Abstract
Background. The aim of this study was to determine the prevalence and outcome of radiation-induced sarcomas (RISs) among sarcoma patients referred to the Norwegian Radium Hospital (NRH). Material and Methods. Ninety patients were identified from the institutional sarcoma data base. Medical records and histological and cytological material from both primary and secondary tumours were reviewed. Results. RIS represented 3.0 % of the sarcomas in the data base. The median latency time from radiotherapy of the primary tumour to the diagnosis of RIS was 13.6 years (range 2.5-57.8 years). Gynaecological, breast and testicular cancers were the most common primary diagnoses. For the RISs 13 different histological types were identified including 25 malignant fibrous histiocytomas (28% of all) and 22 osteosarcomas (24%). The sarcoma-related 5-year crude survival was 33% (95 % CI 23-43 %). Unfavourable prognostic factors were metastases at presentation, incomplete surgery and presence of tumour necrosis. Conclusion. Radiation-induced sarcoma is rare and harbours an aggressive clinical behaviour. Complete surgical resection is mandatory for cure.
Sarcomas are rare tumours representing approximately 1% of all new cancers Citation[1]. The majority of sarcomas are spontaneous meaning that no etiologic factors are known. However, some sarcomas are related to genetic syndromes. Further, in some patients with sarcoma, clearly defined environmental risk factors have been identified Citation[2]. Radiation-induced sarcoma (RIS) is a well-known treatment complication and constitutes 2.5–5.5% of all sarcomas Citation[3–7]. The post-radiotherapy 10-year cumulative risk for developing RIS is estimated to 0.03–0.8% Citation[3]. Identified risk factors for developing RIS are genetic factors, young age at treatment and treatment-related factors including high radiation dose and simultaneous chemotherapy with alkylating agents Citation[8–10]. In the literature the latency since radiation varies from less than 1 year to more than 50 years, with a reported median interval between 10 and 20 years Citation[3], Citation[5], Citation[6].
The prognosis of RIS is poor compared to other sarcomas. The reported 5-year survival rates vary between 17 and 41% Citation[6], Citation[7], Citation[11–13]. Poor prognostic factors for outcome after diagnosis of RIS are the same as for sarcomas in general: metastases at presentation, tumour-contaminated surgical margins and large tumour size Citation[4].
We searched the institutional sarcoma database for patients with a history of radiotherapy to explore the following features of RIS: its prevalence among patients referred to a cancer centre for treatment of a sarcoma, histological types and prognostic factors for outcome.
The local ethical committee approved the performance of the study and permission was obtained from the Data Inspectorate of Norway.
Materials and methods
Patients
The Norwegian Radium Hospital (NRH) is the primary referral centre for patients with bone and soft tissue tumours for South/East Norway covering a population of 2.6 million. The NRH sarcoma data base covers 3 037 cases referred to the hospital from 1980 to 2005. For each patient, information is provided whether or not radiotherapy has been given prior to the diagnosed sarcoma. A total of 114 patients with a diagnosis of sarcoma localized in soft tissue, viscera or bone between 1980 and 2005 and with a history of previous radiation therapy were identified. The criteria for eligibility for the patients of the current study were, with minor modifications, according to Brady et al. Citation[4]:
Radiation therapy had been administered and a subsequent sarcoma developed in the field of previous radiotherapy.
A latency time of at least two years.
A pathologically proven sarcoma being localised either in bone, soft tissue or viscera with morphology different from that of the first tumour.
Carcinosarcomas and desmoid tumours were excluded.
From the medical records of eligible patients the following variables were extracted: date of birth, gender, date of diagnosis of the first primary tumour and the second tumour, histological type and localisation of the primary tumour, type of initial treatment for the primary tumour including surgery, chemotherapy and radiotherapy, non-neoplastic late effects, post-radiotherapy latency for RIS development, localisation of RIS, size, depth of infiltration, stage, status of the surgical margins (free or contaminated, the former defined by at least a 1 mm microscopic tumour-free margin), and date of last observation. The end of follow-up of surviving patients was set to September 1, 2007. Complete surgical remission was defined as a complete removal of all tumour foci, including metastases, with tumour-free margins during primary treatment of RIS.
Diagnosis and pathological review
All cytological and histological slides of the primary lesions and the subsequent sarcomas were reviewed by the first author. All cases were classified according to the WHO recommendations and graded according to the system used in the Scandinavian Sarcoma Group (SSG), in addition, for the soft tissue sarcomas, the French system (FNCLCC) was applied Citation[1], Citation[14], Citation[15]. Grade 3 or 4 was regarded as a high-grade malignant sarcoma in the Scandinavian system. When necessary and possible, immunohistochemistry was performed to assist tumour classification.
Statistical analysis and response criteria
Data were described with median and range. Continuous variables were categorized as below or above the median with exception of size. Size was categorized as ≥or < 5 cm. The observation time was calculated from the diagnosis of RIS to the date of sarcoma-related death, death due to other diseases without metastases from RIS or to the cut-off date of the study, whatever occurred first.
Crude association between ‘complete surgical remission’ and ‘surgery’ was studied with Pearson χ2 test. Crude survival was calculated using Kaplan-Meier method. The effect of each of possible prognostic factors was modelled using univariate Cox regression analysis. Prognostic factors were included in a multivariate regression analysis if they had a p-value of ≤0.1 in the univariate analysis or if they were thought to have an influence on survival based on a clinical knowledge. Due to non-proportional hazards, a weighted Cox regression model was fitted Citation[16].
Missing values were not replaced by a default setting and a missing variable led to exclusion of the patient in the analyses concerned. P-values less than 0.05 were considered statistically significant.
Statistical analyses were performed using SPSS software (SPSS, Inc., Chicago, IL), weighed regression was performed with R, statistical software (http://www.r-project.org/).
Results
Patient history
Of the 114 sarcoma patients identified in the data base as having a history of previous irradiation 24 were excluded for the following reasons: Lack of primary tumour or radiotherapy data (n = 7), the sarcoma was localised outside the irradiated field (n = 11), Stewart-Treves syndrome (sarcomas arising in a lymphoedematous arm outside the irradiated volume, n = 3), carcinosarcoma as the second tumour (n = 2) and no delay between radiotherapy and the second tumour (n = 1). The remaining 90 patients verified as RISs (61 female and 29 male patients) constitute 3.0% of the sarcoma patients in the NRH data base.
Characterisation of the primary tumour
The year of diagnosis of the primary tumour ranged from 1934 to 2000 and the median age at the time when radiotherapy started was 42.5 years (range 6 months to 74 years). The initial tumour was malignant in 75 cases; borderline in 11 and benign in four (). Our series includes seven patients with bilateral retinoblastoma, but none of the patients had known predisposing factors such as ataxia telangiectasia, neurofibromatosis or xeroderma pigmentosum. Forty-one patients had a family history of cancer, including three retinoblastomas and for two patients, familial adenomatous polyposis coli. None of the patients had been exposed to occupational radiation. Seventy patients had had surgery for the first tumour, in addition to radiotherapy, whereas 20 had no surgery. Nineteen had received chemotherapy; with ten patients having alkylating agents. The median dose delivered was 50 Gy (range 30–192 Gy). Eleven of the 90 patients had brachytherapy in addition to external-beam radiation.
Table I. Diagnosis of primary tumours treated with radiation resulting in radiation-induced sarcomas (n = 90).
Long-term side effects after treatment of the first tumour, other than RIS, were reported in 53 patients (59%). These included skin sequele in 20 patients, lymphoedema (n = 18), pain (n = 11), osteoradionecrosis (n = 5), late radiation enteritis (n = 9), radiation cataract (n = 2), xerostomia/caries (n = 3), dry eye (n = 1), induratio penis plastica (n = 2), urethral or vaginal fibrosis/dyspareuni (n = 4), restricted shoulder mobility (n = 3), spinal cord lesion requiring use of wheel chair (n = 2), peripheral polyneuropathy (n = 1) and cardiovascular complications (n = 5). Nine patients experienced a third, fourth or even a fifth cancer, and in three of these patients the subsequent tumours were located in the radiation field.
Characterisation of RIS
The median age at diagnosis of RIS was 59 years (range 7–89 years). The median latency from radiation therapy to the diagnosis of RIS was 13.6 years (range 2.5–57.8 years). Seventeen patients (19%) had metastases at presentation of RIS ( 4 patients had metastases in lung, 1 in liver, 1 in lymph node and 3 in other sites and 8 patients had metastases at multiple sites).
The site of the sarcoma was soft tissue or viscera in 60 patients, bone in 26 patients and not determinable in four cases (). The most common histologic diagnoses were malignant fibrous histiocytoma (28%), osteosarcoma (24%) and leiomyosarcoma (11%), (). High-grade sarcomas were found in 85 patients according to the grading system used in the SSG and three patients had a low-grade sarcoma and in two cases grading was not possible. Using the FNCLCC grading system for soft tissue sarcomas (n = 60) 53 cases were grade 2 or 3, one case grade 1 and six cases were not possible to grade. The median size of the tumour was 7 cm (range 1–28 cm, n = 88 patients). Fifty-two of the 60 soft tissue tumours showed an infiltrative growth pattern, 8 tumours were not assessable. Vessel infiltration was found in 14 of 67 assessable cases (21%).
Figure 1. The localisation of the radiation-induced sarcoma (n = 90).
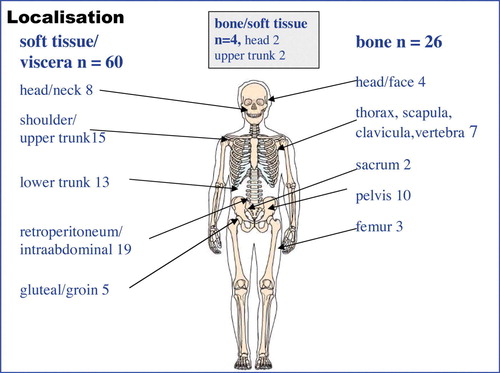
Table II. Distribution of histologic types of radiation induced sarcoma according to localisation (n = 90).
Sixty-five patients were treated with surgery (59 resections and 6 amputations) and of these, 41 specimens (63%) had tumour-free margins at the site of the primary tumour. Two patients of these 41 patients had metastases at diagnosis, and one of these received radical metastasectomy. Thus, 40 patients (44%) were rendered tumour free after surgery following initial treatment. Fifteen patients received additional radiotherapy and 8 had radiotherapy as the only local treatment. Thirty-two patients (36%) received chemotherapy.
Follow up and outcome
No patients were lost to follow-up. Sixty-one patients died from sarcoma and nine died from other causes. Twenty patients were alive at the study's cut-off date, 18 in complete remission and two patients with tumour activity. The median survival time from diagnosis of RIS was 17.4 (range 0.9–330) months. With a median follow-up time in survivors of 81 months (range 17–330 months) the 5-year crude sarcoma-related survival rate was 33% (95% CI 23–43%) (). The crude 5-year survival for the osteosarcoma patients was 11%. Thirty of the 65 patients (46%) treated with surgery experienced a recurrence at the site of surgery after a median time of 10 months (range 0.3–147). In the univariate analysis six factors were significant predictors of improved survival: size <5 cm, surgical treatment, complete surgical remission, no metastases at diagnosis, soft tissue sarcoma and absence of tumour necrosis (). The 5-year crude survival for patients with complete surgical remission was 63% (95% CI 46–80%) compared to 10% (95% CI 2–18%) for those without complete surgical remission (). In the multivariate weighted Cox regression analysis, complete surgical remission (HR = 4.27, 95% CI:[1.94–9.41]), no metastases at diagnosis (HR = 2.6, 95% CI: [1.28–5.30]) and no tumour necrosis (HR = 3.1, 95% CI: [1.45–6.77]) remained to be independent prognostic factors of survival. Surgery was not included into the analysis because this parameter was highly associated with complete surgical remission (p < 0.001).
Figure 2. RIS-related survival in all 90 patients and according to completeness of radical surgery. P-value < 0.001.
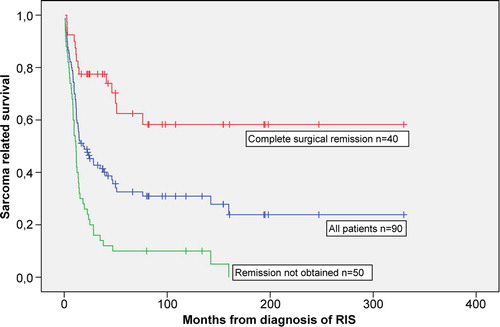
Table III. Survival in 90 patients with radiation-induced sarcoma
Discussion
Radiotherapy is one of the cornerstones in the treatment of cancer. According to international guidelines close to 50% of cancer patients should receive radiotherapy at some time during the course of their disease Citation[17]. RIS is a serious long-term side effect of radiotherapy, and it is important to assess its significance Citation[18]. The current single institution series is one of the largest reported, and our finding that 3.0% of all sarcomas in the NRH database are RIS, is in agreement with the literature Citation[6], Citation[7].
The carcinogenicity of ionizing radiation is partly related to genotoxic stress which can induce sublethal DNA damage, including DNA double-strand break-induced gene translocation, genomic instability and loss of DNA repair Citation[19]. The sarcoma inducing effect of radiotherapy is supported by epidemiological studies Citation[8–10]. The increased risk of sarcoma after radiotherapy was 3.2 higher than baseline risk as compared to 2.0 in patients not receiving radiotherapy or chemotherapy in a Finish study Citation[8]. However, balanced against other aetiological factors like genetic constitution and environmental exposure, the exact contribution of radiotherapy, to secondary carcinogenesis remains unknown. The possibility exists that some tumours may have arisen spontaneously within the radiation field without being aetiologically related to the previous radiotherapy. There is no molecular genetic marker that is specific for therapy-induced cancer except for in therapy-related leukaemia Citation[19].
Lack of any relationship with radiotherapy may apply to gastrointestinal stromal tumours (GISTs). Most RISs in general have a complex karyotype and do not carry specific genetic changes as do e.g. GISTs Citation[2]. Two cases of GIST are included in this series, and these may represent spontaneous tumours which have developed apparently independently of previous irradiation. This is supported by recent reports demonstrating that GISTs may occur as a second malignancy without previous radiation Citation[20], Citation[21].
Sarcomas after radiotherapy of testicular cancer represent a diagnostic challenge. Sarcomatous foci may occur within the primary tumour in testicular germ cell tumours and not at least in late recurrences, which may be misclassified as RIS Citation[22]. This possibility has to be excluded by extensive revision of the primary tumour, as done in our study.
The genetic constitution of the patients can predispose for a second cancer independently of previous radiotherapy, as in patients with previous retinoblastoma or in the Li-Fraumeni syndrome Citation[23]. In our series seven patients had a diagnosis of retinoblastoma. Patients with hereditary retinoblastoma have a predisposition for a second malignancy (particularly sarcomas) and are especially susceptible to the carcinogenic effects of irradiation. The 50 year-cumulative incidence for a second cancer after a diagnosis of retinoblastoma is 36% for hereditary and 5.7% for nonhereditary patients Citation[24]. In hereditary retinoblastoma patients the risk of a subsequent cancer is further increased 3.1 fold following radiotherapy contrary to in non-hereditary patients Citation[24].
Osteosarcoma and malignant fibrous histiocytoma (pleomorphic and spindle cell sarcomas) were the most common subtypes of RIS, and this is in accordance with previous reports Citation[5], Citation[6], Citation[11]. Extraskeletal osteosarcoma is a rare spontaneous tumour constituting 2–4% of all osteosarcomas, but seems to be more common among RIS as 6 of 22 osteosarcoma (27%) were extraskeletal in our series Citation[1], Citation[11]. Two patients had sarcoma development in previously benign giant cell tumours of the sacrum and femur. It is reported that 11% of irradiated giant cell tumours can transform to sarcoma in contrast to less than 1% of giant cell tumours in general Citation[1], Citation[25]. The number of radiation-induced liposarcomas is low, and only one case was observed in our material Citation[26]. This is in contrast to spontaneous sarcomas where liposarcomas belong to the most common histological subtypes Citation[1].
The prognosis of RIS is poor compared to spontaneous sarcoma. In this series we report a crude sarcoma-related 5-year-survival of 33%. In contrast, the crude 5-year survival of all sarcomas treated at our centre is 65% Citation[27]. The multivariate analyses revealed that complete surgical remission was critical for outcome, and as many as 50 of 90 patients were never rendered disease-free after primary therapy. In fact, our multivariate analysis showed that patients who had not obtained a complete surgical remission had a more than four times higher risk of sarcoma-related death compared to the group which had obtained complete surgical remission. Among patients with localized disease and a soft-tissue tumour of RIS (n = 60) 53% were operated with free margins. This observation is in contrast to spontaneous soft tissue sarcomas treated at NRH, from 1995 to 1999, in this group 76% of the patients obtained complete remission Citation[27]. The propensity for central rather than peripheral localization of RIS probably contributes to the unfavourable outcome due to poor surgical accessibility. In addition, the presence of recognized aggressive tumour-related factors is high in our study compared to spontaneous sarcomas Citation[28]. Thus 85 of 88 of assessable tumours were high-grade malignant, and 52/60 showed an infiltrative growth pattern. In contrast, the median size reported was not different from sarcomas in general Citation[27], Citation[28]. Tumour necrosis, a known unfavourable factor in sarcoma, was present in 55 of 83 of evaluable RISs as judged by light microscopy.
The five-year disease-free survival rates varies 17–41% in different series with RIS Citation[6], Citation[7], Citation[11–13]. The crude 5-year survival for the osteosarcoma (11%) patients seems particularly poor compared to osteosarcoma in general (59%) Citation[29]. Bielack et al. have reported that secondary osteosarcoma have a prognosis approaching that of “conventional” osteosarcoma provided that local tumour control is achieved and if treated with combined modality therapy Citation[30]. Only seven of our 22 osteosarcomas obtained complete surgical remission and three of these were living 33–330 months from diagnosis of RIS.
In addition to unfavourable tumour sites and tumour-related factors, previous therapy may reduce treatment possibilities as repeated high-dose radiotherapy often is impossible and/or due to limited bone marrow function. The low 5-year survival of 33% suggests that a future trial should explore a strategy with an intensified preoperative chemotherapy to facilitate radical surgery which is essential for improvement of survival. Due to the high incidence of multifocal RIS after breast cancer treatment one may consider to remove the whole irradiated area and not only the tumour Citation[31]. Reirradiation with modern technique should also be considered Citation[32].
The strength in our study is the size of the series and that most patients were recruited from a well-defined geographic area, good follow-up data and that the histological material is revised, both from the primary and the secondary tumours. An important limitation is the lack of a control group of spontaneous sarcomas. Such comparison will be an aim for a future project.
In conclusion, RIS is a rare but serious complication after radiotherapy and displays a high number of aggressive tumour characteristics, which are associated with a poor prognosis. Radical surgery is difficult to accomplish, but appears mandatory for cure.
Acknowledgements
The contributions of consulting pathologist Anna Elisabeth Stenwig for valuable discussions and diagnostic support, chief physicist Vidar Jetne for reviewing radiotherapy treatment charts and Frances Dodman for linguistic corrections are highly appreciated.
References
- Fletcher CD, Unni KK, Mertens FE. World Health Organization classification of tumours. Pathology and genetics of tumours of soft tissue and bone. IARC Press, Lyon 2002
- Helman LJ, Meltzer P. Mechanisms of sarcoma development. Nat Rev Cancer 2003; 3: 685–94
- Mark RJ, Poen J, Tran LM, Fu YS, Selch MT, Parker RG. Postirradiation sarcomas. A single-institution study and review of the literature. Cancer 1994; 73: 2653–62
- Brady MS, Gaynor JJ, Brennan MF. Radiation-associated sarcoma of bone and soft tissue. Arch Surg 1992; 127: 1379–85
- Murray EM, Werner D, Greeff EA, Taylor DA. Postradiation sarcomas: 20 cases and a literature review. Int J Radiat Oncol Biol Phys 1999; 45: 951–61
- Cha C, Antonescu CR, Quan ML, Maru S, Brennan MF. Long-term results with resection of radiation-induced soft tissue sarcomas. Ann Surg 2004; 239: 903–9
- Huvos AG, Woodard HQ, Cahan WG, Higinbotham NL, Stewart FW, Butler A, et al. Postradiation osteogenic sarcoma of bone and soft tissues. A clinicopathologic study of 66 patients. Cancer 1985; 55: 1244–55
- Virtanen A, Pukkala E, Auvinen A. Incidence of bone and soft tissue sarcoma after radiotherapy: A cohort study of 295,712 Finnish cancer patients. Int J Cancer 2006; 118: 1017–21
- Menu-Branthomme A, Rubino C, Shamsaldin A, Hawkins MM, Grimaud E, Dondon M, et al. Radiation dose, chemotherapy and risk of soft tissue sarcoma after solid tumours during childhood. Int J Cancer 2004; 110: 87–93
- Hawkins MM, Wilson LM, Burton HS, Potok MHN, Winter DL, Marsden HB, et al. Radiotherapy, alkylating agents, and risk of bone cancer after childhood cancer. J Natl Cancer Inst 1996; 88: 270–8
- Wiklund TA, Blomqvist CP, Raty J, Elomaa I, Rissanen P, Miettinen M. Postirradiation sarcoma. Analysis of a nationwide cancer registry material. Cancer 1991; 68: 524–31
- Lagrange JL, Ramaioli A, Chateau MC, Marchal C, Resbeut M, Richaud P, et al. Sarcoma after radiation therapy: Retrospective multiinstitutional study of 80 histologically confirmed cases. Radiation Therapist and Pathologist Groups of the Federation Nationale des Centres de Lutte Contre le Cancer. Radiology 2000; 216: 197–205
- Thijssens KM, van Ginkel RJ, Suurmeijer AJ, Pras E, van der Graaf WTA, Hollander M, et al. Radiation-induced sarcoma: A challenge for the surgeon. Ann Surg Oncol 2005; 12: 237–45
- Angervall L, Kindblom LG, Rydholm A, Stener B. The diagnosis and prognosis of soft tissue tumors. Semin Diagn Pathol 1986; 3: 240–58
- Guillou L, Coindre JM, Bonichon F, Bui NB, Terrier P, Collin F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997; 15: 350–62
- Schemper M. Cox analysis of survival data with non-proportional hazard functions. The Statistician 1992; 41: 455–65
- Lindholm C, Cavallin-Stahl E, Ceberg J, Frodin JE, Littbrand B, Moller TR. Radiotherapy practices in Sweden compared to the scientific evidence. Acta Oncol 2003; 42: 416–29
- Brenn T, Fletcher CD. Postradiation vascular proliferations: An increasing problem. Histopathology 2006; 48: 106–14
- Allan JM, Travis LB. Mechanisms of therapy-related carcinogenesis. Nat Rev Cancer 2005; 5: 943–55
- Grobmyer SR, Luther N, Antonescu CR, Singer S, Brennan MF. Multiple primary soft tissue sarcomas. Cancer 2004; 101: 2633–5
- Tornoczky T, Kover E. Multiple soft tissue sarcomas. Cancer 2005; 104: 440–1
- Oldenburg J, Alfsen GC, Waehre H, Fossa SD. Late recurrences of germ cell malignancies: A population-based experience over three decades. Br J Cancer 2006; 94: 820–7
- Moppett J, Oakhill A, Duncan AW. Second malignancies in children: The usual suspects?. Eur J Radiol 2001; 38: 235–48
- Kleinerman RA, Tucker MA, Tarone RE, Abrahamson DH, Seddon JM, Stovall M, et al. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: An extended follow-up. J Clin Oncol 2005; 23: 2272–9
- Leggon RE, Zlotecki R, Reith J, Scarborough MT. Giant cell tumor of the pelvis and sacrum: 17 cases and analysis of the literature. Clin Orthop 2004; 423: 196–207
- Orosz Z, Rohonyi B, Luksander A, Szanto J. Pleomorphic liposarcoma of a young woman following radiotherapy for epithelioid sarcoma. Pathol Oncol Res 2000; 6: 287–91
- Saeter G, Hall KS, Bruland OS, Solheim OP, Hoie J, Follerås G, et al. Bein- og blotvevsarkomer behandlet ved Det Norske Radiumhospital 1980–99. Tidsskr Nor Laegeforen 2002; 122: 2089–94
- Engellau J, Bendahl PO, Persson A, Domanski HA, Akerman M, Gustafson P, et al. Improved prognostication in soft tissue sarcoma: Independent information from vascular invasion, necrosis, growth pattern, and immunostaining using whole-tumor sections and tissue microarrays. Hum Pathol 2005; 36: 994–1002
- Aksnes LH, Hall KS, Folleraas G, Stenwig AE, Bjerkehagen B, Taksdal I, et al. Management of high-grade bone sarcomas over two decades: The Norwegian Radium Hospital experience. Acta Oncol 2006; 45: 38–46
- Bielack SS, Kempf-Bielack B, Heise U, Schwenzer D, Winkler K. Combined modality treatment for osteosarcoma occurring as a second malignant disease. Cooperative German-Austrian-Swiss Osteosarcoma Study Group. J Clin Oncol 1999; 17: 1164–74
- Holt GE, Thomson AB, Griffin AM, Bell R, Wunder J, Rougraff B, et al. Multifocality and multifocal postradiation sarcomas. Clin Orthop Relat Res 2006; 450: 67–75
- Kasperts N, Slotman BJ, Leemans CR, de Bree R, Doornaert P, Langendijk JA. Results of postoperative reirradiation for recurrent or second primary head and neck carcinoma. Cancer 2006; 106: 1536–47