
Abstract
Background: IgG4-related disease (IgG4-RD) is a recently recognized fibro-inflammatory disorder, which may affect many organs, and often comes to clinical attention due to tumor-like organ swelling or is identified incidentally by specific biopsy findings. Typical histopathology of IgG4-RD is lymphoplasmacytic infiltration rich in IgG4 + plasma cells (PCs), storiform fibrosis, and obliterative phlebitis. Patients with sicca symptoms can be misdiagnosed as primary Sjögren’s syndrome (pSS) instead of IgG4-RD because of clinical and histopathological similarities. Moreover, an association with lymphoma development is described in both diseases. This study investigated signs of IgG4-RD in a population-based cohort of patients diagnosed with pSS complicated by lymphoma.
Methods: Patients with pSS and lymphoma diagnoses and available lymphoma specimens were identified by linkage with the Swedish Patient Register 1964–2007 and the Cancer Register 1990–2007 (n = 79). Clinical data and lymphomas were reviewed and the diagnoses evaluated. All lymphoma tissues and available minor salivary gland biopsies (n = 11) were immunostained for IgG4 + PCs and evaluated for other histopathological signs of IgG4-RD. In a case with specific findings of IgG4-RD, other available tissue specimens of the same patient were investigated for IgG4-RD.
Results: Only one patient of 79 (1.3%) had >10 IgG4 + PCs/high power field (HPF) in the lymphoma tissue, an unspecified low-grade B-cell lymphoma localized in the submandibular gland. This patient also had other histopathological features of IgG4-RD in the lymphoma and a surgical lung biopsy taken five years before lymphoma diagnosis and, therefore, fulfilled the criteria for IgG4-RD. Occasional IgG4 + PCs (<10/HPF) without signs of IgG4-RD were observed in another six lymphomas. No IgG4 + PCs were identified in the minor salivary gland biopsies.
Conclusion: Histopathological findings of IgG4-RD may co-exist with low malignant B-cell lymphoma in patients with initially suspected pSS and may be associated with an underlying IgG4-RD.
IgG4-related disease (IgG4-RD), first described in Japan at the beginning of this century, is an inflammatory fibrotic condition characterized by a diffuse lymphoplasmacytic infiltration rich in IgG4 + plasma cells (PCs). The inflammation typically causes tumor-like swellings, has been described in most organs, and if not treated may lead to fibrosis and dysfunction of the involved organs [Citation1].
Recommendations regarding the nomenclature of IgG4-RD [Citation2] and an international consensus statement on the pathology and diagnosis of the disease have recently been established [Citation3]. The diagnosis is based on the characteristic histopathological findings, an increased number of IgG4 + PCs in the tissue, and an elevated IgG4+/IgG + PC ratio (>0.4) (Supplementary Table SI, available online at http://www.informahealthcare.com) in combination with compatible clinical symptoms and imaging data [Citation1,Citation3]. Elevated IgG4 concentrations in serum, observed in 50–70% of patients, may support the diagnosis [Citation4–6].
Salivary and lacrimal glands are among the sites that may be involved in IgG4-RD [Citation1,Citation7], and the disease displays both clinical and histopathological similarities with primary Sjögren’s syndrome (pSS). The patients with IgG4-related sialadenitis may have continuous or recurrent painless swelling of the parotid and submandibular glands with mild symptoms of dry mouth, hypergammaglobulinemia, and a minor salivary gland biopsy may reveal lymphocytic infiltration as in patients with pSS.
The association with lymphoma development has also been described in both diseases. In pSS, an increased risk of B-cell lymphoma is well established. These lymphomas are typically of the extranodal marginal zone of mucosa-associated lymphoid tissue (MALT) type [Citation8], often situated in major salivary glands. In IgG4-RD there are emerging case reports of patients with lymphoma development and, similar to pSS, MALT lymphoma has been the most commonly reported lymphoma subtype. A majority of these cases have been diagnosed in ocular adnexa of patients with IgG4-related dacryoadenitis [Citation9–12], but other sites and lymphoma subtypes have also been reported [Citation13,Citation14]. A frequent finding in these lymphomas has been a varying infiltration of IgG4 + PCs in the lymphoma tissue, and it has been proposed that the lymphomas may arise in the setting of IgG4-RD. However, it has been described that cancer tissue also in patients without other signs of IgG4-RD may be infiltrated by IgG4 + PCs to various degrees [Citation15–17]. Regarding the presence of IgG4 + PCs in lymphoma tissue in patients without IgG4-RD, the information is very limited, but one study indicates low occurrence [Citation9].
In this study, we retrospectively examined immunostains for IgG4 + PCs and histopathological features of IgG4-RD in the lymphoma tissue of a large Swedish population-based cohort of patients with pSS diagnosis complicated by lymphoma development. The aim was to identify the proportion of patients possibly misdiagnosed as pSS instead of IgG4-RD in this setting, to describe lymphoma characteristics in these patients, and to estimate the occurrence of IgG4 + PCs in lymphoma tissue in patients diagnosed with pSS.
Methods
Study population
A national population-based cohort of individuals with an International Classification of Diseases (ICD) diagnosis code for ‘Sjögren’s syndrome/sicca syndrome’ was identified using the Swedish Patient Register (includes inpatient care since 1964 and outpatient, non-primary care since 2001) 1964–2007. Through linkage with the Cancer Register (covers>95% of all incident cancer in Sweden) [Citation18], we identified SS patients diagnosed with a lymphoma or myeloma at ≥18 years of age 1990–2007 (n = 224) (ICD-codes in online Supplementary Table SII, available online at http://www.informahealthcare.com). As the diagnosis code for SS has not been used for pSS exclusively, the basis for the SS diagnosis was evaluated from collected medical records of all patients (LV). All available information from the patients’ medical records, laboratory test results, and pathology reports was extracted using an extensive and detailed questionnaire.
The pSS diagnoses were validated according to the 2002 American-European Consensus Group (AECG) criteria for pSS [Citation19]. Patients could be included in the study if they: 1) fulfilled the AECG criteria; or 2) did not fulfill the AECG criteria, but the available information in the medical records provided by the treating physician supported the diagnosis of pSS, and there was no other explanation for the SS-diagnosis code than pSS. The patients in Group 2 typically were not fully examined according to the AECG criteria or this information could not be found in the medical records, but they could fulfill other classification criteria in use during the study period. The lymphoma tissues of these patients were reviewed by a hematopathologist (CS) to confirm the lymphoma diagnosis.
Patients with a clear explanation for the SS-diagnosis code other than pSS (e.g., secondary SS, sicca after radiation therapy) and no support for a pSS diagnosis were excluded from further analyses.
In this study, we finally included all patients with a diagnosis of pSS (Groups 1 and 2 above), a confirmed lymphoma by review and available lymphoma biopsy materials for further analyses (n = 79). Of the 79 patients, 44 fulfilled the AECG criteria for pSS. Ethical approval was obtained from the Regional Ethical Review Board, Uppsala, Sweden.
Histopathology and immunohistochemistry
Lymphoma subtypes were classified according to the latest World Health Organization (WHO) classification [Citation20] after additional immunostainings if needed. All lymphoma tissues were immunostained for IgG4 (clone HP6025, Genetex, CA, USA). Specimens with identified IgG4 + PCs were investigated for other histopathological features of IgG4-RD. Further stainings for IgG and CD138 as a PC marker were performed in tissues with signs of inflammation, storiform fibrosis and >10 IgG4 + PCs/high power field (HPF). In cases with features of IgG4-RD in the lymphoma tissue, other archived tissue specimens from the same patient were collected and examined for histopathological features of IgG4-RD and IgG4+, IgG+, CD138 + PCs. Additionally, all available minor salivary gland biopsies (n = 11) obtained as part of the initial pSS investigation of these 79 patients were stained for IgG4 + PCs. Sections of tonsil were used as positive controls.
LV and CS evaluated the histopathology specimens and immunostains independently or together to achieve consensus, using a light microscope with a 40× field objective. The counting of IgG4+, IgG + and CD138 + PCs was performed in the three HPF with the highest density of stained cells, and the average numbers were calculated. Definitions of IgG4-RD were based on the consensus statement on the pathology of IgG4-RD, including the categorization of histological features ‘probable’ or ‘highly suggestive’ of IgG4-RD [Citation3] (Supplementary Table SI).
Results
Of the 79 cases with lymphoma and a pSS diagnosis, one (1.3%) had probable histological features of IgG4-RD in the lymphoma biopsy material from the submandibular gland (, ). This patient also had histological findings highly suggestive of IgG4-RD in a lung biopsy () performed five years before lymphoma diagnosis, and review of the medical records revealed that the patient did not formally fulfill the AECG criteria for pSS, but instead had a medical history compatible with IgG4-RD. Altogether this patient fulfilled the proposed criteria for IgG4-RD and is described in more detail below.
Figure 1. Unspecified low-grade B-cell lymphoma with features of IgG4-related disease in the resected submandibular gland. (a) Abundant infiltration by small lymphocytic and plasma cells, and features of storiform fibrosis (hematoxylin and eosin staining, magnification ×100). (b) IgG4+ plasma cells located in the periphery of the lymphoma tissue (magnification ×400). (c) IgG + plasma cells (magnification ×400). (d) CD138+ plasma cells (magnification ×400).
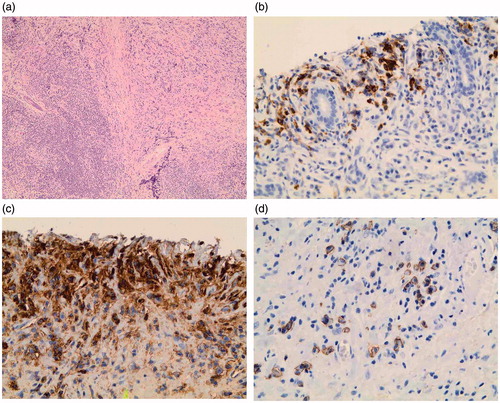
Figure 2. The lung tissue biopsied before lymphoma diagnosis shows dense lymphoplasmacytic infiltration rich in IgG4 + plasma cells, germinal center, and storiform fibrosis (magnification ×100).
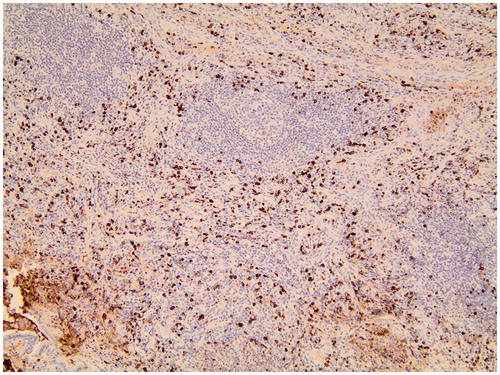
Table 1. IgG4 + plasma cells in the lymphoma tissue of 79 patients with an initial primary Sjögren’s syndrome diagnosis.
In another six lymphoma cases, occasional IgG4 + PCs (<10 cells/HPF) were observed (). Of these, two were situated in lymph nodes, two in parotid glands and one each in bone marrow and stomach. None of them had any other histopathological features of IgG4-RD in the lymphoma tissue. Five of these six patients fulfilled the AECG criteria for pSS, and all six patients had one or more extraglandular pSS manifestations (Raynaud, arthritis/arthralgia, rash, skin vasculitis, alveolitis, lymphadenopathy). Altogether, none of the 44 patients who fulfilled the AECG criteria for pSS had any specific findings for IgG4-RD in the lymphoma tissue, and similarly, no specific IgG4-RD findings and no IgG4 + PCs were found in the available minor salivary gland biopsies from 11 of these patients.
Except for salivary gland and lymph node swellings no other typical clinical manifestations of IgG4-RD (e.g., type 1 autoimmune pancreatitis or retroperitoneal fibrosis) were reported in the medical records of these 79 patients.
The case with findings of IgG4-RD
This was a 60-year-old man with a recently diagnosed biopsy-verified idiopathic pulmonary fibrosis who was referred to a rheumatologist in 1995 with a six-month history of arthralgia and sicca symptoms. Schirmer’s test showed reduced tear flow in both eyes (3 and 4 mm/5 min) and the unstimulated salivary flow rate was decreased (0.3 ml/15 min). Laboratory tests showed mild anemia, elevated ESR (>90 mm/h), increased plasma-IgG levels of 38.9 g/l (reference 6–16) and IgA 4.7 g/l (reference 0.7–3.7), a positive antinuclear antibody (ANA) test without positivity for specific antibodies, and no rheumatoid factor. No diagnostic minor salivary gland biopsy was performed. A diagnosis of suspected pSS was made by the treating physician, and no specific treatment was initiated. In autumn 1999, the patient noticed a rapidly growing lump in the right jaw angle. Clinical examination and computer tomography revealed enlargement of the right submandibular gland and a left submandibular lymph node. Part of the right submandibular gland was resected, and a diagnosis of a low-grade B-cell lymphoma was made with involvement also of the bone marrow. The remainder of the physical and radiographic examination was unremarkable, and no immediate lymphoma treatment was regarded necessary. The lung fibrosis progressed and oxygen therapy and corticosteroids (prednisone 15 mg/day) were initiated in autumn 2000 by a pulmonologist. Additionally, cyclophosphamide was given for six months, followed by azathioprine, both without effect, and the patient died three months later from respiratory failure caused by the lung fibrosis. There were no signs of progress of the lymphoma.
Evaluation of archived biopsies of the case with IgG4-RD
Review of the lymphoma tissue and the original slides of immunohistochemistry from the right submandibular gland from 1999 showed that the tumor cells expressed the B-cell markers CD20, CD79a, CD23 and monoclonal kappa light chain, but were negative for CD5 and CD10. Although the immunohistochemistry was consistent with a diagnosis of MALT lymphoma, the morphology with an extensive patchy fibrosis of the gland was not. Therefore, the final lymphoma diagnosis at review remained unspecified low-grade B-cell lymphoma.
The further evaluation of the submandibular gland showed storiform fibrosis, dense lymphoplasmacytic infiltration, and remnants of germinal centers (). The mean value of IgG4 + PCs/HPF was 60 (range 40–70) (). Immunostainings showed probable features of IgG4-RD, with an IgG4+/IgG + ratio of 0.62, and an IgG4+/CD138 + ratio of 0.60 (). The IgG4+, as the IgG+, and CD138 + PCs were located in the periphery of the sections outside the lymphoma tissue.
The surgical specimen of the lung tissue from 1995 initially diagnosed as idiopathic pulmonary fibrosis was available for further analyses. The review of the lung tissue biopsy was histologically highly suggestive of IgG4-RD. Hematoxylin and eosin staining showed dense lymphoplasmacytic infiltration with areas of storiform fibrosis. The mean value of IgG4 + PCs/HPF was 167 (range 120–200) (), the IgG4+/IgG + PC ratio was 0.92, and the IgG4+/CD138 + PC ratio was 0.94.
Discussion
In this population-based study of 79 lymphoma patients with pSS diagnosis (44 fulfilling and 35 not fulfilling the AECG criteria for pSS), we found seven cases with infiltration by IgG4 + PCs (8.9%) in the lymphoma tissue, but only one patient fulfilling the histological criteria for IgG4-RD [Citation3]. Histopathological findings of IgG4-RD were present both in the submandibular gland with co-existing lymphoma and also in the previous surgical lung tissue biopsy without lymphoma.
This patient was thus, retrospectively evaluated, misdiagnosed as suspected pSS instead of IgG4-RD. At review he did not formally fulfill the AECG criteria for pSS. He had ocular and oral sicca symptoms confirmed by objective methods, was ANA positive, but had no anti-SSA/Ro52 antibodies, and no salivary gland biopsy had been performed at diagnosis in 1995. In the AECG criteria anti-SSA/Ro and/or anti-SSB/La antibodies and/or a positive biopsy are required. He did, however, fulfill the European classification criteria for pSS which were in use during the 1990s, as symptoms and signs of sicca are sufficient to be classified as pSS according to these criteria [Citation21]. He also had no other explanation for the SS-diagnosis code than pSS at medical chart review, and filled the requisites to be included in this study (Group 2).
In the remaining patients, no or sporadic IgG4 + PCs without specific histopathological findings for IgG4-RD were detected in the lymphoma tissues.
To be noted, in all patients strictly fulfilling the AECG criteria for pSS no specific findings for IgG4-RD were identified neither in the lymphoma specimens nor in the 11 available diagnostic minor salivary glands. Our results thus suggest that infiltration by IgG4 + PCs can be found in the lymphoma tissues in patients with a pSS diagnosis, but underlying misdiagnosed IgG4-RD with other specific histopathological features, and clinical manifestations of IgG4-RD is rare (1.3%) in these patients.
Although there are no other studies of features of IgG4-RD in lymphomas in patients with pSS, some studies have searched for undiagnosed IgG4-RD in patients with pSS or sicca symptoms by other approaches. In one study within the Sjögren’s International Collaborative Clinical Alliance (SICCA) research registry of patients with sicca symptoms, one of 2594 labial salivary gland biopsies had histopathological findings consistent with IgG4-RD [Citation22]. This patient also had other findings supporting a diagnosis of systemic IgG4-RD. A limitation is that staining for IgG4 was not performed on all salivary gland biopsies. In a second study, 17 of 55 labial salivary gland biopsies from patients with xerostomia showed positive immunostaining for IgG4 (defined as IgG4+/IgG + PC ratio ≥0.4), including four patients fulfilling the AECG criteria for pSS, but it remains unclear if other histopathological features of IgG4-RD were present in these patients [Citation23]. In a third study, serum concentrations of IgG4 were investigated in 133 patients with pSS [Citation24], and increased IgG4 levels were present in 7.5% of the patients, but were not significantly different from controls with rheumatoid arthritis and systemic lupus erythematosus. IgG4 + PCs were detected in four of six minor salivary gland biopsies from pSS patients with elevated serum IgG4 and in none of four biopsies from patients with normal serum IgG4, but the authors could not verify whether the high-IgG4 group represented misclassified IgG4-RD patients. Altogether, these studies indicate that undiagnosed IgG4-RD seems to be rare among patients with sicca symptoms or pSS in these US-European settings, but epidemiology data is still too scarce to allow comparisons with, e.g., Japan or other Asian countries.
We are aware of one other study reporting the expression of IgG4 in lymphoma tissue in patients without underlying IgG4-RD [Citation9]. In this study, a high infiltration by IgG4 + PCs was present in three lymphomas in ocular adnexa of patients with known IgG4-related dacryoadenitis. However, in 16 patients with ocular adnexal lymphomas and 25 gastric MALT lymphomas without suspicion of IgG4-RD, IgG4 + PCs were ‘absent or sparse’, a finding in line with our results.
We used the criteria from the consensus statement on the pathology of IgG4-RD from 2012 to define the findings in the investigated tissues [Citation3]. The findings in the submandibular gland with lymphoma fulfilled all requirements except the number of required IgG4 + PCs (>100/HPF in salivary gland) to be classified as histologically highly suggestive of IgG4-RD and were therefore instead categorized as probable histological features of IgG4-RD. The proposed cutoff for IgG4 + PCs varies from organ to organ (>10–>200/HPF), and whether the cutoff at a site with a simultaneous lymphoma could be lower than in a solitary sialadenitis was not discussed in the consensus document, but it is acknowledged that the extent of fibrosis may affect the cutoff point. The findings in the lung tissue (>50 IgG4 + PCs/HPF required for surgical biopsy) fulfilled all criteria of ‘histologically highly suggestive of IgG4-RD.’
The current knowledge of the clinical picture of IgG4-RD is rapidly expanding, and our patient had a medical history well in line with what is described in IgG4-RD. So far, it is reported that IgG4-RD mainly affects middle-aged to elderly males. Common early symptoms are arthralgia, atopic symptoms, eosinophilia, lymphadenopathy, swelling of tear and salivary (particularly submandibular) glands, usually with mild sicca symptoms. Swelling of the submandibular salivary glands is described to be more common in IgG4-RD than in pSS [Citation7,Citation25]. Typical laboratory findings except elevated serum IgG4 levels are hypergammaglobulinemia, and about 16–20% are positive for ANA or rheumatoid factor, but are usually negative for anti-SSA and anti-SSB [Citation25–27].
Other specific organs to be more frequently involved are the pancreas, biliary tree, kidneys, thyroid gland, lungs, and aorta [Citation1,Citation7], often with a presence of tumor-like swellings in affected organs, and it is not uncommon that a malignant disease is initially suspected.
Observations in small case series suggest that lung involvement in IgG4-RD occurs in 12–54% of patients. IgG4-related lung disease may manifest as solid lung nodules, ground-glass opacities, lung infiltrates, an interstitial lung disease with honeycombing and fibrosis, bronchiectasis, bronchial thickening, central airway stenosis, nodular pleural lesions and pleural effusion [Citation1,Citation28,Citation29]. The fibro-inflammatory lung disease may progress to severe respiratory insufficiency and lead to death as in the case we describe.
Corticosteroids are the first-line treatment for most patients with IgG4-RD with starting doses Prednisone of 30–40 mg/day, and gradual tapering to maintenance doses of 2.5–10 mg/day [Citation1,Citation30], and can be effective as the only therapy also in IgG4-related lung disease. A poor response to corticosteroids on the lung symptoms was noted in our case, but could have been due to an inadequate dose or the advanced fibrotic lung disease already when the therapy was started.
Case reports suggest effectiveness also of other immunomodulatory drugs, including mycophenolate mofetil, methotrexate [Citation31], cyclosporine, azathioprine [Citation32], and cyclophosphamide [Citation33]. Recent studies suggest that B-cell depletion may induce a prompt clinical response in IgG4-RD [Citation34,Citation35], and new international guidelines have recently been proposed for the management of the disease [Citation36].
The strengths of this study are the identification of pSS patients with lymphoma from nationwide health registers with high coverage combined with verification of diagnoses from chart review and review of lymphoma tissues, detailed clinical data from medical records, additional immunostainings, and evaluation for IgG4-RD in lymphoma tissues from all the patients performed in agreement with the recent consensus statement [Citation3].
Limitations include the retrospective design with in some cases incomplete information in the medical records and the inconsistency of how pSS has been diagnosed in different time periods. During the study period, a number of different classification criteria for pSS were in use, mirroring the difficulties to find consensus about the classification of pSS. We choose to use the currently widespread AECG classification from 2002 as our criteria-based definition of pSS. This allowed us to identify a well defined group of patients of relevance to contemporary pSS patients, although a disadvantage of using the criteria from 2002 was that not all patients were fully examined according to these criteria. To be noted, the patient with IgG4-RD did not formally fulfill the AECG criteria. This observation as well as a recent report [Citation37], rather support that IgG4-RD should be looked for in patients not strictly fulfilling stringent pSS classification criteria.
Another limitation is the lack of information about serum IgG4 to support the IgG4-RD diagnosis, as IgG4 was not measured in patients during the study period (before IgG4-RD was described). However, as serum IgG4 is present in only a proportion of patients [Citation5] and is not a part of the golden standard for diagnosis of IgG4-RD [Citation3], this information would not have changed the results of the study.
We also cannot exclude that there may be patients with IgG4-RD in the studied population without features of IgG4-RD in the lymphoma tissue but only in other organs, and therefore not detected by the approach of this study.
In conclusion, this study suggests that presence of IgG4 + PCs and histopathological findings of IgG4-RD in lymphoma tissue in patients with an initial pSS diagnosis are rare, but if present may be associated with an underlying IgG4-RD. The identified case also highlights that IgG4-RD may co-exist with lymphoma in the same tissue. Pathologists and oncologists should pay attention to uncommon lymphoma histopathological features with intense inflammation, rich infiltration by PCs and storiform fibrosis, as it may indicate underlying IgG4-RD, which is manageable with immunomodulators.
Vasaitis_Supplementary_Table_I.docx
Download MS Word (13.3 KB)Acknowledgments
The work was supported by the ALF funding from County Council of Uppsala, Sweden, the Lions Cancer Research Foundation, Uppsala University Hospital, Uppsala, Sweden, the Swedish Rheumatism Association, the Swedish Society of Medicine, the Gustav V:s 80 year foundation, and scholarships from Agnes & Mac Rudbergs and Gustav Prims Reumatikerfond foundations. The funders had no impact on the performance of the study.
We sincerely thank Ass. Prof. Fredrik Granath, Prof. Johan Askling and Ass. Prof. Karin Ekström-Smedby at the Clinical Epidemiology Unit, Karolinska Institutet, Stockholm, Sweden for helpful cooperation in particular in register-associated matters.
Disclosure statement
The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
References
- Vasaitis L. IgG4-related disease: a relatively new concept for clinicians. Eur J Intern Med 2016;27:1–9.
- Stone JH, Khosroshahi A, Deshpande V, Chan JK, Heathcote JG, Aalberse R, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheumatol 2012;64:3061–7.
- Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 2012;25:1181–92.
- Sah RP, Chari ST. Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Curr Opin Rheumatol 2011;23:108–13.
- Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis 2015;74:14–8.
- Wallace ZS, Deshpande V, Mattoo H, Mahajan VS, Kulikova M, Pillai S, et al. IgG4-related disease: clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol 2015;67:2466–75.
- Brito-Zeron P, Ramos-Casals M, Bosch X, Stone JH. The clinical spectrum of IgG4-related disease. Autoimmun Rev 2014;13:1203–10.
- Ekstrom Smedby K, Vajdic CM, Falster M, Engels EA, Martinez-Maza O, Turner J, et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the interlymph consortium. Blood 2008;111:4029–38.
- Cheuk W, Yuen HK, Chan AC, Shih LY, Kuo TT, Ma MW, et al. Ocular adnexal lymphoma associated with IgG4+ chronic sclerosing dacryoadenitis: a previously undescribed complication of IgG4-related sclerosing disease. Am J Surg Pathol 2008;32:1159–67.
- Kubota T, Moritani S, Yoshino T, Nagai H, Terasaki H. Ocular adnexal marginal zone B cell lymphoma infiltrated by IgG4-positive plasma cells. J Clin Pathol 2010;63:1059–65.
- Nakayama R, Matsumoto Y, Horiike S, Kobayashi S, Nakao R, Nagoshi H, et al. Close pathogenetic relationship between ocular immunoglobulin G4-related disease (IgG4-RD) and ocular adnexal mucosa-associated lymphoid tissue (MALT) lymphoma. Leuk Lymphoma 2014;55:1198–202.
- Naresh KN, Barwick T, Karadimitris A. IgG4 positive mucosa associated lymphoid tissue lymphoma of the orbit-lesson of the month. Histopathology 2014;65:718–21.
- Miki K, Orita Y, Sato Y, Sugitani I, Noyama M, Fuji S, et al. Mucosa-associated lymphoid tissue lymphoma of the thyroid with abundant IgG4-positive plasma cells. Auris Nasus Larynx 2013;40:587–90.
- Venkataraman G, Rizzo KA, Chavez JJ, Streubel B, Raffeld M, Jaffe ES, et al. Marginal zone lymphomas involving meningeal dura: possible link to IgG4-related diseases. Mod Pathol 2011;24:355–66.
- Zhang L, Notohara K, Levy MJ, Chari ST, Smyrk TC. IgG4-positive plasma cell infiltration in the diagnosis of autoimmune pancreatitis. Mod Pathol 2007;20:23–8.
- Deshpande V, Chicano S, Finkelberg D, Selig MK, Mino-Kenudson M, Brugge WR, et al. Autoimmune pancreatitis: a systemic immune complex mediated disease. Am J Surg Pathol 2006;30:1537–45.
- Strehl JD, Hartmann A, Agaimy A. Numerous IgG4-positive plasma cells are ubiquitous in diverse localised non-specific chronic inflammatory conditions and need to be distinguished from IgG4-related systemic disorders. J Clin Pathol 2011;64:237–43.
- Barlow L, Westergren K, Holmberg L, Talback M. The completeness of the Swedish cancer register: a sample survey for year 1998. Acta Oncol 2009;48:27–33.
- Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, et al. Classification criteria for Sjögren's syndrome: a revised version of the European criteria proposed by the American-European consensus group. Ann Rheum Dis 2002;61:554–8.
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: ARC Press; 2008.
- Vitali C, Bombardieri S, Moutsopoulos HM, Balestrieri G, Bencivelli W, Bernstein RM, et al. Preliminary criteria for the classification of Sjögren's syndrome. Results of a prospective concerted action supported by the European community. Arthritis Rheum 1993;36:340–7.
- Baer AN, Gourin CG, Westra WH, Cox DP, Greenspan JS, Daniels TE. Rare diagnosis of IgG4-related systemic disease by lip biopsy in an international Sjögren syndrome registry. Oral Surg Oral Med Oral Pathol Oral Radiol 2013;115:e34–9.
- Hermet M, Andre M, Kemeny JL, Le Guenno G, Dechelotte P, Guettrot-Imbert G, et al. Is IgG4-related disease a cause of xerostomia? a cohort study of 60 patients. Int J Rheumatol 2012;2012:303506.
- Mavragani CP, Fragoulis GE, Rontogianni D, Kanariou M, Moutsopoulos HM. Elevated IgG4 serum levels among primary Sjögren's syndrome patients: do they unmask underlying IgG4-related disease? Arthritis Care Res (Hoboken) 2014;66:773–7.
- Takahashi H, Yamamoto M, Tabeya T, Suzuki C, Naishiro Y, Shinomura Y, et al. The immunobiology and clinical characteristics of IgG4 related diseases. J Autoimmun 2012;39:93–6.
- Yamamoto M, Yajima H, Takahashi H, Yokoyama Y, Ishigami K, Shimizu Y, et al. Everyday clinical practice in IgG4-related dacryoadenitis and/or sialadenitis: results from the SMART database. Mod Rheumatol 2015;25:199–204.
- Inoue D, Yoshida K, Yoneda N, Ozaki K, Matsubara T, Nagai K, et al. IgG4-related disease: dataset of 235 consecutive patients. Medicine (Baltimore) 2015;94:e680.
- Inoue D, Zen Y, Abo H, Gabata T, Demachi H, Kobayashi T, et al. Immunoglobulin G4-related lung disease: CT findings with pathologic correlations. Radiology 2009;251:260–70.
- Matsui S, Taki H, Shinoda K, Suzuki K, Hayashi R, Tobe K, et al. Respiratory involvement in IgG4-related Mikulicz's disease. Mod Rheumatol 2012;22:31–9.
- Kamisawa T, Shimosegawa T, Okazaki K, Nishino T, Watanabe H, Kanno A, et al. Standard steroid treatment for autoimmune pancreatitis. Gut 2009;58:1504–7.
- Della-Torre E, Campochiaro C, Bozzolo EP, Dagna L, Scotti R, Nicoletti R, et al. Methotrexate for maintenance of remission in IgG4-related disease. Rheumatology (Oxford) 2015;54:1934–6.
- Hart PA, Kamisawa T, Brugge WR, Chung JB, Culver EL, Czako L, et al. Long-term outcomes of autoimmune pancreatitis: a multicentre, international analysis. Gut 2013;62:1771–6.
- Kanda H, Koya J, Uozaki H, Tateishi S, Sato K, Hagino N, et al. Membranous nephropathy with repeated flares in IgG4-related disease. Clin Kidney J 2013;6:204–7.
- Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore) 2012;91:57–66.
- Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis 2015;74:1171–7.
- Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, et al. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol 2015;67:1688–99.
- Moriyama M, Tanaka A, Maehara T, Furukawa S, Nakashima H, Nakamura S. T helper subsets in Sjögren's syndrome and IgG4-related dacryoadenitis and sialoadenitis: a critical review. J Autoimmun 2014;51:81–8.