quantitation, the native peptide and the internal standard are immunoaffinity purified by anti-peptide antibodies prior to quantitation by MS, thereby increasing the sensitivity of the assay by 120-fold Citation[40].
Considerations for designing clinical proteomic assays
In clinical analytical practice it will be important to have reliable, cost-effective, high throughput assays. These assays do not need to be the same as those used to initially identify a diagnostic analyte. Whatever the analytical modality used, we can expect that multiple, rather than single, analytes will be required to achieve sufficient specificity and robustness. This will put additional emphasis on developing cost-effective, high throughput assays.
The most reliable analytes would be those that are causally involved in the disease process. Their measurement should therefore be our ultimate goal. A comprehensive set of analytes that are causally involved in the disease process may not currently be identified, for any number of intellectual or practical reasons. In their place a limited number of “marker” proteins that may not be causally related to the disease process, but only correlate with it, can be profitably used. It should be kept in mind that use of such “marker” proteins would likely increase the number of analytes that need to be incorporated to create a reliable assay. Appropriate statistical analysis of the multiple correlating analytes will need to be applied.
Summary
Tissue samples need to be handled reproducibly in ways that preserve the proteome prior to analysis. There exists a need to develop standard operating procedures and quality assurance manuals to document the handling of tissue samples. The proteins need to be extracted maximally and reproducibly. Protein networks that are altered in disease can be analyzed by existing proteomic techniques, if suitable samples and reagents are available. The proteome's sensitivity to proteolysis, its diverse solubility properties, complexity and concentration range makes comprehensive proteomic analysis, by a single analytical modality, unrealistic at present. The current challenge is to select the appropriate set of analytical tools to specifically dissect the most relevant alterations in important disease conditions. We hope the present review can provide basic information to help in those decisions.
Conflict of interest notification page
The authors have no personal or financial relationships that might bias their work. The supporting source has no role in the writing of the report or in the decision to submit the report for publication.
The authors have the pleasure to thank the Wallenberg Consortium North/ Swegene for support, and Bodil Eriksson for contributing .
References
- Qualman SJ, France M, Grizzle WE, LiVolsi VA, Moskaluk CA, Ramirez NC, et al. Establishing a tumour bank: Banking, informatics and ethics. Br J Cancer 2004; 90: 1115–9
- Vortmeyer AO, Weil RJ, Zhuang Z. Proteomic applications for differential diagnosis of histologically identical tumors. Neurology 2003; 61: 1626–7
- Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2005.
- Anderson NL, Anderson NG. The human plasma proteome: History, character, and diagnostic prospects. Mol Cell Proteomics 2002; 1: 845–67
- Craven RA, Banks RE. Use of laser capture microdissection to selectively obtain distinct populations of cells for proteomic analysis. Methods Enzymol 2002; 356: 33–49
- Franzen B, Linder S, Okuzawa K, Kato H, Auer G. Nonenzymatic extraction of cells from clinical tumor material for analysis of gene expression by two-dimensional polyacrylamide gel electrophoresis. Electrophoresis 1993; 14: 1045–53
- Omenn GS. The Human Proteome Organization Plasma Proteome Project pilot phase: Reference specimens, technology platform comparisons, and standardized data submissions and analyses. Proteomics 2004; 4: 1235–40
- Omenn GS, States DJ, Adamski M, Blackwell TW, Menon R, Hermjakob H, et al. Overview of the HUPO Plasma Proteome Project: Results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics 2005; 5: 3226–45
- Banks RE, Stanley AJ, Cairns DA, Barret JH, Clarke PC, Thompson D, et al. Influences of blood sample processing on low-molecular-weight proteome identified by surface-enhanced laser desorption/ionization mass spectrometry. Clin Chem 2005; 51: 1637–49
- Hulmes JD, Bethea D, Ho K, Huang S-P, Ricci DL, Opiteck GJ, et al. An investigation of plasma collection, stabilization, and storage procedures for proteomic analysis of clinical samples. Clin Proteomics J 2004; 1: 17–31
- Rai AJ, Gelfand CA, Haywood BC, Warunek DJ, Yi J, Schuchard MD, et al. HUPO Plasma Proteome Project specimen collection and handling: Towards the standardization of parameters for plasma proteome samples. Proteomics 2005; 5: 3262–77
- Tammen H, Schulte I, Hess R, Menzel C, Kellmann M, Mohring T, et al. Peptidomic analysis of human blood specimens: Comparison between plasma specimens and serum by differential peptide display. Proteomics 2005; 5: 3414–22
- Muller A, MacCallum RM, Sternberg MJE. Structural characterization of the human proteome. Genome Research 2002; 12: 1625–41
- Pieper R, Gatlin CL, Makusky AJ, Russo PS, Schatz CR, Miller SS, et al. The human serum proteome: Display of nearly 3700 chromatographically separated protein spots on two-dimensional electrophoresis gels and identification of 325 distinct proteins. Proteomics 2003; 3: 1345–64
- Shmueli O, Horn-Saban S, Chalifa-Caspi V, Shmoish M, Ophir R, Benjamin-Rodrig H, et al. GeneNote: Whole genome expression profiles in normal human tissues. C R Biol 2003; 326: 1067–72
- Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, et al. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci USA 2002; 99: 4465–70
- Righetti PG, Castagna A, Herbert B, Reymond F, Rossier JS. Prefractionation techniques in proteome analysis. Proteomics 2003; 3: 1397–407
- Perrot M, Sagliocco F, Mini T, Monribot C, Schneider U, Shevchenko A, et al. Two-dimensional gel protein database of Saccharomyces cerevisiae (update 1999). Electrophoresis 1999; 20: 2280–98
- Washburn MP, Wolters D, Yates JR 3rd. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol 2001; 19: 242–7
- Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: The yeast proteome. J Proteome Res 2003; 2: 43–50
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, et al. Global analysis of protein expression in yeast. Nature 2003; 425(6959)737–41
- Jacobs JM, Mottaz HM, Yu LR, Anderson DJ, Moore RJ, Chen WN, et al. Multidimensional proteome analysis of human mammary epithelial cells. J Proteome Res 2004; 3: 68–75
- Humphery-Smith I. A human proteome project with a beginning and an end. Proteomics 2004; 4: 2519–21
- Nilsson P, Paavilainen L, Larsson K, Odling J, Sundberg M, Andersson AC, et al. Towards a human proteome atlas: High-throughput generation of mono-specific antibodies for tissue profiling. Proteomics 2005.
- Uhlen M, Ponten F. Antibody-based proteomics for human tissue profiling. Mol Cell Proteomics 2005; 4: 384–93
- Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, et al. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol 2002; 20: 301–5
- Salomon AR, Ficarro SB, Brill LM, Brinker A, Phung QT, Ericson C, et al. Profiling of tyrosine phosphorylation pathways in human cells using mass spectrometry. Proc Natl Acad Sci USA 2003; 100: 443–8
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002; 298(5600)1912–34
- Wang WQ, Sun JP, Zhang ZY. An overview of the protein tyrosine phosphatase superfamily. Curr Top Med Chem 2003; 3: 739–48
- McCluskey A, Sim AT, Sakoff JA. Serine-threonine protein phosphatase inhibitors: Development of potential therapeutic strategies. J Med Chem 2002; 45: 1151–75
- Honkanen RE, Golden T. Regulators of serine/threonine protein phosphatases at the dawn of a clinical era?. Curr Med Chem 2002; 9: 2055–75
- Qian WJ, Goshe MB, Camp DG 2nd, Yu LR, Tang K, Smith RD. Phosphoprotein isotope-coded solid-phase tag approach for enrichment and quantitative analysis of phosphopeptides from complex mixtures. Anal Chem 2003; 75: 5441–50
- Steinberg TH, Agnew BJ, Gee KR, Leung WY, Goodman T, Schulenberg B, et al. Global quantitative phosphoprotein analysis using Multiplexed Proteomics technology. Proteomics 2003; 3: 1128–44
- Guy GR, Philip R, Tan YH. Analysis of cellular phosphoproteins by two-dimensional gel electrophoresis: Applications for cell signaling in normal and cancer cells. Electrophoresis 1994; 15: 417–40
- Wulfkuhle JD, Aquino JA, Calvert VS, Fishman DA, Coukos G, Liotta LA, et al. Signal pathway profiling of ovarian cancer from human tissue specimens using reverse-phase protein microarrays. Proteomics 2003; 3: 2085–90
- Lowenthal MS, Mehta AI, Frogale K, Bandle RW, Araujo RP, Hood BL, et al. Analysis of albumin-associated peptides and proteins from ovarian cancer patients. Clin Chem 2005; 51: 1933–45
- Adkins JN, Varnum SM, Auberry KJ, Moore RJ, Angell NH, Smith RD, et al. Toward a human blood serum proteome: Analysis by multidimensional separation coupled with mass spectrometry. Mol Cell Proteomics 2002; 1: 947–55
- Cho SY, Lee EY, Lee JS, Kim HY, Park JM, Kwon MS, et al. Efficient prefractionation of low-abundance proteins in human plasma and construction of a two-dimensional map. Proteomics 2005; 5: 3386–96
- Tang HY, Ali-Khan N, Echan LA, Levenkova N, Rux JJ, Speicher DW. A novel four-dimensional strategy combining protein and peptide separation methods enables detection of low-abundance proteins in human plasma and serum proteomes. Proteomics 2005; 5: 3329–42
- Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J Proteome Res 2004; 3: 235–44
- Wulfkuhle JD, Sgroi DC, Krutzsch H, McLean K, McGarvey K, Knowlton M, et al. Proteomics of human breast ductal carcinoma in situ. Cancer Res 2002; 62: 6740–9
- Petricoin EF 3rd, Ornstein DK, Paweletz CP, Ardekani A, Hackett PS, Hitt BA, et al. Serum proteomic patterns for detection of prostate cancer. J Natl Cancer Inst 2002; 94: 1576–8
- Adam BL, Qu Y, Davis JW, Ward MD, Clements MA, Cazares LH, et al. Serum protein fingerprinting coupled with a pattern-matching algorithm distinguishes prostate cancer from benign prostate hyperplasia and healthy men. Cancer Res 2002; 62: 3609–14
- Kozak KR, Amneus MW, Pusey SM, Su F, Luong MN, Luong SA, et al. Identification of biomarkers for ovarian cancer using strong anion-exchange ProteinChips: Potential use in diagnosis and prognosis. Proc Natl Acad Sci USA 2003; 100: 12343–8
- Petricoin EF, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM, et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet 2002; 359(9306)572–7
- Diamandis EP. Analysis of serum proteomic patterns for early cancer diagnosis: Drawing attention to potential problems. J Natl Cancer Inst 2004; 96: 353–6
- Puente XS, Sanchez LM, Overall CM, Lopez-Otin C, et al. Human and mouse proteases: A comparative genomic approach. Nature Rev Gen 2003; 4: 544–58
- Kanduc D, Mittelman A, Serpico R, Sinigaglia E, Sinha AA, Natale C, et al. Cell death: Apoptosis versus necrosis (review). Int J Oncol 2002; 21: 165–70
- Olivieri E, Herbert B, Righetti PG. The effect of protease inhibitors on the two-dimensional electrophoresis pattern of red blood cell membranes. Electrophoresis 2001; 22: 560–5
- Mykles DL. Proteinase families and their inhibitors. Methods Cell Biol 2001; 66: 247–87
- Rivard AL, Gallegos RP, Bianco RW, Liao K. The basic science aspect of donor heart preservation: A review. J Extra-Corp Technol 2004;269–74.
- Cosi C, Marien M. Decreases in mouse brain NAD+ and ATP induced by 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP): Prevention by the poly(ADP-ribose) polymerase inhibitor, benzamide. Brain Res 1998; 809: 58–67
- Ansari KA, Rand A, Hendrickson H, Bentley MD. Qualitative and quantitative studies on human myelin basic protein in situ with respect to time interval between death and autopsy. J Neuropathol Exp Neurol 1976; 35: 180–90
- Berlet HH, Volk B. Studies of human myelin proteins during old age. Mech Ageing Dev 1980; 14: 211–22
- Goggins M, Scott JM, Weir DG. Regional differences in protein carboxymethylation in post-mortem human brain. Clin Sci (Lond) 1998; 94: 677–85
- Naser NA, Naser SA. Clinical Chemistry Laboratory Manual 1998: Mosby Inc.
- Ludwig JA, Weinstein JN. Biomarkers in cancer staging, prognosis and treatment selection. Nat Rev Cancer 2005; 5: 845–56
- Mustard JF, Kinlough-Rathbone RL, Packham MA. Isolation of human platelets from plasma by centrifugation and washing. Methods Enzymol 1989; 169: 3–11
- Lewis MR, Callas PW, Jenny NS, Tracy RP. Longitudinal stability of coagulation, fibrinolysis, and inflammation factors in stored plasma samples. Thromb Haemost 2001; 86: 1495–500
- Chaurand P, Schwartz SA, Billheimer D, Xu BJ, Crecelius A, Caprioli R. Integrating histology and imaging mass spectrometry. Anal Chem 2004; 76: 1145–55
- Schwartz SA, Weil RJ, Thompson RC, Shyr Y, Moore JH, Toms SA, et al. Proteomic-based prognosis of brain tumor patients using direct-tissue matrix-assisted laser desorption ionization mass spectrometry. Cancer Res 2005; 65: 7674–81
- Schwartz SA, Weil RJ, Johnson MD, Toms SA, Caprioli RM. Protein profiling in brain tumors using mass spectrometry: Feasibility of a new technique for the analysis of protein expression. Clin Cancer Res 2004; 10: 981–7
- Dill KA. Dominant forces in protein folding. Biochemistry 1990; 29: 7133–55
- Kauzmann W. Some factors in the interpretation of protein denaturation. Adv Protein Chem 1959; 14: 1–63
- Scopes RK. Making an extract, in protein purification: Principles and practice. 1987.
- Dignam JD. Preparation of extracts from higher eukaryotes. Methods Enzymol 1990; 182: 194–203
- Bollag DM, Edelstein SJ. Protein Methods. New YorkChichester, Brisbane, Toronto, Singapore: Wiley-Liss; 1991.
- Deutscher MP. Maintaining protein stability. Methods Enzymol 1990; 182: 83–9
- Nielsen UB, Cardone MH, Sinskey AJ, MacBeath G, Sorger PK. Profiling receptor tyrosine kinase activation by using Ab microarrays. Proc Natl Acad Sci USA 2003; 100: 9330–5
- Harder A, Wildgruber R, Nawrocki A, Fey SJ, Larsen PM, Gorg A. Comparison of yeast cell protein solubilization procedures for two-dimensional electrophoresis. Electrophoresis 1999; 20: 826–9
- Newcombe J, Eriksson B, Ottervald J, Yang Y, Franzen B. Extraction and proteomic analysis of proteins from normal and multiple sclerosis postmortem brain. J Chromatogr B Analyt Technol Biomed Life Sci 2005; 815: 191–202
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227(259)680–5
- Laemmli UK, Favre M. Maturation of the head of bacteriophage T4. I. DNA packaging events. J Mol Biol 1973; 80: 575–99
- Steinberg TH, Jones LJ, Haugland RP, Singer VL. SYPRO orange and SYPRO red protein gel stains: One-step fluorescent staining of denaturing gels for detection of nanogram levels of protein. Anal Biochem 1996; 239: 223–37
- Steinberg TH, Haugland RP, Singer VL. Applications of SYPRO orange and SYPRO red protein gel stains. Anal Biochem 1996; 239: 238–45
- Mackintosh JA, Choi HY, Bae SH, Veal DA, Bell PJ, Ferrari BC, et al. A fluorescent natural product for ultra sensitive detection of proteins in one-dimensional and two-dimensional gel electrophoresis. Proteomics 2003; 3: 2273–88
- Steinberg TH, Pretty On Top K, Berggren KN, Kemper C, Jones L, Diwu Z, et al. Rapid and simple single nanogram detection of glycoproteins in polyacrylamide gels and on electroblots. Proteomics 2001; 1: 841–55
- Burnette WN. “Western blotting”: Electrophoretic transfer of proteins from sodium dodecyl sulfate–polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem 1981; 112: 195–203
- Bjellqvist B, Ek K, Righetti PG, Gianazza E, Gorg A, Westermeier R, et al. Isoelectric focusing in immobilized pH gradients: Principle, methodology and some applications. J Biochem Biophys Methods 1982; 6: 317–39
- O'Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem 1975; 250: 4007–21
- O'Farrell PZ, Goodman HM, O'Farrell PH. High resolution two-dimensional electrophoresis of basic as well as acidic proteins. Cell 1977; 12: 1133–41
- Sabounchi-Schutt F, Astrom J, Olsson I, Eklund A, Grunewald J, Bjellqvist B. An immobiline DryStrip application method enabling high-capacity two-dimensional gel electrophoresis. Electrophoresis 2000; 21: 3649–56
- Li J, Zhang Z, Rosenzweig J, Wang YY, Chan DW. Proteomics and bioinformatics approaches for identification of serum biomarkers to detect breast cancer. Clin Chem 2002; 48: 1296–304
- Michaud GA, Salcius M, Zhou F, Bangham R, Bonin J, Guo H, et al. Analyzing antibody specificity with whole proteome microarrays. Nat Biotechnol 2003; 21: 1509–12
- Chalmers MJ, Quinn JP, Blakney GT, Emmett MR, Mischak H, Gaskell SJ, et al. Liquid chromatography-Fourier transform ion cyclotron resonance mass spectrometric characterization of protein kinase C phosphorylation. J Proteome Res 2003; 2: 373–82
- Goshe MB, Veenstra TD, Panisko EA, Conrads TP, Angell NH, Smith RD. Phosphoprotein isotope-coded affinity tags: Application to the enrichment and identification of low-abundance phosphoproteins. Anal Chem 2002; 74: 607–16
- Zhou H, Watts JD, Aebersold R. A systematic approach to the analysis of protein phosphorylation. Nat Biotechnol 2001; 19: 375–8
- Alban A, David SO, Bjorkesten L, Andersson C, Sloge E, Lewis S, et al. A novel experimental design for comparative two-dimensional gel analysis: Two-dimensional difference gel electrophoresis incorporating a pooled internal standard. Proteomics 2003; 3: 36–44
- Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 2003; 426(6966)570–4
- Blagoev B, Kratchmarova I, Ong SE, Nielsen M, Foster LJ, Mann M. A proteomics strategy to elucidate functional protein-protein interactions applied to EGF signaling. Nat Biotechnol 2003; 21: 315–8
- Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol 1999; 17: 994–9
- Gygi SP, Rist B, Griffin TJ, Eng J, Aebersold R. Proteome analysis of low-abundance proteins using multidimensional chromatography and isotope-coded affinity tags. J Proteome Res 2002; 1: 47–54
- Ranish JA, Yi EC, Leslie DM, Purvine SO, Goodlett DR, Eng J, et al. The study of macromolecular complexes by quantitative proteomics. Nat Genet 2003; 33: 349–55
- Wingren C, Ingvarsson J, Lindstedt M, Borrebaeck CA. Recombinant antibody microarrays–a viable option?. Nat Biotechnol 2003; 21: 223
- Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci USA 2003; 100: 6940–5
Appendix 1 Abbreviations
AQUA Absolute quantitation (Absolute quantitation of specific peptides by using stable isotope labeled internal standards and MS.).
CID Collision Induced Dissociation spectrum (The spectrum of peptide fragments that emerges from a collision chamber where a specific peptide has been held and physically collided with gas molecules so that it breaks, preferentially at the peptide bonds, generating a series of breakdown products that can be identified by size differences. Can be used to sequence peptides and identify the protein they are derived from (see also MS/MS).
DIGE Differential Gel Electrophoresis (Differential fluorescent pre-labeling of protein extracts prior to electrophoresis, followed by co electrophoresis and separate detection of the proteins of each extract. Allows detection and relative quantitation of each resolved protein-pair.).
ELISA Enzyme Linked Immuno Sorbent Assay (A method to identify and quantify proteins by binding to specific antibodies. Antibody arrays are a version of ELISA that has been miniaturized and analyzes multiple antibody/antigen pairs in parallel.).
ESI ElectroSpray Ionization (Ionization of proteins or peptides in solution emerging as a spray from a capillary. Allows feeding an HPLC eluate or flowthrough directly into a mass spectrometer for real-time analysis.).
HPLC High Pressure Liquid Chromatography system.
HUPO Human Proteome Organization.
ICAT Isotope Coded Affinity Tag (Differential chemical pre-labelling of protein extracts with heavy/light stable isotope reagents at specific amino acid side chains. The proteins are then digested with a sequence-specific protease, such as trypsin. The affinity tag allows enrichment of the chemically labeled peptides only, increasing the sensitivity of detection. Allows detection and relative quantitation of specific peptides, representing most proteins, on an LC-MS system.).
IMAC Immobilized Metal Affinity Chromatography (Metal ion coordination of specific chemical groups, such as phosphate-groups. Allows for generalized affinity purification of phosphate-containing peptides.).
LC-MS Liquid Chromatography-Mass Spectrometry system (An HPLC coupled directly to a mass spectrometer to monitor the protein or peptide eluate in real time).
MALDI Matrix Assisted Laser Desorption Ionization (A method to at once desorb and ionize an analyte (protein or peptide) for mass spectrometry by the interaction of laser light with a matrix compound in which the analyte is dry-embedded. Compare ESI.).
MS Mass Spectrometry (A technique to separate and detect charged molecules in a magnetic field by their size-to-charge ratio. Heavier or less charged molecules are accelerated less. Allows very precise weighing of an analyte. Proteins can be identified by the precise weight of their sequence-specifically cut peptides or from amino-acid sequencing based on the size difference between peptides differing by one amino acid after CID (see entries for CID and MS/MS). MS constitutes a size separation and detection system in one.).
MS/MS Tandem mass spectrometry (A form of mass spectrometer that separates and then holds specific selected peptides. The held peptides can then be subjected to CID (see that entry) and the resulting peptide fragments subjected to a second round of MS analysis for identification and sequencing.).
PAGE Poly Acrylamide Gel Electrophoresis (Polyacrylamide is one of several matrices that can be used to counter convection during electrophoresis. It is favored since it has a low non-specific binding ability to proteins, and can be cast with different concentrations and with different amounts of cross-linker to generate a wide spectrum of pore-sizes. The specific pore size determines the range of protein molecular weights that are optimally resolved.).
PPP Plasma Proteome Project (Within HUPO).
SDS Sodium Dodecyl Sulfate (An ionic detergent that very effectively denatures proteins. In so doing SDS inhibits enzymatic and binding activities. Its binding is roughly proportional along the length of the protein, making the charge proportional to the size of the protein. Most proteins, except highly charged proteins, migrate in SDS-PAGE approximately as expected for their size, unless posttranslationally modified.).
SELDI Surface Enhanced Laser Desorption Ionization (A version of MALDI marketed by Ciphergen, Inc. It is composed of a chip with a chromatographic matrix, such as an ion-exchange matrix, that selectively binds subsets of a protein extract. The bound proteins are ionized, desorbed and analyzed in a mass spectrometer.).
SILAC Stable Isotope Labeling by Amino acids in Cellculture. (Differential metabolic labeling of cells in culture by culturing in medium containing either light or heavy isotope labeled essential amino acids. The two separate differentially labeled proteins are extracted, mixed and specific proteins or protein complexes purified and identified. Subsequently the ratio of the two stable isotopes between the two extracts is determined by MS.).
SISCAPA Stable Isotope Standards and Capture by Anti-Peptide Antibodies (Mass spectrometric quantitation of peptides and proteins using stable isotope labeled internal standards and capture by anti-peptide antibodies.).
2-D PAGE Two-dimensional Poly Acrylamide Gel Electrophoresis (High resolution electrophoretic method to resolve protein extracts based on the two independent parameters isoelectric point and protein size, in a polyacrylamide matrix. The proteins are detected by general or specific reagents applied following the electrophoresis.).